一种环保型正交保护的二氨基二酸类化合物、其制备方法及其应用与流程
一、技术领域
本发明涉及一种环保型正交保护的二氨基二酸类化合物、其制备方法及其应用。
二、
背景技术:
二硫键广泛存在于折叠的蛋白质结构中,能促进蛋白质的三级结构的稳定。但是一些具有潜在药用和科研研究价值的蛋白质因其天然二硫键容易在还原性化学条件和生理环境下断裂,从而影响蛋白质的结构和生理功能。目前用二氨基二酸骨架替换二硫键来解决这类问题是一个行之有效的策略。二氨基二酸模拟二硫键不仅能提高多肽药物的抗降解性而且能在一定程度增强生理活性。在多肽中嵌入二硫键骨架类似物时,需要对二氨基二酸两端进行正交保护,以适应fmoc法合成多肽的化学条件。
正交保护的二氨基二酸类化合物有多种,目前已经报道的二氨基二酸的种类及其应用有:
文献(angew.chem.int.ed.2013,52,9558–9562)首次报道将二氨基二酸作为替换二硫键的骨架嵌入多肽中,开发了用alloc和allyl保护的c-s骨架、s-c骨架型二氨基二酸以及pnb和pnz保护的c-s骨架、s-c骨架型二氨基二酸。但二氨基二酸正交保护基的脱除需要重金属钯,易造成环境污染,而且蛋白质易吸附钯离子从而对自身活性产生影响。
文献(angew.chem.int.ed.2015,54,1–7)首次证实c-s、s-c型骨架二氨基二酸对多肽生物活性有不同的影响。因此完善和开发环保型二氨基二酸种类对多肽蛋白质的研究具有较为重要的意义。
文献(tetrahedronletters58(2017)1677–1680)开发了一种正交保护基脱除环保型的c-s骨架二氨基二酸。
三、
技术实现要素:
本发明旨在提供一种环保型正交保护的二氨基二酸类化合物、其制备方法及其应用。本发明二氨基二酸类化合物能绿色高效脱除正交保护基,增加了二氨基二酸骨架种类。
本发明环保型正交保护的二氨基二酸类化合物,在s-c结构中,硫元素在远dmab/ivdde保护端的氨基酸衍生物侧链上,其结构式具体为:
其中dmab、ivdde的结构表达如下:
本发明环保型正交保护的二氨基二酸类化合物的制备方法,包括如下步骤:
步骤1:将半胱氨酸衍生物和烯丙基溴溶于有机溶剂中,随后加入弱碱盐,常温下搅拌反应2-10h;反应结束后加入乙酸乙酯,然后用饱和食盐水洗涤有机相,向所得有机相中加入无水硫酸钠干燥,过滤并浓缩有机相,柱层析分离(洗脱液为石油醚:乙酸乙酯=3:1或5:1,v/v)获得中间产物ⅰ;
各原料的添加量以半胱氨酸或其衍生物为基准,烯丙基溴当量为1-2,弱碱盐当量为1-2。
所述弱碱盐为碳酸氢钠或碳酸氢钾。
所述有机溶剂为dmf或dcm;有机溶剂添加量以半胱氨酸衍生物为基准,为1.5-3ml/mmol。
所述半胱氨酸衍生物的通式为:
其中r1为trt、mmt或acm等。
本步骤的反应过程如下:
步骤2:保护基保护
2a、将高丝氨酸和弱碱盐溶于水中,在冰浴条件下缓慢加入二恶烷并搅拌,随后再加入boc酸酐,室温下搅拌反应2-10h,反应结束后将反应液用乙醚洗涤,随后将水相ph值调至2-4,用乙酸乙酯萃取水相后合并有机相,饱和食盐水洗涤有机相,无水硫酸钠干燥后旋蒸除去溶剂,得到n端boc保护的高丝氨酸残基;
以高丝氨酸为基准,boc酸酐的当量为1-3,弱碱盐的当量为2-6,水的添加量为1.5-4ml/mmol,二恶烷的添加量为0.8-1.5ml/mmol。
所述弱碱盐为碳酸氢钠或碳酸氢钾。
本步骤的反应过程如下:
