一种用于检测棕榈疫霉菌的引物对及其检测方法与流程
本发明涉及一种用于检测棕榈疫霉菌的引物对及其检测方法,属于作物病害检测、鉴定及防治技术领域。
背景技术:
棕榈疫霉菌(Phytophthora palmivora)属于卵菌门(Oomycota)、卵菌纲(Oomycetes)、霜霉目(Peronosporales)、腐霉科(Pythiaceae)、疫霉属(Phytophthora)。棕榈疫霉菌是农业生产上的一种危害极为严重的病原菌。棕榈疫霉菌寄主范围十分广泛,能侵染包括果树、林木、花卉等在内的百余种植物,其可侵染植物根、茎、叶、花及果实等各个部位,在气候潮湿的条件下发病更为严重。由其侵染引起的植物病害分布广泛,常给农业作物(如菠萝、番木瓜、榴莲、木薯和橡胶树等)造成重大损失,如由棕榈疫霉菌侵染引起的榴莲根茎腐烂病是一种危害榴莲生产的重要病害,严重年份果园发病达70%,其中果实上的发病率达40%-50%,受棕榈疫霉侵染的榴莲果实果皮变褐,果肉颜色呈深黄色,香味变淡,食用性降低。目前对棕榈疫霉菌引起的病害还没有快速有效的防治措施,控制该病菌病害最有效的途径就是加强检疫,防止病原菌的传播和阻碍其扩散方式。新西兰和澳大利亚曾分别对泰国榴莲进行风险评估,新西兰将榴莲上的棕榈疫霉列为控制性有害生物,澳大利亚把榴莲上的棕榈疫霉视为有风险的潜在有害生物。在我国,棕榈疫霉虽然未列入检疫性有害生物名录,但在引进种子、苗木检疫中,棕榈疫霉往往被列为禁止携带的有害生物。所以建立棕榈疫霉菌的快速分子检测技术对于保护我国农林业生产的安全及棕榈疫霉菌病害的早期控制具有重要意义。
棕榈疫霉菌传统的检测方法是采用选择性培养基从土壤、植物组织或水体中分离菌株,再对这些菌株的形态等进行鉴定,来确定是否存在病原菌,或者用肉眼和借助显微镜技术对发病症状进行判定。传统的检测方法不仅费时、准确度低,而且要求检测人员要有丰富的经验,最重要一点是易遗漏潜育期或隐症的病害,以致延误病害的防治,导致病害的暴发,因此传统病原学检测方法已不能满足现代植物病理学研究的需要。
随着分子生物学技术的发展,应用PCR技术对病原菌进行特异、灵敏的快速分子检测的成功例子已越来越多。国内外研究人员主要利用rDNA/ITS序列设计特异引物来对植物病原菌进行快速检测、鉴定。然而疫霉属卵菌的分子检测技术研究结果表明,rDNA/ITS在疫霉属的近缘种之间变化很小,从ITS序列上设计引物很难将两个相似度高的种区分开;因此要对疫霉属病原菌进行高灵敏性和特异性检测,应从疫霉菌其它基因上设计引物进行检测。已有研究表明,疫霉属中Ras 家族相关的Ypt1编码基因含有多个外显子和内含子,外显子具有保守性,而这些内含子在不同种之间具有多变性,非常适合于设计引物进行疫霉属卵菌近缘种的特异性分子检测、鉴定。目前尚无针对Ypt1基因设计的特异性引物检测棕榈疫霉菌的报道。
本发明根据棕榈疫霉菌Ypt1基因序列信息设计特异引物,不仅能特异鉴定该病原菌,而且可直接用于发病组织和土壤中病菌的检测,这将对棕榈疫霉菌病害的早期诊断具有重要意义。
技术实现要素:
本发明的目的是针对现有技术中对棕榈疫霉菌检测和鉴定主要基于形态学特征,方法耗时长、程序繁琐、经验性强、准确度低,难以做到对病害发生的及时监测和控制病原菌的传播、流行的问题,以及现有分子检测中rDNA-ITS序列差异很小,以ITS为靶标设计的引物难以将一些近缘种的病原菌区分开的技术缺欠,提供了棕榈疫霉菌特异性PCR检测引物对及其检测方法;利用本发明所述的PCR检测引物对和检测方法检测棕榈疫霉菌准确性高、特异性强、灵敏度高、易于操作、检测时间短且结果可靠。
