用于检测靶核酸的高灵敏度方法与流程
本发明涉及一种靶核酸的高灵敏度检测方法,其包括组合至少一种选择性抑制非靶核酸的核酸扩增的寡核苷酸和具有错配核酸内切酶活性的多肽。
背景技术:
已知有遗传性或后天性疾病与包括碱基置换、缺失和插入的基因突变有关。在这些突变中,某些突变与疾病直接相关和某些突变与疾病的风险和/或预后有关。在这些疾病中,对于随着疾病的进展而野生型基因变为突变型基因或具有突变型基因的细胞增加的疾病,检测突变型基因的存在是重要的。伴有基因突变的后天性疾病的实例和与疾病有关的基因突变的实例包括:肺癌和egfr基因突变或k-ras基因突变;肺鳞状细胞癌和ddr2基因突变;结肠直肠癌和k-ras基因突变、pik3ca基因突变或b-raf基因突变;及胃癌和kit基因突变或pdgfr基因突变。
也存在与上述的案例相反的情况。例如,在伴有基因突变的后天性疾病治疗过程中,监测到突变型基因减少和野生型基因增加。作为一个实例,提到微小残留病变的检测。
作为一种分析基因突变的方法,限制性片段长度多态性方法(rflp法)已被常规使用。然而,rflp法需要繁琐操作和时间长。另外,依单核苷酸多态性(snp)位点邻近的核苷酸序列而定,rflp法可能不常应用。因此,已有人指出rflp法对临床实验室使用存在许多问题。近年来,已开发了各种更简单和通用的方法,包括侵入物(invader)法、taqmanpcr法、单碱基延伸法、焦磷酸测序(pyrosequencing)法和核酸外切酶循环试验法。然而,在使用常规pcr的情况下,当与野生型基因混合的少量突变型基因小于野生型基因量的5%时,在突变型基因的靶核酸达到可检出量之前扩增反应到达平台期,并且因此靶核酸不能完全检测。
因此,pcr方法对检测作为靶核酸的突变型基因是一种非常有用的工具。然而,在pcr方法中,随着扩增产物的浓度增加,扩增产物的退火与引物退火之间竞争越来越激烈,并且最后引物不能退火且反应达到平台期。结果,含有snp或短缺失或插入的靶核酸的扩增片段与大量过量存在的非靶核酸的扩增片段一同退火。因此,当扩增反应到达平台期时,靶核酸的扩增片段的丰度比降至低于反应开始之前的丰度比。因此,为了检测相同区域的少量snp或缺失或插入,有必要自突变型基因特异性地扩增核酸,同时抑制野生型基因的核酸扩增。
作为选择性扩增突变型基因的核酸同时抑制野生型基因的核酸扩增的方法的实例,富集pcr法(非专利文献1)和限制性核酸内切酶介导的选择性pcr方法(非专利文献2和3及专利文献1)已被报道。在这些方法中,在含有目标基因的突变位点核苷酸序列的引物中引入限制性内切酶的识别/切割位点,并从而选择性扩增突变型基因,同时抑制野生型基因的扩增。然而,这些方法需要选择能够识别和切割待分析的目标基因中的突变位点的限制性内切酶,并且另外,由限制性内切酶识别和切割的基因必须为野生型基因。另外,在pcr循环期间,限制性内切酶不得被灭活。存在的问题是只有有限的限制性内切酶满足这些要求,并且方法适用的对象野生型基因和突变型基因受限。
作为另一种用于扩增野生型基因中核酸和突变型基因中核酸的方法,已建议用3’修饰的寡核苷酸进行突变体富集(memo)的方法(非专利文献4和专利文献2)。这种方法使用含有目标基因的突变位点的3’末端封端引物。3’末端封端引物的特征为与野生型基因100%匹配,而与突变型基因错配。通过使用3’末端封端引物,在野生型基因的核酸扩增中,用于扩增的引物退火和延伸受到抑制。另一方面,在突变型基因的核酸扩增中,由于突变型基因与3’末端封端引物之间错配而导致不稳定,因而不能抑制用于扩增的引物退火和延伸。结果,突变型基因的核酸扩增优先。方法中的检测通过解链曲线分析或测序实施。然而,该方法须使3’末端封端引物杂交,以致仅抑制野生型基因的引物延伸反应,并且要求在核酸扩增反应期间严格控制温度。另外,保持在初始样品中大量存在的野生型基因的初始量,并且不能减少野生型基因的核酸的意外扩增。
最近几年,已经报道了包括使用耐热错配核酸内切酶(专利文献3)的突变检测方法。然而,为抑制野生型基因的核酸扩增并选择性扩增突变型基因以便仅正确检测突变型基因的存在,这些方法仍存在问题。
文献列表
专利文献
专利文献1:wo1996/32500
专利文献2:us2013/0149695a
专利文献3:wo2014/142261
非专利文献
非专利文献1:leukemia,第5卷第2期,第160-161页,1991.
非专利文献2:oncogene,第6卷,第6期,第1079-1083页,1991.
非专利文献3:americanjournalofpathology,第153卷,第373-379页,1998.
非专利文献4:thejournalofmoleculardiagnostics,第13卷,第657-668页,2011。
技术实现要素:
技术问题
本发明的一个目的是提供一种相对于非靶核酸以很少量存在的靶核酸的高灵敏度检测方法。
问题的解决方案
本发明人分析了具有识别并切割错配位点的错配核酸内切酶活性的多肽性质,特别是错配的种类或位置和切割活性之间的相关性。结果,本发明人发现一种切割在其序列中至少一个碱基不同的两种核酸中任一种的方法,而不受不同碱基种类的限制。另外,本发明人发现一种选择性扩增罕见靶核酸的方法,其包括通过上述的切割方法处理含有大量非靶核酸和罕见靶核酸的混合物的样品,从而选择性切割非靶核酸,并因此使得能够选择性扩增靶核酸。另外,本发明人发现一种靶核酸的高灵敏度检测方法,其包括上述的核酸扩增方法。因此,本发明得以完成。
具体地讲,本发明的第一方面涉及一种选择性切割在含有靶核酸和具有与靶核酸同源的核苷酸序列区域的非靶核酸的样品中的非靶核酸的方法,该方法包括使样品与以下接触的步骤:
(i)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配;和
(ii)具有错配核酸内切酶活性的多肽。
在本发明的第一方面,非靶核酸通过碱基置换可具有与靶核酸的核苷酸序列不同的核苷酸序列,并且当寡核苷酸与非靶核酸杂交时寡核苷酸可形成至少一个错配,和当寡核苷酸与靶核酸杂交时可形成与所述至少一个错配对应的错配和至少另一个错配。进一步地,当寡核苷酸与非靶核酸杂交时寡核苷酸可形成一个错配,和当寡核苷酸与靶核酸杂交时可形成与所述一个错配对应的错配和另一个错配。进一步地,当寡核苷酸与靶核酸杂交时形成的两个错配优选地位置连续或间隔不超过5个碱基。
在本发明第一方面的另一个实施方案中,非靶核酸通过碱基插入具有与靶核酸的核苷酸序列不同的核苷酸序列,并且当寡核苷酸与在非靶核酸中含有插入的区域杂交时,寡核苷酸形成至少一个错配。
在另一个实施方案中,非靶核酸通过碱基缺失具有与靶核酸的核苷酸序列不同的核苷酸序列,并且当寡核苷酸与在非靶核酸含有缺失的区域杂交时,寡核苷酸形成至少一个错配。
在本发明的第一方面,具有错配核酸内切酶活性的多肽可为源自耐热微生物的多肽或其突变体。另外,本发明可在酸性高分子物质存在下实施。进一步地,本发明可与增殖细胞核抗原(pcna)的使用组合实施。
本发明的第二方面涉及一种在含有靶核酸和具有与靶核酸同源的核苷酸序列区域的非靶核酸的样品中选择性扩增靶核酸的方法,该方法包括:
(1)通过本发明第一方面的方法切割样品中的非靶核酸的步骤;和
(2)扩增靶核酸的步骤。
在本发明的第二方面,靶核酸的扩增可通过pcr实施。
本发明的第三方面涉及一种在含有靶核酸和具有与靶核酸同源的核苷酸序列区域的非靶核酸的样品中选择性检测靶核酸的方法,该方法包括:
(1)通过本发明第二方面的方法选择性扩增靶核酸的步骤;和
(2)与步骤(1)同时或之后检测靶核酸的步骤。
在本发明的第三方面,靶核酸的检测可通过循环探针法或taqman(注册商标)探针法实施。
本发明的第四方面涉及一种用于本发明的第二方面和第三方面的组合物,该组合物含有:
(a)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,并且当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配;和
(b)至少一对寡核苷酸引物;
(c)具有错配核酸内切酶活性的多肽;和
(d)具有dna聚合酶活性的多肽。
本发明的第五方面涉及一种用于本发明的第二方面和第三方面的试剂盒,该试剂盒含有:
(a)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,并且当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配;和
(b)至少一对寡核苷酸引物;
(c)具有错配核酸内切酶活性的多肽;和
(d)具有dna聚合酶活性的多肽。
在本发明的第四和第五方面,组合物和试剂盒可进一步含有酸性高分子物质。在本发明中,具有错配核酸内切酶活性的多肽和具有dna聚合酶活性的多肽可为耐热多肽或其突变体。组合物和试剂盒可进一步含有增殖细胞核抗原(pcna)。
本发明的第六方面涉及一种在酸性高分子物质存在下选择性切割在含有靶核酸和具有与靶核酸的核苷酸序列至少一个碱基不同的核苷酸序列的非靶核酸的样品中的非靶核酸的方法,该方法包括使样品与以下接触的步骤:
(i)所述酸性高分子物质;
(ii)具有错配核酸内切酶活性的多肽;和
(iii)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配。
在本发明的第六方面,具有错配核酸内切酶活性的多肽可为耐热多肽或其突变体。进一步地,本发明可与增殖细胞核抗原(pcna)的使用组合实施。
发明效果
根据本发明,对于含有大量非靶核酸和罕见靶核酸的混合物的样品,提供一种抑制非靶核酸的核酸扩增和选择性扩增靶核酸的方法。另外,提供一种包括组合上述的核酸扩增方法和靶核酸的特异性实时检测方法,从而使得能够高灵敏度检测罕见突变型基因的方法。
附图说明
图1显示实施例1中制备的含有各种丰度比的非靶核酸和靶核酸的样品的实时检测结果。
图2显示实施例2中制备的含有各种丰度比的非靶核酸和靶核酸的样品的实时检测结果。
图3a显示示出在实施例3中酸性高分子物质(在没有海藻酸钠的情况下)对本发明切割方法的影响的实时检测结果。
图3b显示示出在实施例3中酸性高分子物质(在56μg/ml海藻酸钠存在下)对本发明切割方法的影响的实时检测结果。
图3c显示示出在实施例3中酸性高分子物质(在140μg/ml海藻酸钠存在下)对本发明切割方法的影响的实时检测结果。
图3d显示示出在实施例3中酸性高分子物质(在168μg/ml海藻酸钠存在下)对本发明切割方法的影响的实时检测结果。
图4显示实施例5中制备的含有各种丰度比的非靶核酸和靶核酸的样品的实时检测结果。
具体实施方式
在本发明中,错配指的是在双链核酸中存在的与沃森-克里克碱基对不同的碱基配对,换言之,除g(鸟嘌呤碱基)-c(胞嘧啶碱基)和a(腺嘌呤碱基)-t(胸腺嘧啶碱基)或u(尿嘧啶碱基)的碱基配对以外的组合中的碱基结合。
在本发明中,靶核酸指的是期望检测的任一种核酸(例如组成天然存在的基因的核酸和人工制备的核酸)。本发明可用于区分靶核酸与具有同靶核酸的核苷酸序列相似的核苷酸序列的非靶核酸。本发明中的靶核酸和非靶核酸优选地为(但不限于)dna。
靶核酸和非靶核酸的实例包括(但不限于):突变型基因的核酸和与突变型基因对应的野生型基因的核酸的组合;人工引入突变后的核酸与人工引入突变前的核酸的组合;用亚硫酸氢盐处理的核酸与用亚硫酸氢盐处理前的甲基化核酸的组合;置换、缺失或插入发生后的核酸与突变发生前的核酸的组合;以及自活体、组织、细胞等得到的给予药物前后的核酸的组合。在本发明中,依上述的组合的哪一种核酸被设定为靶核酸而定,组合中的靶核酸和非靶核酸可以彼此替换。
在下文,本发明将详细阐释。
(1)本发明非靶核酸的选择性切割方法
本发明提供一种在含有靶核酸和具有与靶核酸同源的核苷酸序列区域的非靶核酸的样品中仅选择性切割非靶核酸的方法。本文使用的“具有与靶核酸同源的核苷酸序列区域的非靶核酸”意指非靶核酸的核苷酸序列含有与靶核酸的核苷酸序列的至少一部分同源的核苷酸序列。非靶核酸的实例包括:通过置换一个或多个碱基具有与靶核酸的核苷酸序列不同的核苷酸序列的非靶核酸、通过插入一个或多个碱基具有与靶核酸的核苷酸序列不同的核苷酸序列的非靶核酸、和通过缺失一个或多个碱基具有与靶核酸的核苷酸序列不同的核苷酸序列的非靶核酸。在同源序列中置换、插入或缺失的碱基数目不受特别限制。然而,按照本发明,靶核酸和非靶核酸通过在其核苷酸序列中存在至少一个不同的碱基来区分。随着不同碱基的数目增加,靶核酸和非靶核酸之间辨别的准确性得以改善。
在本发明的第一方面,非靶核酸在靶核酸和具有与靶核酸同源的核苷酸序列区域的非靶核酸的混合物中被选择性切割。上述的方法使用寡核苷酸(当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时,比当该寡核苷酸与非靶核酸杂交时形成更多的错配)(在下文,有时称为抑制性寡核苷酸)和具有错配核酸内切酶活性的多肽。当抑制性寡核苷酸与非靶核酸杂交时,只要发生选择性切割非靶核酸,形成的至少一个错配的数目不受特别限制,并且依抑制性寡核苷酸的长度而定,其实例包括1-7、1-5或1-3个错配。当至少一个错配的数目表达为在抑制性寡核苷酸长度的百分数时,例如至少一个错配占抑制性寡核苷酸长度的1-20%、3-15%或4-8%。作为该方面的一个实例,在其核苷酸序列中仅一个碱基不同的靶核酸和非靶核酸的组合被阐释。当抑制性寡核苷酸与靶核酸杂交时,在抑制性寡核苷酸与在靶核酸和非靶核酸之间不同的碱基之间形成错配(在下文,有时称为第一错配),同时形成另一个错配(在下文,有时称为第二错配)。
第一错配的碱基对涉及靶核酸与非靶核酸之间不同的碱基,而不考虑错配的碱基对是否被共存的具有错配核酸内切酶活性的多肽识别和切割。
第二错配为由共存的具有错配核酸内切酶活性的多肽识别和切割的错配的碱基对。例如,当使用具有识别和切割鸟嘌呤碱基-鸟嘌呤碱基、鸟嘌呤碱基-胸腺嘧啶碱基或胸腺嘧啶碱基-胸腺嘧啶碱基的错配核酸内切酶活性的多肽时,抑制性寡核苷酸被设计形成选自上述的错配碱基对的错配。
当抑制性寡核苷酸与非靶核酸杂交时形成第二错配,但不形成第一错配。
设计和使用具有上述性质的抑制性寡核苷酸使得能够通过具有错配核酸内切酶活性的多肽选择性切割非靶核酸。本发明不限于使用能够形成上述的一个或两个错配的抑制性寡核苷酸。只要发生选择性切割期望的核酸,能够形成3个或更多错配的抑制性寡核苷酸就可被设计和使用。在这样的情况下,在靶核酸和非靶核酸中形成的至少一个错配可为第二错配,并且其他的错配可或可不由具有错配核酸内切酶活性的多肽识别和切割。3个或更多错配优选地由具有错配核酸内切酶活性的多肽识别和切割。
用于本发明方法的抑制性寡核苷酸由dna组成,但是dna的一部分可用核苷酸类似物或rna替换。换言之,抑制性寡核苷酸不受特别限制,只要抑制性寡核苷酸具有当与非靶核酸杂交时形成具有至少一个错配的双链核酸的结构,并且错配被共存的具有错配核酸内切酶活性的多肽识别和切割。
例如,当两种核酸之间一个碱基的差异通过使用具有错配核酸内切酶活性的多肽区分时,通常将寡核苷酸设计成与两个核酸中的一个形成原本可通过具有错配核酸内切酶活性的多肽识别的错配,并然后被使用。
然而,依待区分的碱基而定,原本由具有错配核酸内切酶活性的多肽识别的错配可能不会形成。在这样的情况下,如同本发明的方法那样,在核酸中不含待区分碱基的区域内形成错配,并且因此形成的错配与形成/不形成基于上述的不同碱基的错配组合,这使得选择性切割两个核酸中的一个。换言之,本发明使用这样核苷酸序列的寡核苷酸,以致当寡核苷酸与在其核苷酸序列中至少一个碱基不同的两个核酸中的一个杂交时,形成至少一个原本由具有错配核酸内切酶活性的多肽识别的错配,同时当寡核苷酸与两个核酸中的另一个杂交时,形成上述的至少一个错配以及至少一个基于两个核酸的核苷酸序列中不同碱基的错配。当该寡核苷酸(抑制性寡核苷酸)和具有错配核酸内切酶活性的多肽组合使用时,其中形成一个错配的双链核酸由多肽切割,但这样的切割并不发生在其中形成两个或更多个错配的双链核酸上。
因此,本发明方法的另一方面提供一种方法,其特征在于非靶核酸通过碱基置换具有与靶核酸的核苷酸序列不同的核苷酸序列,并使样品与寡核苷酸(当寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当寡核苷酸与靶核酸杂交时形成与所述至少一个错配对应的错配和至少另一个错配)和具有错配核酸内切酶活性的多肽接触。在这方面,寡核苷酸包括下述寡核苷酸,当与非靶核酸或其互补链杂交时其形成一个错配(第二错配),和当与靶核酸或其互补链杂交时形成与上述的错配对应的错配和另一个错配(第一错配)。