2b、将2a所得n端boc保护的高丝氨酸残基和tebac及弱碱盐溶于有机溶剂中,随后缓慢加入叔丁基卤并搅拌,40-70℃无光照条件下反应24-48h;反应结束后用水稀释有机相,加入乙酸乙酯萃取,合并有机相并旋蒸浓缩,水洗并干燥后得到c端tbu保护的高丝氨酸残基(即n-叔丁氧羰基-l-高丝氨酸叔丁酯);
以n端boc保护的高丝氨酸残基为基准,tebac当量为1-3,弱碱盐当量为4-6,叔丁基卤当量为4-10,有机溶剂的添加量为1.5-3ml/mmol。
所述弱碱盐为碳酸氢钠或碳酸氢钾。
所述叔丁基卤为叔丁基氯、叔丁基溴或叔丁基碘;
所述有机溶剂为dmf或dcm等。
本步骤的反应过程如下:
2c、将2b所得c端tbu保护的高丝氨酸残基直接和四溴化碳混合并溶于二氯甲烷(4-8ml/mmol)中,在冰浴条件下缓慢滴加三苯基膦的二氯甲烷溶液,在惰性气体保护下于室温反应1-4h;反应所得混合物用二氯甲烷稀释,随后加入饱和食盐水洗涤,无水硫酸钠干燥,过滤后旋蒸浓缩,浓缩物用体积比1:1的乙酸乙酯/石油醚混合液萃取,沉淀出磷的氧化物,将萃取液浓缩干燥,硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v)得到中间产物ⅱ(即(2s)-3-溴-2-(叔丁氧羰基)氨基-丁酸叔丁酯);
以c端tbu保护的高丝氨酸残基为基准,四溴化碳当量为1-2,三苯基膦的当量为1-2。
本步骤的反应路线为:
步骤3:耦合反应
3a、将中间产物ⅰ加入二氯甲烷、脱除试剂、三异丙基硅烷的混合液中,常温下搅拌反应1-2h,反应结束后向反应液中加入二氯甲烷和水稀释,再加入碳酸氢钠调节ph值为中性,分离留取有机相,洗涤、干燥并浓缩后得到侧链裸露的半胱氨酸残基。
以中间产物ⅰ为基准,三异丙基硅烷的当量为2-4,二氯甲烷的添加量为10-20ml/mmol,三氟乙酸的添加量为20-40ml/mmol。
本步骤的反应过程如下:
其中r1为trt、mmt或acm等。
脱除试剂的选择与中间产物ⅰ的结构有关:当r1为trt时,脱除试剂为三氟乙酸,以中间产物ⅰ为基准添加量为10-20ml/mmol;当r1为mmt时,脱除试剂为体积分数2%的三氟乙酸的dcm溶液,以中间产物ⅰ为基准三氟乙酸的dcm溶液的添加量为10-20ml/mmol;当r1为acm时,脱除试剂为碘的乙酸和水的混合溶液,以中间产物ⅰ为基准碘的添加量为20倍当量,混合溶液中乙酸和水的体积比为1:1,以中间产物ⅰ为基准添加量为20-30ml/mmol。
3b、在氮气保护下,将3a所得侧链裸露的半胱氨酸残基和中间产物ⅱ溶于乙酸乙酯(15-30ml/mmol)中,随后加入四丁基溴化铵的饱和碳酸氢钠溶液,常温下搅拌反应8-12小时,反应结束后用乙酸乙酯萃取反应液,饱和食盐水洗涤有机相,无水硫酸钠干燥,过滤后旋蒸有机相,硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1或2:1,v/v)得到耦合的高丝氨酸端tbu和boc保护的二氨基二酸(即s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-叔丁氧羰基-l-高半胱氨酸叔丁酯);
以侧链裸露的半胱氨酸残基为基准,中间产物ⅱ的当量为1-2、四丁基溴化铵的当量为2-4,饱和碳酸氢钠溶液的添加量为4-6ml/mmol。
本步骤的反应过程如下:
3c、将3b所得化合物溶于三氟乙酸(以3b为基准当量为3-6ml/mmol)的二氯甲烷溶液中,在室温条件下搅拌反应2-12h,反应结束后将反应液浓缩、干燥并溶于乙酸乙酯中,随后依次加入ivdde-oh和tfa(以3b为基准当量为0.1-0.5),50-90℃、惰性气体保护下加热回流反应8-12小时,反应结束后向反应液中加入乙酸乙酯及盐酸,分离有机相并用无水硫酸镁干燥,浓缩后经过硅胶柱分离(洗脱液为石油醚:乙酸乙酯=4:1,v/v)得到高丝氨酸残基端羧酸裸露、氨基ivdde保护的二氨基二酸(即s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸);
以耦合的高丝氨酸端tbu和boc保护的二氨基二酸为基准,ivdde-oh的当量为1-3,首次加入的三氟乙酸的添加量为3-6ml/mmol,第二次加入的三氟乙酸的当量为0.1-0.5,二氯甲烷的添加量为3-6ml/mmol。
所用盐酸浓度为1-3mol/l。
本步骤的反应过程如下:
3d、将3c所得化合物溶于二氯甲烷中,然后加入dmab-oh、dcc和dmap,常温下反应2-12h,反应结束后过滤得到有机相,然后向有机相中加入饱和食盐水洗涤,分离合并有机相后用无水硫酸钠干燥,有机相浓缩后硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v),得到高丝氨酸残基端羧酸dmab保护、氨基ivdde保护的二氨基二酸(即s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯);
以高丝氨酸残基端羧酸裸露、氨基ivdde保护的二氨基二酸为基准,dmab-oh的当量为1-3、dcc的当量为1-2、dmap的当量为0.1-0.5,二氯甲烷的添加量为4-8ml/mmol。
本步骤的反应过程如下:
3e、将3d得到的化合物加入溶解有三苯基磷钯和苯硅烷的二氯甲烷中,常温下搅拌反应1-2h,随后加入甲醇(3-6ml/mmol),常温下搅拌5-30min后将反应液浓缩,硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v)得到目标产物(即s-(r)-((2-(芴甲氧羰基)氨基-2-羧基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯);
以高丝氨酸残基端羧酸dmab保护、氨基ivdde保护的二氨基二酸为基准,三苯基磷钯的当量为2-6、苯硅烷的当量为0.5-1,二氯甲烷的添加量为8-15ml/mmol。
本步骤的反应过程如下:
本发明环保型正交保护的二氨基二酸类化合物的应用,是以本发明二氨基二酸类化合物合成多肽时,首先将二氨基二酸类化合物嵌入多肽序列中,在环化前通过脱保护试剂将dmab和ivdde保护基脱除。
所述脱保护试剂为质量浓度1-3%的nh2nh2溶液和质量浓度15-25%的哌啶溶液。
保护基脱除的反应在常温下进行,反应时间5-10min;每种脱保护试剂分别重复脱除2-4次。
与已有的二氨基二酸种类相比,本发明的有益效果体现在:
1、本发明二氨基二酸用于多肽合成时,高丝氨酸残基端保护基可在1-3%的肼和15-25%哌啶中较为绿色、温和脱除。蛋白质多肽研究需要较为苛刻的生物条件,本发明的脱除条件避免前人工作中保护基脱除时所用钯试剂对多肽造成的污染。
2、本发明合成了带有易绿色脱除的保护基且骨架为s-c的二氨基二酸,增加了二氨基二酸种类,为研究不同骨架对多肽的影响提供了便利。
四、附图说明
图1是n-芴甲氧羰基-s-三苯甲基-l-半胱氨酸烯丙酯的质谱谱图。