为实现上述目的,本发明采用如下技术方案:
1. 通过测定棕榈疫霉菌(Phytophthora palmivora)和其它疫霉菌(Phytophthora spp)的Ypt1基因序列,对疫霉属不同种间Ypt1基因序列进行比对分析,设计出一种用于检测棕榈疫霉菌的引物对,该引物对特异性强,其核苷酸序列为:
上游引物PPF:5'- GCAAAAGTGTTGATGGTTTGG -3',
下游引物PPR:5'- CTTGACTAGGAACGCCTGGA -3';
该引物对的扩增产物大小约为220bp。
2. 本发明的另一目的在于提供一种利用上述引物对检测棕榈疫霉菌检测的方法,包括以下步骤:
(1)提取待测样品基因组DNA。
用于检测病原菌纯培养物时,采用CTAB 法提取基因组DNA,具体方法如下:取少量菌丝粉于1.5mL离心管中(菌丝粉刚盖过半圆形底部为宜),加入900 µL 2%CTAB(十六烷基三甲基溴化铵)提取液(2% CTAB;100 mmol/L Tris-HCl, pH 8.0;20 mmol/L EDTA,pH8.0;1.4 mol/L NaCl)和90 µL SDS(十二烷基苯磺酸钠)【注:CTAB,SDS需60℃预热】,使用振荡器振荡混匀,60℃水浴1h(DNA释放至缓冲液中),12000 r·min-1离心15 min;取上清液700 µL,加等体积酚、氯仿、异戊醇(25:24:1),轻轻振荡混匀,12000 r·min-1离心9 min;取上清液500 µL,加入等体积氯仿再抽提一次,12000 r·min-1离心5 min;取上清液350 µL,加入1/10体积3 mol·L-1 NaAc和2倍体积无水乙醇,-20℃沉淀30 min,12000 r·min-1离心5 min;弃去上清液,加入700µL 冰70%乙醇进行洗涤(稍离心;倾掉上清液),在超净工作台上晾干无酒精味,加入30~60 µL TE(10 mmol/L Tris-HCl,0.1 mmol/L EDTA,pH 8.0) 溶液进行溶解,得到 DNA 溶液,用紫外分光光度计检测 DNA 浓度并稀释至100 ng/µL待用。
用于检测植物组织中存在棕榈疫霉菌时,采用NaOH快速裂解法提取DNA,具体过程如下:向每毫克植物组织中加入10µL 0.5 mol/L NaOH,在研钵中将组织充分磨碎成糊后转入1.5mL离心管中,12,000 rpm离心6 min,取上清液5 µl加入495µL 0.1 mol/L Tris-HCl(pH=8.0)混合均匀,取1.0 µL作为PCR模板进行扩增;
用于检测土壤样品中存在棕榈疫霉菌时,采用土壤DNA提取试剂盒,提取DNA。
(2)PCR扩增反应:以步骤(1)提取的DNA为模板,利用PPF/PPR这一对引物进行PCR扩增。PCR反应体系25 µL,包括2×Taq PCR Master Mix 12.5µL,10 µmol/L的PPF/PPR引物各1.0µL,DNA 模板1.0µL,用无菌超纯水补足至25 µL;扩增参数为:95 ℃预变性5 min,94 ℃变性30 s,60 ℃退火45 s,72 ℃延伸30 s,共35个循环,最后72 ℃延伸10 min。