例如,当突变型基因上的碱基突变位点(例如snp)上的碱基不能形成原本由具有错配核酸内切酶活性的多肽识别的错配时,抑制性寡核苷酸可被设计成具有核苷酸序列,其使得在为靶核酸的突变型基因中snp位点形成错配和形成邻近snp位点的另一个错配。另一个错配与和snp无关的碱基(即突变型基因和野生型基因两者共同的碱基)形成,并且可由具有错配核酸内切酶活性的多肽识别和切割。与snp位点的碱基对应的抑制性寡核苷酸中的碱基与野生型基因中的碱基正确配对。当抑制性寡核苷酸与野生型基因的核酸杂交时形成的双链核酸包含一个错配,其可由具有错配核酸内切酶活性的多肽切割,因此选择性切割野生型基因的核酸。另一方面,抑制性寡核苷酸不能与突变型基因的核酸稳定杂交,因为形成两个错配,并且进一步地,基于多肽的切割受到抑制。因此,本发明的方法的特征在于,由具有错配核酸内切酶活性的多肽选择性切割野生型基因在与共存的突变型基因的碱基突变位点不同的位置上进行。
本发明方法的另一方面提供一种方法,其特征在于非靶核酸通过碱基插入具有与靶核酸的核苷酸序列不同的核苷酸序列,并使样品与寡核苷酸(当寡核苷酸与在非靶核酸的核苷酸序列中含有插入的区域杂交时其形成至少一个错配)和具有错配核酸内切酶活性的多肽接触。在这方面,寡核苷酸(抑制性寡核苷酸)与之杂交的区域优选地选自在靶核酸中不存在和在非靶核酸中存在的区域。或者,寡核苷酸(抑制性寡核苷酸)与之杂交的区域可为延伸跨越靶核酸中不存在的区域和靶核酸中存在的区域的区域。
抑制性寡核苷酸被设计形成至少一个错配,当抑制性寡核苷酸与非靶核酸杂交时,错配由具有错配核酸内切酶活性的多肽识别和切割。因为如此设计的寡核苷酸与没有被插入到非靶核酸的核苷酸序列的靶核酸形成更多的错配,因此形成的双链核酸不稳定,或者寡核苷酸不能与靶核苷酸杂交。如果要形成另外的错配,则错配的位置不受限制。
本发明方法的另一方面提供一种方法,其特征在于非靶核酸通过碱基缺失具有与靶核酸的核苷酸序列不同的核苷酸序列,并使样品与寡核苷酸(当寡核苷酸与在非靶核酸中含有缺失位点的区域杂交时其形成至少一个错配)和具有错配核酸内切酶活性的多肽接触。在这方面,寡核苷酸(抑制性寡核苷酸)与之杂交的区域优选地选自在非靶核酸中含有缺失位点的区域。抑制性寡核苷酸被设计成当抑制性寡核苷酸与非靶核酸杂交时形成至少一个错配,其由具有错配核酸内切酶活性的多肽识别和切割。因为如此设计的寡核苷酸与具有在非靶核酸中缺失的核苷酸序列的靶核酸形成更多的错配,所以形成的双链核酸不稳定,或者寡核苷酸不能与靶核苷酸杂交。如果要形成另外的错配,则错配的位置不受限制。
用于本发明方法的抑制性寡核苷酸的长度优选地为7个或更多个碱基的长度,并且其实例包括(但不特别限于)7-40个碱基,优选地10-30个碱基,和更优选地11-25个碱基范围内的长度。抑制性寡核苷酸的核苷酸序列被设计成当与非靶核酸杂交时形成至少一个错配,和当与靶核酸杂交时比寡核苷酸与非靶核酸杂交时形成更多的错配。此时,选择这样的核苷酸序列,以致当与靶核酸(例如突变型基因的核酸)或其互补链杂交时,与靶核酸和非靶核酸之间不同的碱基形成错配,并且与5’末端侧或3’末端侧方向的不同碱基位置优选地相距最多5个碱基,更优选地相距最多2个碱基,或者紧挨着形成至少另一个错配碱基对。换言之,优选地使用使两个错配位置间隔不超过5个碱基的核苷酸序列。另外,寡核苷酸被优选地设计在邻近寡核苷酸的中心碱基的区域形成两个错配。进一步地,当该核苷酸序列的寡核苷酸与非靶核酸(例如野生型基因的核酸)杂交时,寡核苷酸不与靶核酸和非靶核酸之间不同的碱基形成错配。当寡核苷酸与靶核酸或其互补链杂交时,与靶核酸和非靶核酸之间不同的碱基形成的错配可或可不由具有错配核酸内切酶活性的多肽识别和切割。当寡核苷酸与靶核酸或非靶核酸或其互补链杂交时,与除了靶核酸和非靶核酸之间不同的碱基外的碱基形成的错配中至少一个为由具有错配核酸内切酶活性的多肽识别和切割的错配。这样设计的抑制性寡核苷酸使得本发明的方法与采用限制性内切酶等的常规技术相比较能够检测很宽范围靶标内的突变。寡核苷酸的3’末端可被修饰以致抑制寡核苷酸通过dna聚合酶的延伸反应,本发明不特别限制于此。修饰的实例包括氨基化。
在本发明的方法中,抑制性寡核苷酸与具有对识别和切割错配的碱基对最佳的错配核酸内切酶活性的多肽组合使用。多肽的优选实例包括(但不限于)得自激烈火球菌(pyrococcusfuriosus)的多肽pf0012(wo2014/142261)和该肽的突变体(有时统称为nucs蛋白)。源自深海火球菌(pyrococcusabyssi)(genbank收录号q9v2e8)、thermococcusbarophilus(rsfseqid:yp_004072075)和詹氏甲烷球菌(methanocaldococcusjannaschii)(rsfseqid:np247194)的上述多肽的同源物及其突变体也可优选地用于本发明的方法。
在本发明的一个优选方面,对与本发明方法同时实施的核酸扩增或检测,优选地使用源自耐热微生物的具有错配核酸内切酶活性的多肽。多肽的优选实例包括(但不限于)甚至在热循环比如pcr期间保留活性的多肽以及在50℃或更高,优选地70℃或更高、更优选地90℃或更高不失活的多肽。
本发明的方法可在酸性高分子物质存在下实施。已发现酸性高分子物质具有通过酸性高分子物质的共存控制多肽的错配识别和切割活性的作用。酸性高分子物质在含有少量核酸的样品中发挥更大作用。酸性高分子物质的优选实例包括多聚阴离子。酸性高分子物质的优选实例也包括具有糖骨架的酸性多糖和具有线性碳链的酸性多糖。作为酸性多糖,可使用一种或多种选自以下的物质:含硫酸岩藻糖的多糖、硫酸葡聚糖、卡拉胶、肝素、硫酸鼠李聚糖、硫酸皮肤素(硫酸软骨素b)、硫酸乙酰肝素、透明质酸、海藻酸、果胶、聚谷氨酸、聚丙烯酸、聚硫酸乙烯酯、聚硫酸苯乙烯酯及其盐类,以及不同于靶核酸和非靶核酸的核酸。
本发明的方法可进一步与增殖细胞核抗原(pcna)的使用组合实施。
(2)本发明靶核酸的选择性扩增方法
本发明核酸的选择性扩增方法为在含有具有彼此相似的核苷酸序列区域的两种核酸(靶核酸和非靶核酸)的样品中选择性扩增两种核酸中的一种(靶核酸)的方法,其特征在于包括:(1)通过本发明非靶核酸的选择性切割方法切割样品中的非靶核酸的步骤,和(2)扩增靶核酸的步骤。上述的两个步骤可分别和依序实施,或者可同时进行。在后者情况下,非靶核酸的切割可发生在核酸扩增反应期间。
在本发明靶核酸的选择性扩增方法中,非靶核酸由具有错配核酸内切酶活性的多肽选择性切割,并且非靶核酸的丰度比降低,并且结果,靶核酸得到选择性扩增。在核酸扩增的步骤中,可采用已知的核酸扩增方法,并且优选地使用产生使抑制性寡核苷酸与非靶核酸杂交的条件的核酸扩增方法。更优选地,pcr方法可用于本发明的方法中,但本发明不限于使用pcr方法。
在本发明中,核酸扩增可在酸性高分子物质存在下实施。当上述的步骤(1)和(2)两者同时实施时,核酸扩增效率的提高和非靶核酸通过具有错配核酸内切酶活性的多肽识别和切割的效率提高可通过酸性高分子物质的作用同时达到。酸性高分子物质在含有少量核酸的样品中发挥更大作用。酸性高分子物质的优选实例包括以上在本发明非靶核酸的选择性切割方法的阐释中提及的物质。
进一步地,本发明的方法可与增殖细胞核抗原(pcna)的使用组合实施。
(3)本发明靶核酸的选择性检测方法
本发明靶核酸的选择性检测方法的特征在于包括:(1)通过本发明靶核酸的选择性扩增方法选择性地扩增靶核酸的步骤,和(2)与以上步骤(1)同时或之后检测靶核酸的步骤。当检测方法应用于含有大量非靶核酸和很少量靶核酸的样品时,初始样品中的非靶核酸与靶核酸的丰度比和检测步骤中的丰度比反转。换言之,通过扩增靶核酸同时抑制非靶核酸的扩增,可以高灵敏度检测靶核酸。在本发明的检测方法中,步骤(2)可在步骤(1)之后实施,或者步骤(1)和步骤(2)两者可同时实施。步骤(2)的检测方法可为用于检测或定量扩增产物的方法,或为用于特异性检测扩增产物的核苷酸序列的方法。例如,对于终点检测,可使用直接测序法或使用插入剂(intercalator)的高分辨率解链曲线分析(hrm)方法。对于实时检测,可使用采用序列特异性探针的循环探针法(例如wo2003/074696)或taqman(注册商标)测定方法(例如wo92/02638)。更优选地,使用可与pcr方法组合的循环探针方法检测。
用于pcr方法的引物对可被设计使得含有靶核酸和非靶核酸之间不同的核苷酸序列的靶核酸和非靶核酸区域(即抑制性寡核苷酸可与之杂交的区域)扩增。可通过已知方法进行引物的设计和合成。
