一种唑菌胺酯新晶型及其制备方法与流程
本发明涉及一种唑菌胺酯新晶型及其制备方法。
[
背景技术:
]
唑菌胺酯有无定型和结晶型,结晶型唑菌胺酯为白色或浅米色无味结晶体,易溶于丙酮、乙酸乙酯、甲苯、二氯甲烷、环己酮、乙腈,可溶于甲醇、乙醇、异丙醇、正丁醇、正辛醇等有机溶剂,微溶于正庚烷,不溶于水(1.9mg/L,20℃)。
中国专利公开号CN101203136B公开了唑菌胺酯的四种晶型(晶型Ⅰ、晶型Ⅱ、晶型Ⅲ和晶型Ⅳ),晶型Ⅳ是其中最稳定的固态形式。晶型Ⅰ由熔融后的唑菌胺酯缓慢冷却后制备,熔点范围是55-56度。晶型Ⅱ熔点范围是57-58度。晶型Ⅲ熔点范围是59-60度。晶型Ⅳ熔点范围是62-72度,尤其是65-67度。
本发明的研究目的是从唑菌胺酯晶型研究入手,通过晶型筛选技术、晶型生物活性评价技术,在相同有效物质不同晶型状态层面上寻找、发现、开发唑菌胺酯的优势晶型物质状态,为从唑菌胺酯晶型物质基础上申请国家或国际的知识产权发明专利保护提供科学数据。
本发明成功制备了一种新的唑菌胺酯晶型,其在高湿度下的吸湿性明显低于已知唑菌胺酯晶型,可提高唑菌胺酯稳定性。
[
技术实现要素:
]
本发明的目的是制备得到一种新的唑菌胺酯晶型,其在高湿度下的吸湿性明显低于已知唑菌胺酯晶型,可提高唑菌胺酯稳定性。
本发明的唑菌胺酯晶型物质,具有如下特征:
1、粉末X射线衍射
仪器:锐影X射线衍射仪(荷兰帕纳科)
靶:Cu-Kα辐射
波长:
X-射线光管电压:45kV
X-射线光管电管流:40mA
步长:0.01313°
扫描速度:0.041683°/s
扫描范围:5°-40°
结果表明:在8.4116°、8.9640°、9.4669°、11.8177°、11.9942°、12.8544°、14.5789°、14.7938°、15.4163°、15.4940°、15.7852°、16.2994°、16.8570°、17.8786°、18.8565°、19.1538°、20.4913°、21.6443°、22.0675°、22.8191°、23.7159°、24.1817°、25.1631°、25.6891°、26.2381°、26.9101°、27.6011°、28.3907°、29.3377°、30.2081°、30.7281°、31.4032°、33.7657°、35.8967°、36.8632°、37.4886°、38.7344°处有特征峰。
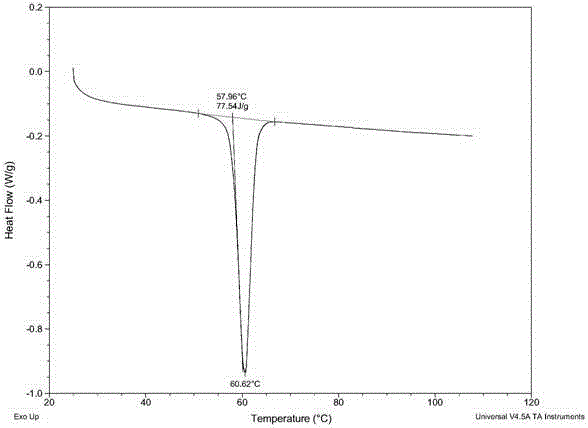
2、差示扫描量热法(DSC)
仪器:DSC Q2000差式扫描量热仪(美国,TA仪器)
温度范围:25℃-160℃
升温速度:10℃/min
结果表明:唑菌胺酯新晶型的熔融起始温度(onset temperature)在57.96℃。
3、热重分析法(TGA)
仪器:TGA Q500热重分析仪(美国,TA仪器)
温度范围:30℃-400℃
升温速度:10℃/min
结果表明:唑菌胺酯新晶型在30-110℃范围内失重0.23%。
本发明的另一目的是提供一种制备唑菌胺酯新晶型的方法。
将不同于本发明唑菌胺酯新晶型的其他唑菌胺酯晶型(中国专利公开号CN101203136B公开的晶型Ⅰ、晶型Ⅱ、晶型Ⅲ和晶型Ⅳ)在常温固体状态下溶于有机溶剂中;
在-80℃-0℃(优选为-80℃--24℃)下结晶,结晶时间为8-36小时;
过滤得到固体,在室温下干燥,即得唑菌胺酯新晶型。
所述有机溶剂为乙腈、二氯甲烷、氯乙烷、环己酮中的一种或两种以上的混合溶剂;优选为二氯甲烷、环己酮。
本发明中公开的唑菌胺酯新晶型与已有专利报道的唑菌胺酯晶型的粉末X射线衍射、DSC均不同,因此所述固体形态是一种完全不同于现有技术的唑菌胺酯的晶型形态。同时该新晶型在高湿度下的吸湿性明显低于已知唑菌胺酯晶型,可提高唑菌胺酯稳定性。
[附图说明]
图1是本发明唑菌胺酯新晶型的XRPD衍射图谱;
图2是本发明唑菌胺酯新晶型的DSC图;
图3是本发明唑菌胺酯新晶型的TGA图。
[具体实施方式]
检测方法
1、粉末X射线衍射
仪器:锐影X射线衍射仪(荷兰帕纳科)