(3)电泳检测:取步骤(2)的PCR扩增产物5.0 µL用1.5%琼脂糖凝胶进行电泳分离,4-5V/cm,电泳40min后经溴化乙锭染色于紫外灯下观察,根据扩增产物的有无及其片段大小对结果进行判断,如果能特异性地扩增出约220bp的产物,即可判断所述的检测样品中存在棕榈疫霉菌,否则所述的检测样品中未存在该菌。
本发明的显著优点
本发明与现有技术相比,具有以下的技术优势和有益效果:
1.特异性强、准确性高:本发明能准确地检测、鉴定出棕榈疫霉菌,只有棕榈疫霉菌的菌株能扩增出大小约为220bp的条带,而其它菌株均扩增不出条带;说明本发明的引物对具有很强的特异性。使用本发明的检测方法对不同地理来源的棕榈疫霉菌和携带棕榈疫霉菌的植物组织和土壤进行了测试验证,只有棕榈疫霉菌和携带该病菌的样品能特异性地扩增出一条220bp的电泳条带,说明本发明所设计的引物对能准确诊断出棕榈疫霉菌。
2.重复性好、灵敏度高:多次试验证实本发明具有很好的重复性,目标物DNA只需要100 pg/µL,就可以检测、鉴定出棕榈疫霉菌;
3.操作简便、快速:应用本发明方法,对待测样品基因组DNA进行提取、PCR扩增和常规的琼脂糖电泳后即可判定结果,整个检测过程采用DNA快速提取方法,操作简单,无需对病原菌进行分离培养,大大缩短了检测时间,一般整个检测过程可在6小时内完成。
附图说明
图1为本发明所述引物对的特异PCR扩增电泳图,图中:泳道M为2000bp Marker,泳道1-3为棕榈疫霉菌,泳道4-8分别为辣椒疫霉菌、晚疫病菌、大豆疫霉菌、芋疫霉菌和豇豆疫霉菌,泳道9为阴性对照;
图2为本发明引物对的灵敏性检测扩增电泳图,图中泳道M为2000bp Marker,泳道1为100 ng,泳道2为10 ng,泳道3为1 ng,泳道4为100 pg,泳道5为10 pg,泳道6为1 pg,泳道7为100 fg,泳道8为10 fg,泳道9为1 fg,泳道10-11为阴性对照,泳道12为阳性对照;
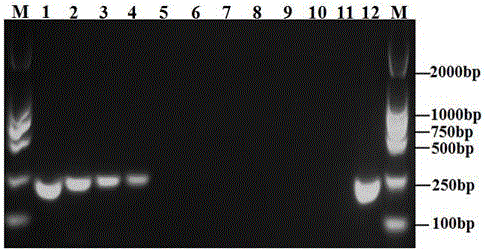
图3为本发明检测方法对番木瓜果实组织和土壤样品中棕榈疫霉菌检测结果电泳图,图中泳道M为2000bp Marker,泳道1为阴性对照,泳道2为阳性对照,泳道3为健康番木瓜果实,泳道4为人工接种发病的番木瓜果实,泳道5为高压灭菌土壤,泳道6-7为携带棕榈疫霉菌的土壤。
具体实施方式
以下结合具体实施例对本发明作进一步阐述, 但不用来限制本发明的范围。以下实施例均按照常规实验条件,或已发表相关文献中所述的操作技术规程,或按照厂商所建议的实验条件。
实施例 1:棕榈疫霉菌PCR检测引物对的设计及引物对的特异性验证
1.棕榈疫霉菌基因组DNA的提取
采用CTAB 法提取不同地区来源的棕榈疫霉菌基因组 DNA,具体方法如下:取少量菌丝粉于1.5 mL离心管中(菌丝粉刚盖过半圆形底部为宜),加入900 µL 2%CTAB(十六烷基三甲基溴化铵)提取液(2% CTAB;100 mmol/L Tris-HCl, pH 8.0;20 mmol/L EDTA,pH8.0;1.4 mol/L NaCl)和90 µL SDS(十二烷基苯磺酸钠)【注:CTAB,SDS需60℃预热】,使用振荡器振荡混匀,60℃水浴1h(DNA释放至缓冲液中),12000 r·min-1离心15 min;取上清液700 µL,加等体积酚、氯仿、异戊醇(25:24:1),轻轻振荡混匀,12000 r·min-1离心9 min;取上清液500 µL,加入等体积氯仿再抽提一次,12000 