当检测通过循环探针法实施时,检测系统可含有(但不限于)具有核糖核酸酶h活性的多肽。具有核糖核酸酶h活性的多肽优选地为耐热的。多肽的优选实例包括(但不限于)源自以下的核糖核酸酶h:热坚芽孢杆菌(bacilluscaldotenax)、激烈火球菌、掘越氏火球菌(pyrococcushorikoshii)、闪烁古生球菌(archaeoglobusfulgidus)、栖热球菌(thermococcuslitoralis)、速生热球菌(thermococcusceler)、海栖热袍菌(thermotogamaritima)和深处古生球菌(archaeoglobusprofundus)。
循环探针法能够检测与非靶核酸有区别的靶核酸。可用于这样检测的探针(嵌合寡核苷酸探针)可通过已知方法,例如在wo2012/014988中公开的方法设计。嵌合寡核苷酸探针的优选实例包括含有夹在两部分之间的rna部分的那些,其中两部分中的一个用荧光物质标记,和另一个用使自荧光物质产生的荧光猝灭的物质(猝灭剂)标记。物质组合的优选实例包括:6-fam(6-羧基荧光素)和dabcyl(4-二甲基氨基偶氮苯-4’-砜)、rox(6-羧基-x-若丹明)和dabcyl、6-fam和eclipse(由epochbiosciences制备)、rox和eclipse、tet(四氯荧光素)和dabcyl、以及tet和eclipse。
用于本发明靶核酸的选择性检测方法的抑制性寡核苷酸优选地被设计具有对本发明非靶核酸的选择性切割方法阐释的抑制性寡核苷酸的性质,并且当与用于靶核酸检测方法的寡核苷酸探针杂交时,不被共存的具有错配核酸内切酶活性的多肽识别和切割。
在采用pcr的本发明检测方法中,方法可与阳性对照核酸的扩增和检测组合。阳性对照核酸优选地为样品中最初存在的与待检测的靶基因不同的基因(例如管家基因等)的核酸。当最初存在于样品中的基因用作阳性对照核酸时,组合使用用于扩增基因中任何区域的引物对和用于检测的探针。还可制备人工核酸,并将其预先加入到样品中。例如,人工核酸可为与靶核酸和非靶核酸的扩增区域相同区域的核酸,或者与靶核酸和非靶核酸的扩增区域不同区域的核酸。当已知量的人工核酸作为阳性对照核酸预先加入到样品中时,如果人工核酸能够通过采用与用于靶核酸和非靶核酸的扩增区域相同的引物扩增,则使用相同的引物对,并且如果人工核酸为与靶核酸和非靶核酸的扩增区域不同的区域,则使用用于扩增人工核酸区域的引物对。作为用于检测阳性对照核酸的探针,使用能够选择性地检测阳性对照核酸的探针。同时实施本发明靶核酸的检测和阳性对照核酸的检测,使得能够通过本发明的检测系统证实存在或不存在扩增异常,和基于扩增曲线的比较来半定量分析靶核酸的初始量。
本发明的检测方法可在酸性高分子物质存在下实施。酸性高分子物质在含有少量核酸的样品中发挥更大作用。酸性高分子物质的优选实例包括以上在本发明非靶核酸的选择性切割方法的阐释中提及的物质。进一步地,本发明的方法可与增殖细胞核抗原(pcna)的使用组合实施。
关于具有特定核苷酸序列的核酸,例如与其中已知存在基因突变的基因对应的核酸,本发明的检测方法能够区别性地检测野生型基因和突变型基因。当实施本发明的检测方法,其中具有突变型基因的核苷酸序列的核酸被设定为靶核酸和具有野生型基因的核苷酸序列的核酸设定为非靶核酸时,少量的突变等位基因能够在大量过量的正常等位基因(即具有野生型核苷酸序列的dna)存在下进行检测。例如,本发明的方法可用于检测循环肿瘤dna,或检测包含在母亲血液中的少量胎儿dna序列。突变的实例包括微缺失和点突变。
具有特定核苷酸序列的核酸的优选实例包括(但不限于)含有选自以下的至少一种单核苷酸多态性的核酸:用作肿瘤标志物的单核苷酸多态性突变、与用于癌症治疗的药物治疗作用相关的单核苷酸多态性突变及已知与细胞癌变相关的单核苷酸多态性突变。snp的实例包括经常在肿瘤细胞中发现的那些snp和已知与用于治疗癌症的药物治疗作用或致癌相关的那些snp。这样的后天性基因突变的实例包括k-ras基因、b-raf基因和表皮生长因子受体(egfr)基因的突变。k-ras基因的体细胞突变在结肠直肠癌、肺腺癌、甲状腺癌等中经常被发现。b-raf基因的体细胞突变在结肠直肠癌、恶性黑色素瘤、乳头状甲状腺癌、非小细胞肺癌、肺腺癌等中经常被发现。egfr基因的体细胞突变在各种实体肿瘤中经常被发现。已知当癌组织中的egfr基因具有特定的单核苷酸多态性突变时,使用egfr抑制剂比如吉非替尼(gefitinib)或厄洛替尼(erlotinib)治疗癌症很可能是有效的。与此相比,已知当癌组织中的k-ras基因具有单核苷酸多态性突变时,癌症很可能对egfr抑制剂有抗性。
本发明的检测方法也可用于甲基化分析。例如,将自生物体来源的样品提取的甲基化dna设定为野生型基因,和将用硫酸氢盐处理含有野生型基因的组合物后得到的dna设定为突变型基因。按照本发明的检测方法,少量的甲基化等位基因能够在大量过量的非甲基化等位基因存在下进行检测,或者少量的非甲基化等位基因能够在大量过量的甲基化等位基因存在下进行检测。
作为亚硫酸氢盐处理,可采用用于检测甲基化dna的已知的亚硫酸氢盐方法。通过处理,非甲基化胞嘧啶变为尿嘧啶,而甲基化胞嘧啶不发生变化。当用亚硫酸氢盐处理的反应溶液经受pcr的扩增时,尿嘧啶变为胸腺嘧啶,并且甲基化胞嘧啶变为胞嘧啶。换言之,在特定位点上在大量过量的非甲基化等位基因存在下检测少量甲基化等位基因和在大量过量的甲基化等位基因存在下检测少量非甲基化等位基因,分别相当于检查在大量过量的胸腺嘧啶存在下的胞嘧啶存在情况和检查在大量过量的胞嘧啶存在下的胸腺嘧啶存在情况。当大量过量的含有胸腺嘧啶或胞嘧啶的dna扩增受到抑制时,易于检查少量甲基化等位基因或非甲基化等位基因的存在情况。
在本发明的检测方法中,通过向反应混合物中加入具有错配核酸内切酶活性的多肽和与野生型基因形成错配的抑制性寡核苷酸,野生型基因的扩增能够被抑制和罕见的突变型基因能够选择性扩增,并因此易于检测。进一步地,如后面在实施例中描述的那样,通过人工设计初始碱基突变位点上的错配和与初始碱基突变位点不同的位点上的至少另一个错配,可广泛设计能够由具有错配核酸内切酶活性的多肽识别和切割的抑制性寡核苷酸,而不必考虑碱基突变位点上碱基的种类。
(4)本发明的组合物
用于本发明靶核酸的选择性扩增方法和/或选择性检测方法的组合物实例包括含有以下的组合物:(a)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配;(b)至少一对寡核苷酸引物;(c)具有错配核酸内切酶活性的多肽或其突变体;和(d)具有dna聚合酶活性的多肽。
组合物可含有酸性高分子物质。酸性高分子物质的优选实例包括以上在本发明非靶核酸的选择性切割方法的阐释中提及的物质。
组合物可进一步含有增殖细胞核抗原(pcna)。
用于本发明靶核酸的选择性检测方法(其中靶核酸的检测通过pcr实施)的组合物可含有具有核糖核酸酶h活性的多肽或其突变体、为嵌合寡核苷酸的循环探针或taqman探针。特别是,具有错配核酸内切酶活性的多肽、具有dna聚合酶活性的多肽和具有核糖核酸酶h活性的多肽可选自野生型多肽或突变型多肽。具有错配核酸内切酶活性的多肽的优选实例包括(但不限于)来自pfu的nucs蛋白的突变体。进一步地,用于检测阳性对照核酸的引物对和嵌合寡核苷酸探针或taqman探针可组合使用。
(5)本发明的试剂盒
用于本发明靶核酸的选择性扩增方法和/或选择性检测方法的试剂盒的实例包括含有以下的试剂盒:(a)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时,比该寡核苷酸与非靶核酸杂交时形成更多的错配;(b)至少一对寡核苷酸引物;(c)具有错配核酸内切酶活性的多肽或其突变体;和(d)具有dna聚合酶活性的多肽或其突变体。
试剂盒可进一步含有酸性高分子物质,并且可优选地使用以合适浓度制备的酸性高分子物质的溶液。酸性高分子物质的优选实例包括以上在本发明非靶核酸的选择性切割方法的阐释中提及的物质。
试剂盒可进一步含有增殖细胞核抗原(pcna)。
用于本发明靶核酸的选择性检测方法(其中靶核酸检测通过pcr实施)的试剂盒可含有具有核糖核酸酶h活性的多肽或其突变体、为嵌合寡核苷酸的循环探针或taqman探针。特别是,具有错配核酸内切酶活性的多肽、具有dna聚合酶活性的多肽和具有核糖核酸酶h活性的多肽可选自野生型多肽或突变型多肽。具有错配核酸内切酶活性的多肽的优选实例包括(但不限于)来自pfu的nucs蛋白的突变体。进一步地,用于检测阳性对照核酸的引物对和嵌合寡核苷酸探针或taqman探针可组合使用。