靶:Cu-Kα辐射
波长:
X-射线光管电压:45kV
X-射线光管电管流:40mA
步长:0.01313°
扫描速度:0.041683°/s
扫描范围:5°-40°
结果表明:在8.4116°、8.9640°、9.4669°、11.8177°、11.9942°、12.8544°、14.5789°、14.7938°、15.4163°、15.4940°、15.7852°、16.2994°、16.8570°、17.8786°、18.8565°、19.1538°、20.4913°、21.6443°、22.0675°、22.8191°、23.7159°、24.1817°、25.1631°、25.6891°、26.2381°、26.9101°、27.6011°、28.3907°、29.3377°、30.2081°、30.7281°、31.4032°、33.7657°、35.8967°、36.8632°、37.4886°、38.7344°处有特征峰;如图1所示。
2、差示扫描量热法(DSC)
仪器:DSC Q2000差式扫描量热仪(美国,TA仪器)
温度范围:25℃-160℃
升温速度:10℃/min
结果表明:唑菌胺酯新晶型的熔融起始温度(onset temperature)在57.96℃,如图2所示。
3、热重分析法(TGA)
仪器:TGA Q500热重分析仪(美国,TA仪器)
温度范围:30℃-400℃
升温速度:10℃/min
结果表明:唑菌胺酯新晶型在30-110℃范围内失重0.23%,如图3所示。
本发明的另一目的是提供一种制备唑菌胺酯新晶型的方法。
将不同于本发明唑菌胺酯新晶型的其他唑菌胺酯晶型(比如中国专利公开号CN101203136B记载的晶型Ⅰ、晶型Ⅱ、晶型Ⅲ和晶型Ⅳ)在常温固体状态下溶于有机溶剂中;
在-80℃-0℃(优选为-80℃--24℃)下结晶,结晶时间为8-36小时;
过滤得到固体,在室温下干燥,即得唑菌胺酯新晶型。
所述有机溶剂为乙腈、二氯甲烷、氯乙烷、环己酮中的一种或两种以上的混合溶剂;优选为二氯甲烷、环己酮。
实施例1:唑菌胺酯晶型制备方法
将300mg唑菌胺酯晶型Ⅳ,加入0.5ml二氯甲烷,搅拌至完全溶解,置于-80℃下结晶12小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
实施例2:唑菌胺酯晶型制备方法
将300mg唑菌胺酯晶型Ⅳ,加入0.5ml二氯甲烷,搅拌至完全溶解,置于-24℃下结晶36小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
实施例3:唑菌胺酯晶型制备方法
将200mg唑菌胺酯晶型Ⅳ,加入0.4ml二氯甲烷,搅拌至完全溶解,置于0℃下结晶36小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
实施例4:唑菌胺酯晶型制备方法
称取1.5g唑菌胺酯晶型Ⅳ,加入1ml二氯甲烷,搅拌至完全溶解,置于-24℃下结晶8小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
实施例5:唑菌胺酯晶型制备方法
称取1.5g唑菌胺酯晶型Ⅳ,加入2ml环己酮,搅拌至完全溶解,置于-40℃下结晶36小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
实施例6:唑菌胺酯晶型制备方法
称取1.5g唑菌胺酯晶型Ⅳ,加入1ml环己酮,搅拌至完全溶解,置于-24℃下结晶24小时,过滤分离,固体置于通风的地方干燥,得唑菌胺酯新晶型。
吸湿性测定:
仪器:TGA Q5000 SA动态水分吸附分析仪(美国,TA仪器)
温度:30℃
湿度范围:50%RH-94%RH
升湿速率:0.5%RH/min
测试数据如下表1所示:
表1样品的吸湿性测试数据
唑菌胺酯新晶型对湿度的稳定性有明显提高,表1可见,在高湿度如50-94%RH范围内唑菌胺酯新晶型的吸湿性明显低于已知唑菌胺酯晶型。
- 还没有人留言评论。精彩留言会获得点赞!