r·min-1离心5 min;取上清液350 µL,加入1/10体积3 mol·L-1 NaAc和2倍体积无水乙醇,-20℃沉淀30 min,12000 r·min-1离心5 min;弃去上清液,加入700µL 冰70%乙醇进行洗涤(稍离心;倾掉上清液),在超净工作台上晾干无酒精味,加入30~60 µL TE(10 mmol/L Tris-HCl,0.1 mmol/L EDTA,pH 8.0) 溶液进行溶解,得到 DNA 溶液,用紫外分光光度计检测 DNA 浓度并稀释至100 ng/µL待用。
2.棕榈疫霉菌检测靶标Ypt1基因扩增及测序
以Ypt1基因通用引物(ph1F:5'-CGACCATTGGCGTGGACTTT-3'和Yph2R:5'-ACGTTCTCGCAGGCGTATCT-3')对供试棕榈疫霉菌(P. palmivora)的Ypt1基因进行扩增,PCR反应体系25 µL,包括2×Taq PCR Master Mix(北京天根生化科技有限公司)12.5µL,10 µmol/L的ph1F / Yph2R引物各1.0µL,DNA 模板1.0µL,用无菌超纯水补足至25 µL。扩增反应程序为:94℃预变性5min;94℃变性1 min、58℃退火30 S、72℃延伸1 min,35个循环,最后72℃延伸10 min。将PCR扩增产物送至上海生工生物工程有限公司进行测序。
3.棕榈疫霉菌检测特异引物对的设计
将测序得到的棕榈疫霉菌(P. palmivora)的Ypt1基因序列与GenBank中疫霉属18个不同种的Ypt1基因序列进行同源性比较分析,根据棕榈疫霉菌与其它种间的差异位点(在BioEdit中比对),用Primer Primer5软件设计了棕榈疫霉菌(P. palmivora)的特异性引物,上游引物PPF:5'- GCAAAAGTGTTGAT- GGTTTGG -3',下游引物PPR:5'- CTTGACTAGGAACGCCTGGA -3',引物由上海生工生物工程有限公司合成。
4.棕榈疫霉菌PCR检测方法的建立及引物特异性PCR验证
在已设计特异引物对的基础上,通过PCR反应体系和扩增参数的优化,建立的棕榈疫霉菌PCR检测方法,PCR反应体系25 µL,包括2×Taq PCR Master Mix(北京天根生化科技有限公司)12.5µL,10 µmol/L的PPF/PPR引物各1.0µL,DNA 模板1.0µL,用无菌超纯水补足至25 µL。扩增反应程序为:94℃预变性5min;94℃变性1 min、60℃退火45 s、72℃延伸30 s,35个循环,最后72℃延伸10 min。以供试的棕榈疫霉菌及其它病原菌的基因组DNA为模板,采用已建立好的棕榈疫霉菌PCR检测扩增体系和扩增程序对棕榈疫霉菌引物对PPF/PPR进行特异性检验。取5 µL PCR产物进行1.5%琼脂糖电泳检测,经溴化乙锭染色后于紫外灯下观察,根据DNA条带的有无及大小对棕榈疫霉菌引物对的特异性进行验证。
5.引物特异性验证结果
PCR扩增结果表明,引物PPF/PPR可特异性地从供试的棕榈疫霉菌基因组DNA中扩增出大小约为220bp的条带(图1),而其它病原菌及阴性对照均无扩增条带。说明该对引物可以将棕榈疫霉菌与其它病原菌区分开来,具有种的特异性,可用于棕榈疫霉菌快速可靠的检测和鉴定。
实施例2:棕榈疫霉菌引物对灵敏度检测