(6)本发明非靶核酸在酸性高分子物质存在下的选择性切割方法
本发明非靶核酸的切割方法的另一方面包括一种在酸性高分子物质存在下,于含有靶核酸和具有与靶核酸同源区域的非靶核酸的样品中选择性切割非靶核酸的方法,该方法包括使样品与以下接触的步骤:(i)所述酸性高分子物质;(ii)具有错配核酸内切酶活性的多肽;和(iii)寡核苷酸,当该寡核苷酸与非靶核酸杂交时其形成至少一个错配,和当该寡核苷酸与靶核酸杂交时比该寡核苷酸与非靶核酸杂交时形成更多的错配。
在该方法中,酸性高分子物质、具有错配核酸内切酶活性的多肽和寡核苷酸的优选实例包括以上在本发明非靶核酸的选择性切割方法的阐释中提及的那些。本发明的方法可进一步与增殖细胞核抗原(pcna)的使用组合实施。
实施例
在下文,本发明将通过实施例(本发明不限于此)进行更具体阐释。
实施例1:很少量的混合突变型基因egfrt790m的特异性扩增
(1)用于检测的模板制备
自以genbank收录号nc_000007公开的人类egfr基因的cds序列信息,人工合成与后面描述的引物egfr_t790m_f和egfr_t790m_r对应的核苷酸序列和含有在引物核苷酸序列之间区域的核苷酸序列的dna。此时,把egfr基因中第2369位核苷酸的碱基c转变成t。通过这样的碱基置换,人工合成dna中与第790位氨基酸对应的苏氨酸(t)的密码子被甲硫氨酸(m)的密码子置换。因此得到的人工合成基因命名为egfrcodon790dna,并且用作突变型基因。这样的突变命名为egfr_t790m。在这种情况下,错配的碱基对可被设计在t790m位点上形成。作为具有野生型基因的核苷酸序列的dna,自人细胞hl60制备基因组dna。
在该实施例中,靶核酸为egfr_t790mdna,其在egfr第790位密码子具有突变,和非靶核酸为hl60基因组dna。
(2)用于扩增和抑制性寡核苷酸的引物制备
通过常规方法制备两种具有在seqidno:1和2显示的核苷酸序列的引物,egfr_t790m_f和egfr_t790m_r。然后,通过常规方法制备具有在seqidno:3显示的核苷酸序列的寡核苷酸,其可与含有egfr基因第2369位核苷酸的碱基的区域杂交。为防止自寡核苷酸3’末端的延伸反应,3’末端的羟基用氨基修饰。寡核苷酸命名为抑制性寡核苷酸t790m_nucg-1。当寡核苷酸与egfr野生型基因的负链杂交时,在与第2369位核苷酸(正链)上的c互补的g位上形成g-g错配。另一方面,当寡核苷酸与其中第2369位核苷酸上的碱基由t置换的突变型基因的负链杂交时,在与t互补的a位上形成g-a错配。
(3)用于特异性突变型基因检测的探针设计和制备
嵌合寡核苷酸探针作为用于特异性检测egfrcodon790dna的探针被合成,其中寡核苷酸探针具有自egfrcodon790dna的与egfr基因上的第2369号核苷酸对应的位点3’方向延伸1个碱基和在5’方向延伸8个碱基的区域的负链核苷酸序列,并且自核苷酸序列的5’末端起第4个dna由rna置换。另外,寡核苷酸的5’末端结合于eclips猝灭剂,和寡核苷酸的3’末端结合于fam染料。因此制备的嵌合寡核苷酸探针命名为t790m_detect-1。t790m_detect-1的核苷酸序列显示在seqidno:4。
因为t790m_detect-1与其中第2369位核苷酸上的碱基被t置换的突变型egfr基因完全杂交,所以t790m_detect-1的rna部分被共存的核糖核酸酶h切割。与此相比,当t790m_detect-1与野生型egfr基因杂交时形成错配,并因此t790m_detect-1不经历核糖核酸酶h的切割。
(4)伴随野生型基因扩增抑制的特异性突变检测方法的研究
通过使hl60基因组dna与在实施例1-(1)中制备的egfrcodon790dna以50000个拷贝:500个拷贝(=100:1)、50000个拷贝:50个拷贝(=1000:1)和50000个拷贝:5个拷贝(=10000:1)的比率混合,制备dna样品。hl60基因组dna的50000个拷贝的dna量约为185ng。作为对照,制备仅含有hl60基因组dna的样品。这些样品用作模板。
源于激烈火球菌的具有错配的核苷酸特异性双链切割活性的多肽(在下文,称为nucs蛋白),其用作具有错配核酸内切酶活性的多肽,为其中引入w77f的多肽pf0012的突变体,其通过在wo2014/142261的制备实施例1-3和实施例1中描述的方法制备。
伴随野生型基因扩增抑制的特异性突变检测如以下描述的那样实施。使用cycleavepcrreactionmix(由takarabioinc.制备),制备最终体积25μl的反应混合物,其含有上述的样品、112nmnucs蛋白、0.2μmt790m_nucg-1、各为0.2μm的egfr_t790m引物对和0.2μmt790m_detect-1。通过热循环仪tp900(由takarabioinc.制备)实施实时pcr检测。在95℃下初始变性处理10秒,然后95℃5秒、55℃10秒和72℃20秒的45次循环的反应条件下实施pcr。随着时间推移观察荧光强度。同时,不含nucs蛋白的反应混合物作为对照制备,并且实施相同的测量。结果显示在图1中。
具体地讲,图1显示用于egfr基因中t970m突变的高灵敏度检测方法的结果。纵轴显示荧光强度,横轴显示pcr循环的数目。显示突变型基因的拷贝数目(0、5、50和500个拷贝)的荧光曲线。如自图1(b)可见的那样,当反应混合物不含有nucs蛋白时,未观察到基于嵌合寡核苷酸探针t790m_detect-1切割的荧光值增加。这表明来自egfrcodon790dna的dna扩增受到共存的hl60基因组dna抑制。
与此相比,如自图1(a)可见的那样,当使用含有nucs蛋白的反应混合物实施pcr时,随着时间的推移,甚至在相对于hl60基因组dna的50000个拷贝含有egfrcodon790dna的5个拷贝的样品中观察到荧光值增加。换言之,自egfrcodon790dna扩增的dna作为模板被t790m_detect-1特异性检测。该结果显示,nucs与抑制性寡核苷酸的组合抑制作为模板的hl60基因组dna的dna扩增,并因此使得能够检测egfrcodon790dna。
实施例2:具有l858r突变的egfr基因的特异性检测
(1)用于检测的模板制备
自以genbank收录号nc_000007公开的人egfr基因的cds序列信息,人工合成与后面描述的引物egfr_l858r_f和egfr_l858r_r对应的核苷酸序列和含有在引物的核苷酸序列之间区域的核苷酸序列的dna。此时,把egfr基因中第2573位核苷酸的碱基t转变成g。通过这样的碱基置换,人工合成dna中与第858位氨基酸对应的亮氨酸(l)的密码子被精氨酸(r)的密码子置换。因此得到的人工合成基因命名为egfrcodon858dna,并且用作突变型基因。这样的突变命名为egfr_l858r。在这种情况下,错配的碱基对不可被设计在l858r位点上形成。作为野生型基因,使用如在实施例1中制备的人细胞hl60基因组dna。
在该实施例中,靶核酸为egfr_l858rdna,其在egfr第858位密码子具有突变,并且非靶核酸为hl60基因组dna。
(2)用于扩增和抑制性寡核苷酸的引物制备
通过常规方法制备两种具有在seqidno:5和6显示的核苷酸序列的引物,egfr_l858r_f和egfr_l858r_r。然后,制备具有在seqidno:7显示的核苷酸序列的寡核苷酸,其可与含有egfr基因第2573位核苷酸的区域杂交。为防止自寡核苷酸3’末端的延伸反应,3’末端的羟基用氨基修饰。寡核苷酸命名为抑制性寡核苷酸egfr_l858r_sup_p3。egfr_l858r_sup_p3的核苷酸序列显示在seqidno:7。当寡核苷酸与egfr野生型基因的正链杂交时,在第2575位核苷酸的g位上形成g-t错配。另一方面,当寡核苷酸与其中第2573位核苷酸上的碱基被g置换的突变型基因的正链杂交时,在g位上形成g-a错配,并且另外,也在置换位点的3’方向上第二位(第2575位核苷酸)的g位上形成g-t错配。
(3)用于特异性突变型基因检测的探针设计和制备