图2是s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-叔丁氧羰基-l-高半胱氨酸叔丁酯的质谱图。
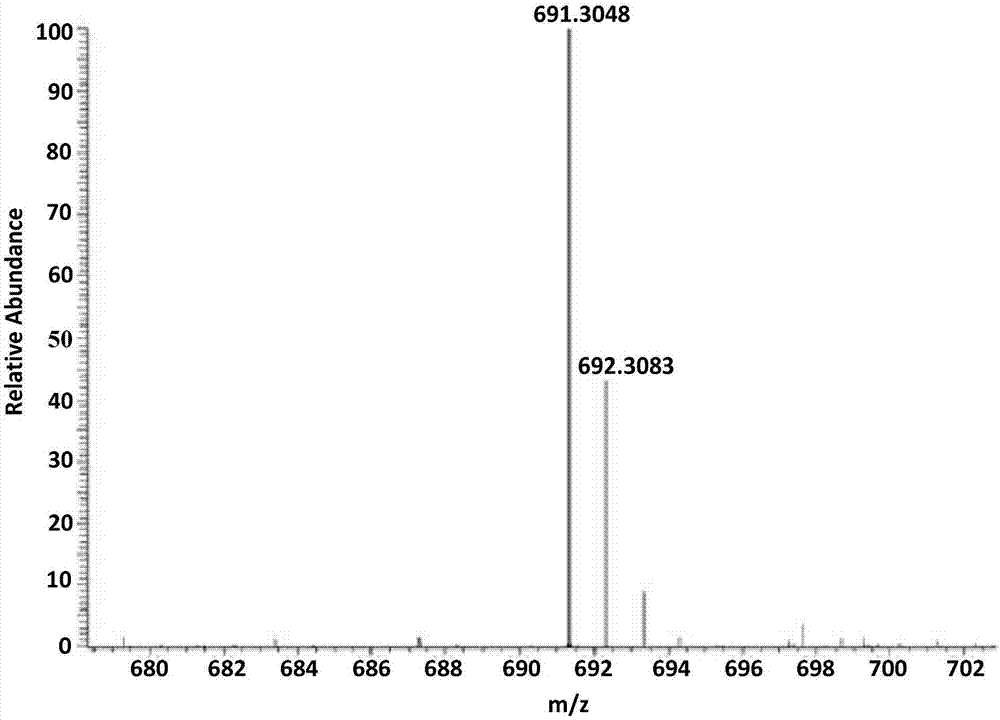
图3是s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸的质谱图。
图4是s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯的质谱图。
图5是s-(r)-((2-(芴甲氧羰基)氨基-2-羧基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯的质谱图。
图6是本发明环保型正交保护的二氨基二酸类化合物在合成多肽时脱除保护基的示意图。
五、具体实施方式
为了便于理解本发明,下面结合具体的实施例对本发明的实施过程作进一步说明,这些描述只是为了进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
实施例1:n-芴甲氧羰基-s-三苯甲基-l-半胱氨酸烯丙酯的制备
将8.4mmol半胱氨酸或其衍生物(用fmoc-cys(trt)或侧链trt保护的半胱氨酸表示)溶于20mldmf中,随后加入碳酸氢钠10mmol和烯丙基溴10mmol并搅拌反应过夜;反应结束后加入乙酸乙酯,然后用饱和食盐水洗涤有机相,所得有机相中加入无水硫酸钠干燥,过滤并浓缩有机相,柱层析分离(石油醚/乙酸乙酯=5:1,v/v)得到4.7mmol中间产物ⅰ。
esi-ms(positive):648.2179(observed,m+na);625.2287(calculated,m).
1hnmr(400mhz,cdcl3)δ7.76(dd,j=7.4,3.7hz,2h),7.60(dd,j=7.2,2.6hz,2h),7.40(dd,j=5.4,3.4hz,8h),7.29(dd,j=12.8,7.3hz,6h),7.24(d,j=2.2hz,2h),7.22–7.17(m,3h),5.87(dq,j=10.9,5.7hz,1h),5.35–5.21(m,3h),4.65–4.53(m,2h),4.44–4.30(m,3h),4.22(t,j=7.0hz,1h),2.66(qd,j=12.5,5.5hz,2h).
实施例2:n-叔丁氧羰基-l-高丝氨酸叔丁酯的制备
将10mmol高丝氨酸、40mmol碳酸氢钠溶于30ml水中,再缓慢加入10ml二恶烷并搅拌5min,冰浴条件下加入15mmolboc酸酐,随后在室温下搅拌反应12h;反应结束后将反应液用乙醚洗涤两次,用2m的盐酸将水相ph调至2,乙酸乙酯萃取水相后合并有机相,饱和食盐水洗涤有机相,无水硫酸钠干燥后旋蒸除去溶剂,得到n端boc保护的高丝氨酸残基;
将所得n端boc保护的高丝氨酸残基和10mmoltebac、54mmol碳酸钾溶于25mldmf中,随后缓慢加入90mmol叔丁基溴并搅拌,50℃无光照条件下反应48h;反应结束后用75ml水稀释有机相,加入乙酸乙酯萃取,合并有机相并旋蒸浓缩至50ml,水洗并干燥有机相,得到c端tbu保护的高丝氨酸残基。
1hnmr(400mhz,cdcl3)δ5.37(s,1h),4.36(s,1h),3.74–3.62(m,2h),2.27–2.07(m,2h),1.47(s,9h),1.45(s,9h).