用无菌超纯水对棕榈疫霉菌基因组DNA进行稀释,配制成10倍数量级的系列浓度备用。使用本发明所述引物对PPF/PPR对不同系列浓度的基因组DNA进行PCR扩增,评估该引物对的灵敏性,扩增反应体系和反应程序如下:PCR反应体系25 µL,包括2×Taq PCR Master Mix(北京天根生化科技有限公司)12.5µL,10 µmol/L的PPF/PPR引物各1.0µL,DNA模板1.0µL,用无菌超纯水补足至25 µL。扩增反应程序为:94℃预变性5min;94℃变性1 min、60℃退火45 S、72℃延伸1 min,35个循环,最后72℃延伸10 min。
检测灵敏度结果表明,当以本发明所述引物PPF/PPR进行常规PCR扩增时,反应灵敏度可以达到100pg(图2),说明本发明引物对具有较高的灵敏度。
实施例3:番木瓜果实发病组织中棕榈疫霉菌的检测
人工接种发病组织的获得:将番木瓜(品种:台农杂交5号)果实用70%的乙醇表面消毒,果实表面用无菌打孔器(Φ5mm)打深约5mm的孔,每孔内接种一块直径5mm、厚约2mm的棕榈疫霉菌丝块,用无菌湿棉团覆盖接种处。接种的果实置于25℃下保湿,待果实发病后,取发病组织备用。
发病果实组织基因组DNA的提取:采用NaOH快速裂解法提取DNA,具体过程如下:向每毫克植物组织中加入10µL 0.5 mol/L NaOH,在研钵中将组织充分磨碎成糊后转入1.5mL离心管中,12,000 rpm离心6 min,取上清液5 µl加入495µL 0.1 mol/L Tris-HCl(pH=8.0)混合均匀,取1.0 µL作为PCR模板进行扩增。
PCR扩增检测:利用本发明所述引物PPF/PPR进行PCR扩增。PCR反应体系25 µL,包括2×Taq PCR Master Mix(北京天根生化科技有限公司)12.5µL,10 µmol/L的PPF/PPR引物各1.0µL,DNA 模板1.0µL,用无菌超纯水补足至25 µL;扩增参数为:95 ℃预变性5 min,94 ℃变性30 s,60 ℃退火45 s,72 ℃延伸30 s,共35个循环,最后72 ℃延伸10 min。
检测结果:取PCR扩增产物5.0 µL用1.5%琼脂糖电泳分离,电压为4-5V/cm,电泳结束经溴化乙锭染色后于紫外灯下观察,根据扩增产物的有无及其片段大小对结果进行判断,如果能特异性地扩增出约220bp的产物,即可判断发病组织中带有棕榈疫霉菌。检测结果(图3)表明,人工接种发病的果实中可检测出棕榈疫霉菌,而健康组织及阴性对照则无特异性条带出现,说明该套技术能用于植物组织中棕榈疫霉菌的快速分子检测。
实施例4:土壤中棕榈疫霉菌的检测
携带棕榈疫霉菌土壤的制备:将供试的棕榈疫霉菌菌株培养于黑麦培养基平板上,待菌丝长满平板后,用适量的无菌水将平板中的菌丝及孢子囊洗下,双层纱布过滤去除菌丝,得到的滤液为棕榈疫霉菌孢子囊悬浮液,将孢子囊悬浮液与适量土壤混合,混合后的土壤在室温下风干,得到携带棕榈疫霉菌土壤。
带菌土壤基因组DNA的提取:采用Sigma公司的土壤DNA提取试剂盒(Sigma,DNB100,Soil DNA Isolation Kit)提取土壤中的总DNA,取1.0 µL作为PCR模板进行扩增。
PCR扩增检测:利用本发明所述引物对PPF/PPR进行PCR扩增。PCR反应体系25 µL,包括2×Taq PCR Master Mix(北京天根生化科技有限公司)12.5µL,10 µmol/L的PPF/PPR引物各1.0µL,DNA 模板1.0µL,用无菌超纯水补足至25 µL;扩增参数为:95 ℃预变性5 min,94 ℃变性30 s,60 ℃退火45 s,72 ℃延伸30 s,共35个循环,最后72 ℃延伸10 min。