嵌合寡核苷酸探针作为用于特异性检测egfrcodon858dna的探针被合成,其中寡核苷酸探针具有自egfrcodon858dna的与egfr基因上的第2573号核苷酸对应的位点3’方向延伸1个碱基和在5’方向延伸9个碱基的区域的互补链的核苷酸序列,并且自核苷酸序列的5’末端起第3个dna被rna置换。另外,寡核苷酸的5’末端结合于eclips猝灭剂,和寡核苷酸的3’末端结合于fam染料。因此制备的嵌合寡核苷酸探针命名为egfr_l858r_p2。egfr_l858r_p2的核苷酸序列显示在seqidno:8。
因为egfr_l858r_p2与其中第2573位核苷酸上的碱基t被g置换的突变型egfr基因完全杂交,egfr_l858r_p2的rna部分通过共存的核糖核酸酶h切割。与此相比,当egfr_l858r_p2与野生型egfr基因杂交时形成错配,并因此egfr_l858r_p2不经历核糖核酸酶h的切割。
(4)伴随野生型基因扩增抑制的特异性突变检测方法的研究
通过使hl60基因组dna与egfrcodon858dna以50000个拷贝:500个拷贝(=100:1)、50000个拷贝:50个拷贝(=1000:1)和50000个拷贝:5个拷贝(=10000:1)的比率混合,制备dna样品。作为对照,制备仅含有hl60基因组dna的样品。hl60基因组dna的50000个拷贝的dna量约为185ng。
伴随野生型基因扩增抑制的特异性突变检测如以下描述的那样实施。使用cycleavepcrreactionmix(由takarabioinc.制备),制备最终体积25μl的反应混合物,其含有上述的dna样品、112nmnucs蛋白、0.2μmegfr_l858r_sup_p3、各为0.2μm的egfr_l858r引物对和0.2μmegfr_l858r_sup_p2。通过热循环仪tp900(由takarabioinc.制备)实施实时pcr检测。在95℃下初始变性处理10秒,然后95℃5秒、55℃10秒和72℃20秒的45次循环的反应条件下实施pcr。随着时间推移观察荧光强度。同时,不含nucs蛋白的反应混合物组作为对照制备,并且进行相同的测量。结果显示在图2中。
具体地讲,图2显示用于egfr基因中l858r突变的检测方法的结果。纵轴显示荧光强度,和横轴显示pcr循环的数目。显示突变型基因的拷贝数目(0、5、50和500个拷贝)的荧光曲线。如自图2(b)可见的那样,当反应混合物不含有nucs蛋白时,未观察到基于嵌合寡核苷酸探针egfr_l858r_p2切割的荧光值增加。因此,这显示来自egfrcodon858dna的dna扩增被共存的hl60基因组dna抑制。
与此相比,如自图2(a)可见的那样,当使用含有nucs蛋白的反应混合物实施pcr时,甚至在相对于野生型基因的50000个拷贝含有突变型基因的5个拷贝的样品中观察到荧光值增加,即嵌合寡核苷酸egfr_l858r_p2的切割。换言之,其表明,egfr_l858r突变型基因能够在大量过量的共存野生型基因存在下进行检测。
该结果显示,即使当核酸含有不能形成通过多肽识别的错配的碱基置换时,用于基于核酸中碱基置换的存在或不存在区别性地选择性切割两个核酸中的一个的系统,也能够通过组合本发明提供的抑制性寡核苷酸和具有错配核酸内切酶活性的多肽(nucs蛋白)来构建。进一步地,其表明,通过组合上述的组合与核酸扩增反应,以高丰度比存在的来自野生型基因的核酸扩增受到抑制,并且来自突变型基因的核酸被优先扩增,从而可以高灵敏度检测突变型基因。
实施例3:在酸性高分子物质存在下的靶核酸检测方法研究
(1)在酸性高分子物质存在下的突变检测方法研究
如在实施例2(4)中描述的那样制备一系列dna样品。以同样的方式,通过使hl60基因组dna与egfrcodon858dna以5000个拷贝:500个拷贝(=10:1)、5000个拷贝:50个拷贝(=100:1)和5000个拷贝:5个拷贝(=1000:1)的比率混合,制备dna样品。作为对照,制备不含模板dna的样品。hl60基因组dna的5000个拷贝的dna量约为18.5ng。
使用cycleavepcrreactionmix(由takarabioinc.制备),制备最终体积25μl的反应混合物,其含有上述的dna样品、112nmnucs蛋白、0.2μmegfr_l858r_sup_p3、各为0.2μm的egfr_l858r引物、0.2μmegfr_l858r_sup_p2和最终浓度为56μg/ml、140μg/ml或168μg/ml的海藻酸钠。为了比较,制备除了不含海藻酸钠以外相同组成的反应混合物。
通过热循环仪tp900(由takarabioinc.制备)实施实时pcr检测。在95℃下初始变性处理10秒,然后95℃5秒、55℃10秒和72℃20秒的45次循环的反应条件下实施pcr。随着时间推移观察荧光强度。
(2)酸性高分子物质作用的分析
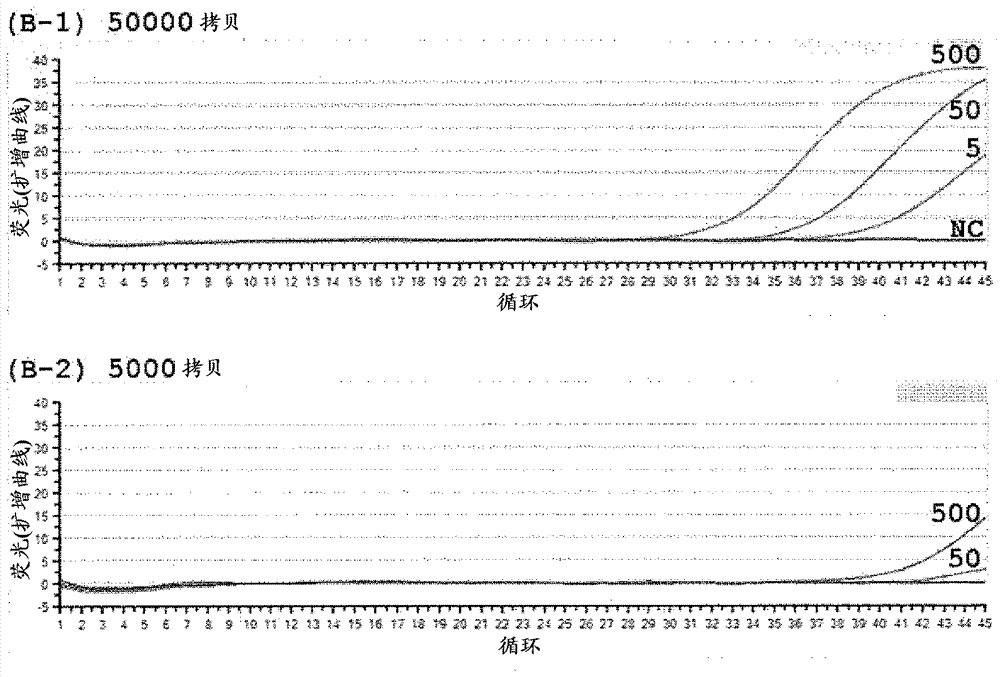
实时pcr检测的结果显示在图3中。具体地讲,图3显示酸性高分子物质在本发明突变型基因检测方法中的作用结果。纵轴显示荧光强度,和横轴显示pcr循环的数目。显示无模板dna(n.c.)的荧光曲线和突变型基因的拷贝数目(5、50和500个拷贝)。图3(a-1)、(b-1)、(c-1)和(d-1)显示其中存在hl60基因组dna的50000个拷贝情况下的结果,和图3(a-1)、(b-2)、(c-2)和(d-2)显示其中存在hl60基因组dna的5000个拷贝情况下的结果。图3(a)、(b)、(c)和(d)分别显示不存在海藻酸钠和存在56μg/ml、140μg/ml和168μg/ml海藻酸钠下的作用。结果发现,任何浓度下的海藻酸钠与不存在海藻酸钠相比较均增加检测效率。特别是,在少量模板dna(hl基因组dna的5000个拷贝)的情况下显示作用增加。因此发现,即使当来自样品的dna量小时,突变型基因也能够通过组合使用酸性高分子物质和本发明检测方法进行有效检测。
实施例4:酸性高分子物质对具有错配核酸内切酶活性的多肽的核酸切割反应的作用
酸性高分子物质的作用被进一步研究。如以下描述的那样制备反应混合物。使用cycleavepcrreactionmix(由takarabioinc.制备),制备最终体积25μl的反应混合物,其含有egfrcodon790dna的5个拷贝(0.16fg)、112nmnucs蛋白、0.2μmt790m_mucg-1、各为0.2μm的egfr_t790m引物对、0.2μmt790m_detect-1和最终浓度为140μg/ml的海藻酸钠。该反应混合物命名为反应混合物4。进一步制备:反应混合物1(其除了不含nucs蛋白和海藻酸钠以外与反应混合物4相同)、反应混合物2(其除了不含nucs蛋白以外与反应混合物4相同)和反应混合物3(其除了不含海藻酸钠以外与反应混合物4相同)。在如实施例1中描述的条件下实施实时pcr检测。
结果,在反应混合物1、反应混合物2和反应混合物4中观察到突变型基因的扩增,尽管这些反应混合物含有非常少的突变型基因拷贝数目。与此相比,在反应混合物3中未观察到扩增。
其表明,意想不到的杂交能够在反应混合物3的抑制性寡核苷酸与突变型基因之间发生(因为反应混合物中不存在野生型基因),并然后能够经nucs蛋白切割。与此相比,表明在反应混合物4中酸性高分子物质与nucs蛋白相互作用,并从而抑制意想不到的切割。
因此发现,酸性高分子物质经与nucs蛋白相互作用而具有控制nucs蛋白的错配识别和切割活性的作用。
实施例5:egfr_外显子19缺失突变基因的特异性检测
研究使用本发明的方法检测egfr基因中外显子19的缺失突变。在该实施例中,egfr_19_dele746-a750dna为靶核酸和人基因组dna(由takarabioinc.制备)为非靶核酸。
(1)用于检测的模板制备
自以genbank收录号nc_000007公开的人egfr的cds序列信息,人工合成与引物egfr_19_dele746-a750_f和egfr_19_dele746-a750_r对应的核苷酸序列和含有在引物的核苷酸序列之间区域的核苷酸序列并缺失第2236位-第2250位的核苷酸的dna。通过这样的碱基缺失,在人工合成的dna中缺失egfr中第746位-第750位氨基酸。因此得到的人工合成基因命名为egfr_19_dele746-a750dna,并且用作突变型基因。
(2)用于扩增的引物和抑制性寡核苷酸的制备