实施例3:(2s)-3-溴-2-(叔丁氧羰基)氨基-丁酸叔丁酯的制备
将实施例2制备得到的4mmolc端tbu保护的高丝氨酸残基直接和5mmol四溴化碳混合并溶于25ml二氯甲烷溶液中,冰浴条件下缓慢滴加5mmol三苯基膦和10ml二氯甲烷的混合液,在室温和氮气氛围下反应2h;二氯甲烷稀释反应所得混合物,饱和食盐水洗涤,无水硫酸钠干燥,过滤后旋蒸浓缩至10ml,浓缩后所得溶液用体积比1:1的乙酸乙酯和石油醚萃取,沉淀出磷的氧化物,将萃取液浓缩干燥后通过硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v)得到3.1mmol中间产物ⅱ;
1hnmr(400mhz,cdcl3)δ5.11(d,j=6.5hz,1h),4.28(d,j=4.9hz,1h),3.49–3.37(m,2h),2.41–2.08(m,2h),1.48(s,9h),1.45(s,9h).
实施例4:s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-叔丁氧羰基-l-高半胱氨酸叔丁酯的制备
将2.2mmol中间产物ⅰ溶于40ml二氯甲烷中,随后加入66ml三氟乙酸和6.6mmol三异丙基硅烷,常温下搅拌反应1h;反应结束后加入二氯甲烷和水稀释,再加入碳酸氢钠调节ph值为中性,分离得到有机相并洗涤、干燥、浓缩,得到侧链裸露的半胱氨酸残基;
在氮气保护下,将所得侧链裸露的半胱氨酸残基和2mmol中间产物ⅱ溶于50ml乙酸乙酯中,随后加入溶有2.6g四丁基溴化铵的饱和碳酸氢钠溶液(10ml),氮气保护下搅拌反应过夜;反应结束后用乙酸乙酯萃取反应液,饱和食盐水洗涤有机相,无水硫酸钠干燥,过滤后旋蒸有机相,硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1或2:1,v/v)得到1.1mmol耦合的高丝氨酸端tbu和boc保护的二氨基二酸。
esi-ms(positive):641.2891(observed,m+na);640.2818(calculated,m).
1hnmr(400mhz,cdcl3)δ7.77(d,j=7.5hz,2h),7.61(d,j=7.4hz,2h),7.40(t,j=7.4hz,2h),7.32(t,j=7.4hz,2h),5.93(dq,j=11.0,5.8hz,1h),5.73(d,j=7.4hz,1h),5.32(dd,j=33.9,13.5hz,2h),5.12(d,j=7.6hz,1h),4.74–4.57(m,3h),4.47–4.33(m,2h),4.24(t,j=7.2hz,2h),3.03(d,j=3.9hz,2h),2.70–2.47(m,2h),2.12–2.01(m,1h),1.86(dt,j=14.3,8.0hz,1h),1.46(d,j=4.9hz,9h),1.44(s,9h).
实施例5:s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸的制备
将实施例4中得到的耦合的高丝氨酸端tbu和boc保护的二氨基二酸1mmol溶于5ml二氯甲烷中,加入5ml三氟乙酸,在室温条件下搅拌反应12h;反应结束后将反应液浓缩、干燥并溶于5ml乙酸乙酯中,随后依次加入2mmolivdde-oh和0.1mmoltfa,85℃、氮气保护条件下加热回流过夜,反应结束后加入乙酸乙酯稀释,1mhcl溶液洗涤,分离有机相并加入无水硫酸镁干燥,浓缩后经过硅胶柱分离(洗脱液为石油醚:乙酸乙酯=4:1,v/v),得到0.4mmol产物。
esi-ms(positive):691.3048(observed,m+h);690.2975(calculated,m).
1hnmr(400mhz,cdcl3)δ14.12(d,j=8.4hz,1h),7.76(d,j=7.5hz,2h),7.60(d,j=7.3hz,2h),7.40(t,j=7.4hz,2h),7.31(t,j=7.4hz,2h),5.98–5.83(m,1h),5.82–5.73(m,1h),5.41–5.24(m,2h),4.75–4.59(m,4h),4.39(d,j=7.0hz,2h),4.23(t,j=7.0hz,1h),3.12–3.03(m,2h),2.68(d,j=7.0hz,3h),2.37(d,j=14.2hz,4h),2.28–2.13(m,3h),1.61(d,j=6.8hz,1h),1.06–0.89(m,12h).