检测结果:取PCR扩增产物5.0 µL用1.5%琼脂糖电泳分离,电压为4-5V/cm,电泳结束经溴化乙锭染色后于紫外灯下观察,根据扩增产物的有无及其片段大小对结果进行判断,如果能特异性地扩增出约220bp的产物,即可判断土壤样品中存在棕榈疫霉菌。检测结果(图3)表明,携带棕榈疫霉菌的土壤中均可检测出该菌,而高压灭菌的土壤及阴性对照则无特异性条带出现,说明该套技术能用于土壤中棕榈疫霉菌的快速分子检测。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
SEQUENCE LISTING
<110> 福建省农业科学院植物保护研究所
<120> 一种用于检测棕榈疫霉菌的引物对及其检测方法
<130> 4
<160> 4
<170> PatentIn version 3.3
<210> 1
<211> 21
<212> DNA
<213> PPF
<400> 1
gcaaaagtgt tgatggtttg g 21
<210> 2
<211> 20
<212> DNA
<213> PPR
<400> 2
cttgactagg aacgcctgga 20
<210> 3
<211> 20
<212> DNA
<213> ph1F
<400> 3
cgaccattgg cgtggacttt 20
<210> 4
<211> 20
<212> DNA
<213> Yph2R
<400> 4
acgttctcgc aggcgtatct 20
- 还没有人留言评论。精彩留言会获得点赞!
- 一种促使辣椒疫霉菌产生游动孢子囊的培养基的制作方法
- 四氟苯氧基烟碱胺类化合物、其制备方法及用作杀菌的用途的制作方法
- 一种芹菜为原料制作适宜恶疫霉菌产生孢子囊的液体培养基的制作方法
- 一种三七为原料制作适宜恶疫霉菌产生游动孢子的液体培养基的制作方法
- 来自辣椒疫霉菌的阿魏酸酯酶pcfae1及其编码基因与应用的制作方法
- 来自辣椒疫霉菌的阿魏酸酯酶pcfae2及其编码基因与应用的制作方法
- 来自辣椒疫霉菌的果胶裂解酶pcpel16及其编码基因与应用的制作方法
- 来自辣椒疫霉菌的果胶裂解酶pcpel20及其编码基因与应用的制作方法
- 来自辣椒疫霉菌的果胶裂解酶pcpel15及其编码基因与应用的制作方法
- 来自辣椒疫霉菌的果胶裂解酶pcpel18及其编码基因与应用的制作方法
- 一种荧光定量PCR技术检测意大利蜜蜂王浆主蛋白MRJP 2基因表达的方法与流程
- 一种用于对不同银杏进行基因分型的SNP引物及检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种基于荧光PCR检测ESR1基因突变的引物及探针的制造方法与工艺
- 转基因甜菜GTSB77品系特异性实时荧光PCR检测引物、探针、方法和试剂盒与流程
- 一种基于单核苷酸多态性的葡萄球菌种水平鉴定和菌株分型一体化方法与流程
- 一套毛花猕猴桃和中华猕猴桃物种间杂交子代的鉴定引物的制造方法与工艺
- 检测黄杉大小蠹的引物、试剂盒及检测方法与流程
- 环介导等温扩增技术检测铜绿假单胞菌的引物和试剂盒的制造方法与工艺
- 一种用于鉴定浙贝母的引物对及其应用的制造方法与工艺