通过常规方法制备两种具有在seqidno:9和10显示的核苷酸序列的引物,egfr_19_dele746-a750_f和egfr_19_dele746-a750_r。然后,制备具有在seqidno:11显示的核苷酸序列的寡核苷酸,其可与egfr基因上的缺失区域杂交。为防止自寡核苷酸3’末端的延伸反应,3’末端的羟基用氨基修饰。寡核苷酸命名为抑制性寡核苷酸egfr_19_dele746-a750oligo-7。当寡核苷酸与egfr野生型基因的正链杂交时,在第2245位核苷酸的g位上形成g-c错配。另一方面,寡核苷酸不与缺失突变型基因的正链杂交。
(3)用于特异性突变型基因检测的探针的设计和制备
嵌合寡核苷酸探针作为用于特异性检测egfr_19_dele746-a750dna的探针被合成,其中寡核苷酸探针具有该dna中与egfr基因的第2228-2235位核苷酸和第2251-2253位核苷酸对应的区域的互补链的核苷酸序列,并且自核苷酸序列的5’末端起第3个dna被rna置换。另外,寡核苷酸的5’末端结合于eclips猝灭剂,和寡核苷酸的3’末端结合于fam染料。因此制备的嵌合寡核苷酸探针命名为egfr_2236-2250del。egfr_2236-2250del的核苷酸序列显示在seqidno:12。
因为egfr_2236-2250del与其中第2236-2250位核苷酸的核苷酸序列缺失的突变型egfr基因完全杂交,所以egfr_2236-2250del的rna部分通过共存的核糖核酸酶h切割。与此相比,egfr_2236-2250del不与野生型egfr基因杂交,并因此egfr_2236-2250del不经历核糖核酸酶h的切割。
(4)伴随野生型基因扩增抑制的特异性突变检测方法研究
通过使人基因组dna与egfr_19_dele746-a750dna以50000个拷贝:500个拷贝(=100:1)、50000个拷贝:50个拷贝(=1000:1)和50000个拷贝:5个拷贝(=10000:1)的比率混合,制备dna样品。作为对照,仅使用人基因组dna。人基因组dna的50000个拷贝的dna量约为185ng。
伴随野生型基因扩增抑制的特异性突变检测如以下描述的那样实施。使用cycleavepcrreactionmix,制备最终体积25μl的反应混合物,其含有上述的dna样品、687.5nmnucs蛋白、0.2μmegfr_2236-2250del、0.2μmegfr_19_dele746-a750引物对、0.2μmegfr_19_dele746-a750oligo-7和最终浓度为140μg/ml的海藻酸钠。通过热循环仪tp900实施实时pcr检测。在95℃下初始变性处理10秒,然后95℃5秒、55℃10秒和72℃20秒的45次循环的反应条件下实施pcr。随着时间推移观察荧光强度。同时,不含nucs蛋白的反应混合物组作为对照制备,并且进行相同的测量。结果显示在图4中。
具体地讲,图4显示用于egfr基因外显子19缺失突变的检测方法的结果。纵轴显示荧光强度,和横轴显示pcr循环的数目。显示突变型基因的拷贝数目(0、5、50和500个拷贝)的荧光曲线。当含有nucs蛋白的反应混合物用于pcr时,甚至在相对于野生型基因的50000个拷贝共存突变型基因的5个拷贝的样品中观察到荧光值增加,即嵌合寡核苷酸egfr_2236-2250del的切割。与此相比,当反应混合物不含nucs蛋白时,未观察到基于嵌合寡核苷酸探针egfr_2236-2250del切割的荧光值增加。换言之,其表明,来自egfr_19_dele746-a750dna的dna扩增由于人基因组dna的共存而受到抑制。
该结果显示,甚至在大量过量的野生型基因存在下,通过组合本发明提供的抑制性寡核苷酸与具有错配核酸内切酶活性的多肽(nucs蛋白),能够以高灵敏度检测期望的突变型基因,即使突变型基因含有缺失型基因突变时也没有问题。
实施例6:k-ras基因第12位密码子和第13位密码子的突变分析
研究了采用本发明方法检测k-ras基因中第12位密码子和第13位密码子的点突变。在该实施例中,k-ras基因的第12位密码子点突变体和第13位密码子点突变体为靶核酸,和hl60基因组dna为非靶核酸。
(1)用于检测的模板制备
自以genbank收录号nc_000012.12公开的人k-ras基因的信息,化学合成具有在seqidno:13和14显示的核苷酸序列的引物k-rasg12_f和k-rasg12_r。进一步地,人工合成具有引物的核苷酸序列之间区域的核苷酸序列的dna,其中第12位密码子特定的氨基酸通过第12位密码子的点突变自甘氨酸变为丝氨酸、自甘氨酸变为半胱氨酸、自甘氨酸变为精氨酸、自甘氨酸变为天冬氨酸、自甘氨酸变为缬氨酸和自甘氨酸变为丙氨酸。通过常规方法,使这些人工合成dna的各自插入到t-载体pmd20(由takarabioinc.制备)的t-克隆位点。因此制备的质粒命名为k-rasg12sdna、k-rasg12cdna、k-rasg12rdna、k-rasg12ddna、k-rasg12vdna和k-rasg12adna,并且用作突变型基因。
以同样的方式,人工合成具有与k-rasg12_f和k-rasg12_r的引物对对应的核苷酸序列之间区域的核苷酸序列的dna,其中第13位密码子特定的氨基酸通过第13位密码子的点突变自甘氨酸变为丝氨酸、自甘氨酸变为半胱氨酸、自甘氨酸变为精氨酸、自甘氨酸变为天冬氨酸和自甘氨酸变为丙氨酸。通过常规方法,使因此得到的人工合成dna各自插入到t-载体pmd20的t-克隆位点。因此制备的质粒命名为k-rasg13sdna、k-rasg13cdna、k-rasg13rdna、k-rasg13ddna和k-rasg13adna,并且用作突变型基因。
(2)抑制性寡核苷酸的制备
制备具有在seqidno:15显示的核苷酸序列的寡核苷酸,其可与k-ras基因中含有第12位密码子和第13位密码子的区域杂交。为防止自寡核苷酸3’末端的延伸反应,3’末端的羟基用氨基修饰。寡核苷酸命名为抑制性寡核苷酸k-rasg12/13_s1。当寡核苷酸与具有在seqidno:16显示的核苷酸序列的k-ras第12位密码子野生型基因的正链杂交时,在第35位核苷酸的g位上形成g-g错配。另一方面,当寡核苷酸与k-rasg12sdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第34位核苷酸的a位上形成a-c错配。当寡核苷酸与k-rasg12cdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第34位核苷酸的t位上形成t-c错配。当寡核苷酸与k-rasg12rdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第34位核苷酸的c位上形成c-c错配。当寡核苷酸与k-rasg12ddna的正链杂交时,在第35位核苷酸的a位上形成a-g错配。当寡核苷酸与k-rasg12vdna的正链杂交时,在第35位核苷酸的t位上形成t-g错配。当寡核苷酸与k-rasg12adna的正链杂交时,没有错配形成。
当抑制性寡核苷酸k-rasg12/13_s1与k-ras第13位密码子野生型基因的正链杂交时,在第35位核苷酸的g位上形成g-g错配。另一方面,当寡核苷酸与k-rasg13sdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第37位核苷酸的a位上形成a-c错配。当寡核苷酸与k-rasg13cdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第37位核苷酸的t位上形成t-c错配。当寡核苷酸与k-rasg13rdna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第37位核苷酸的c位上形成c-c错配。当寡核苷酸与k-rasg13ddna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第38位核苷酸的a位上形成a-c错配。当寡核苷酸与k-rasg13adna的正链杂交时,在第35位核苷酸的g位上形成g-g错配,并且另外,在第38位核苷酸的c位上形成c-c错配。
(3)野生型基因扩增抑制的证实