实施例6:s-(r)-((2-(芴甲氧羰基)氨基-2-烯丙氧羰基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯的制备
将实施例5所得产物0.4mmol溶于2ml二氯甲烷中,然后加入1mmoldmab-oh、0.5mmoldcc和0.05mmoldmap,常温下搅拌反应12h;反应结束后过滤混合液得到有机相,饱和食盐水洗涤,分离合并有机相后用无水硫酸钠干燥,浓缩并用硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v),得到产物0.1mmol。
esi-ms(positive):1002.30(observed,m+h);1001.49(calculated,m).
1hnmr(400mhz,cdcl3)δ7.76(d,j=7.5hz,2h),7.60(d,j=6.7hz,2h),7.39(dd,j=12.6,4.9hz,4h),7.30(dd,j=12.2,4.8hz,3h),7.11(d,j=8.2hz,2h),6.01–5.85(m,1h),5.79(d,j=7.9hz,1h),5.35(d,j=16.9hz,1h),5.27(d,j=10.7hz,1h),5.21(t,j=9.1hz,1h),4.67(q,j=12.4hz,3h),4.47–4.33(m,2h),4.23(t,j=7.0hz,1h),3.01–2.90(m,4h),2.63(d,j=3.3hz,2h),2.41(d,j=25.2hz,8h),2.25–2.11(m,2h),1.93–1.79(m,3h),1.61(dd,j=17.7,10.3hz,2h),1.06(d,j=13.2hz,6h),1.02(s,6h),0.95(t,j=5.4hz,3h),0.89(t,j=8.5hz,3h),0.76(d,j=6.4hz,6h).
实施例7:s-(r)-((2-(芴甲氧羰基)氨基-2-羧基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯的制备
将实施例6中所得产物1mmol加入溶解有6mmol三苯基磷钯和0.6mmol苯硅烷的二氯甲烷10ml中,搅拌反应1.5h,随后加入甲醇搅拌反应10min,所得溶液浓缩后用硅胶柱分离(洗脱液为石油醚:乙酸乙酯=5:1,v/v)得到目标产物。
esi-ms(positive):984.4432(observed,m+na);961.4547(calculated,m).
1hnmr(400mhz,cdcl3)δ14.03(d,j=8.4hz,1h),7.65(d,j=7.5hz,2h),7.51(d,j=6.7hz,2h),7.28(t,j=7.7hz,4h),7.22–7.16(m,2h),7.01(d,j=8.3hz,2h),6.22(s,1h),5.11(dd,j=24.9,12.5hz,2h),4.94(s,1h),4.61(s,1h),4.27(dd,j=9.3,6.7hz,1h),4.21–4.08(m,2h),2.98–2.88(m,3h),2.79(d,j=15.0hz,2h),2.53(s,2h),2.41–2.23(m,8h),2.16–2.02(m,2h),1.76(ddd,j=20.2,12.6,7.0hz,3h),1.17(dd,j=13.9,6.3hz,4h),0.99(s,6h),0.92(s,6h),0.86–0.76(m,6h),0.70–0.63(m,6h).
实施例8:s-(r)-((2-(芴甲氧羰基)氨基-2-羧基)乙基)-n-(1-(2-甲基)丙基-2-(4,4-二甲基-2,6-二氧代环己基)乙烯基)-l-高半胱氨酸-(1-(4-((1-(4,4-二甲基-2,6-二氧代环己亚基)-3-甲基丁基)氨基)苯基)甲基)酯的在多肽合成中的应用
fmoc法合成多肽时,在需要替换二硫键的位点或需要人工添加二硫键模拟骨架的位点,嵌入本发明二氨基二酸类化合物的fmoc端,随后继续用spps法合成多肽直至环化前一个氨基酸;然后用用质量浓度20%的哌啶的dmf溶液常温下脱除5min,脱除两次,最后再用质量浓度2%的肼的dmf溶液常温下脱除10min,脱除两次;dmab和ivdde脱除后可继续用spps法合成多肽。
综上所述,本发明提供了一种新型二氨基二酸的合成方法,改善了脱除条件,扩增了二氨基二酸种类。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,也不因各实施例之间的前后顺序对本发明造成任何限制,任何熟悉本领域的技术人员在本发明透露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!