通过使人基因组dna与k-rasg12cdna以50000个拷贝:50个拷贝(=1000:1)的比率混合,制备dna样品。作为对照,仅使用人基因组dna。人基因组dna的50000个拷贝的dna量为约185ng。
也以同样的方式,通过使人基因组dna与k-rasg13cdna以50000个拷贝:50个拷贝(=1000:1)的比率混合来制备dna样品。
制备最终体积25μl的反应混合物,其含有上述的dna样品、1000nmnucs蛋白、各为0.2μm的k-rasg12_f和k-rasg12_r引物对、0.2μmk-rasg12/13_s1和最终浓度为140μg/ml的海藻酸钠。通过热循环仪tp900实施pcr检测。在95℃下初始变性处理10秒,然后95℃5秒、55℃10秒和72℃20秒的45次循环的反应条件下实施pcr。因此得到的扩增片段经takaraminibestdna片段纯化试剂盒版本号4.0(由takarabioinc.制备)纯化,并然后使用具有在seqidno:13显示的核苷酸序列的寡核苷酸进行直接测序。
自直接测序的结果,来自上述的含有k-rasg12cdna的样品的主要扩增产物被鉴定为来自k-rasg12cdna的扩增产物。另外,来自含有k-rasg13cdna的样品的主要扩增产物也被鉴定为来自k-rasg13cdna的扩增产物。因此发现,通过本发明的方法,野生型基因的扩增被抑制和突变型基因被优先扩增。换言之,其表明,突变型基因可通过本发明的方法选择性浓缩。
实施例7:与酸性高分子物质组合的高灵敏度检测方法
如在实施例2中描述的那样,在具有l858r突变的egfr基因的特异性检测方法模型上进一步研究酸性高分子物质的作用。在该实施例中,研究具有糖链骨架的硫酸软骨素b的盐或不具有糖链骨架的聚丙烯酸作为酸性高分子物质的用途。
(1)酸性高分子物质的作用-1
使用cycleavepcrreactionmix,制备最终体积25μl的反应混合物,其含有上述的在实施例3(1)中制备的dna样品、125nmnucs蛋白、0.2μmegfr_l858r_sup_p3、各为0.2μm的egfr_l858r引物对、0.2μmegfr_l858r_sup_p2和最终浓度为30μg/ml、40μg/ml、50μg/ml或60μg/ml的硫酸软骨素b钠盐(由sigma-aldrich制备)。为了比较,也制备除了不含硫酸软骨素b钠盐以外相同组成的反应混合物。
在与实施例3(1)相同的条件下实施实时pcr检测。结果,与反应混合物不含硫酸软骨素b钠盐的情况相比较,当反应混合物含有任何最终浓度的硫酸软骨素b钠盐时,ct值明显减少。因此发现,具有糖链骨架的酸性高分子物质在本发明的方法中是有用的。
(2)酸性高分子物质的作用-2
接着,研究了聚丙烯酸5000(由wakopurechemicalindustries,ltd.制备)作为不具有糖链骨架的酸性高分子物质的用途。除了含有最终浓度1μg/ml、2μg/ml、3μg/ml、4μg/ml或5μg/ml的聚丙烯酸5000替代硫酸软骨素b钠盐以外,制备最终体积25μl的与在以上(1)中描述的相同组成的反应混合物。
作为实时pcr检测的结果,与反应混合物不含聚丙烯酸的情况相比较,当反应混合物含有任何最终浓度的聚丙烯酸时,ct值明显减少。因此发现,不具有糖链骨架的酸性高分子物质在本发明的方法中是有用的。
实施例8:本发明的方法与阳性对照核酸的扩增组合
研究采用本发明的方法定量靶核酸的方法。egfr_l858r用作靶核酸。egfr基因中另一个区域用作阳性对照核酸。具体地讲,通过常规方法,合成具有在seqidno:17和18显示的核苷酸序列的引物egfrl858r_ic2_f和egfrl858r_ic2_r。进一步地,嵌合寡核苷酸探针作为用于检测阳性对照核酸的探针被合成,其中寡核苷酸探针具有在seqidno:19显示的核苷酸序列,并且自核苷酸序列的5’末端起第3个dna被rna置换。另外,寡核苷酸的5’末端结合于eclips猝灭剂,和寡核苷酸的3’末端结合于fam染料。因此制备的嵌合寡核苷酸探针命名为egfrl858r_ic2_p2。
作为模板dna,使用在实施例2中制备的dna样品。如以下描述的那样制备反应混合物。具体地讲,使用cycleavepcrreactionmix,制备最终体积25μl的反应混合物,其含有dna样品、125nmnucs蛋白、0.2μmegfr_l858r_sup_p3、各为0.2μm的egfr_l858r引物对、0.2μmegfr_l858r_sup_p2和最终浓度为56μg/ml的海藻酸钠、及各为0.2μm的egfrl858r_ic2引物对和0.2μmegfrl858r_ic2_p2。在与实施例3中描述的相同条件下实施实时pcr检测。
实时pcr检测显示与实施例3相同的结果,而与同时实施阳性对照核酸的扩增无关。因此发现,反应混合物之间的误差可基于作为指标的阳性对照核酸的扩增量进行校正。也发现,样品中靶核酸的存在能够通过比较样品中扩增的对照核酸的扩增量和靶核酸的扩增量进行定量。因此发现,即使当来自样品的dna量小时,突变型基因也被本发明的检测方法有效检测,并且突变型基因也可通过与阳性对照核酸的扩增比较来定量。
实施例9:本发明的方法与pcna组合
研究了pcna在本发明方法中的作用。反应混合物的组成与在实施例8中描述的相同,除了反应混合物含有作为嵌合寡核苷酸探针的egfrl858r_ic2_p3(其具有在seqidno:20显示的核苷酸序列,并且自核苷酸序列的5’末端起第3个dna被rna置换)、100μg/ml硫酸软骨素b钠盐、8μg/ml通过在wo2007/004654中描述的方法制备的pufpcnad143r突变体(pcna13)和375nm的nucs蛋白。在与实施例3中描述的相同条件下实施实时pcr检测。
作为实时pcr检测的结果,与反应混合物不含pcna的情况相比较,当反应混合物含有pcna时,ct值明显减少,而与同时实施阳性对照核酸的扩增无关。因此发现,使用pcna进一步改善本发明的方法。
工业适用性
根据本发明的高灵敏度突变检测方法,通过人为地允许发生错配碱基对的形成,大量存在的野生型基因的扩增能被消除,很少量存在的突变型基因可被特异性地扩增和检测,而与具有错配核酸内切酶活性的多肽原本能识别的错配类型无关。本发明的方法可用于广泛领域,包括基因技术、生物学、医学和农业。
序列表自由文本
seqidno:1:egfr_t790m_f引物
seqidno:2:egfr_t790m_r引物
seqidno:3:t790m_nucg-1。3’-末端用氨基接头(nh2)修饰。
seqidno:4:t790m_detect-1。5’-末端被加入eclips和3’-末端被加入fam。第4位核苷酸为rna。
seqidno:5:egfr_l858r_f引物
seqidno:6:egfr_l858r_r引物
seqidno:7:egfr_l858r_sup_p3。3’-末端用氨基接头(nh2)修饰。
seqidno:8:egfr_l858r_p2。5’-末端被加入eclips和3’-末端被加入fam。第3位核苷酸为rna。
seqidno:9:egfr_19_dele746-a750_f引物
seqidno:10:egfr_19_dele746-a750_r引物
seqidno:11:egfr_19_dele746-a750oligo-7。3’-末端用氨基接头(nh2)修饰。
seqidno:12:egfr_2236-2250del。5’-末端被加入eclips和3’-末端被加入fam。第3位核苷酸为rna。
seqidno:13:k-rasg12_f引物
seqidno:14:k-rasg12_r引物
seqidno:15:k-rasg12/13_s1。3’-末端用氨基接头(nh2)修饰。
seqidno:16:含有k-ras第12位密码子和第13位密码子区域的seqiddna片段
seqidno:17:egfrl858r_ic2_f引物
seqidno:18:egfrl858r_ic2_r引物
seqidno:19:egfrl858r_ic2_p2。5’-末端被加入eclips和3’-末端被加入fam。第3位核苷酸为rna。
seqidno:20:egfrl858r_ic2_p3。5’-末端被加入eclips和3’-末端被加入fam。第3位核苷酸为rna。
- 还没有人留言评论。精彩留言会获得点赞!