一种N‑甲基‑美达霉素类化合物及其制备方法和应用与流程
本发明涉及生物技术领域,特别是涉及一种n-甲基-美达霉素类化合物及其制备方法和应用。
背景技术:
海洋放线菌与真菌因其产生结构新颖、功能多样的天然活性产物而备受关注。特殊多样的海洋生态环境,赋予了海洋放线菌与真菌复杂独特的代谢途径,其次级代谢产物在结构类型与生物活性等方面都呈现出与陆生放线菌与真菌不同的特点与多样性,成为了发现天然活性产物与新药先导化合物的重要来源。
同时在链霉菌的次级代谢产物中,芳香聚酮类的抗生素是一类结构功能较为多样化的化合物。在链霉菌的生物合成途径中,这类化合物首先是在细胞中聚酮合酶、酮还原酶、芳香化酶以及环化酶等的作用下合成化合物的芳香聚酮碳骨架,并加以氧化、糖基化、甲基化等修饰,形成独特的芳香聚酮类的次级代谢产物。这些多样化的修饰手段使得芳香聚酮类的抗生素拥有了多样化的结构与活性。
在芳香聚酮类的抗生素中,吡喃酮甲萘酚醌类(pyranonaphthoquinones,biqs)代表一类具有相似的芳香聚酮碳骨架的抗生素,包括美达霉素(medermycin)、卡拉真菌素、放线菌紫红素、榴菌素等。其中美达霉素,是一类具有芳香聚酮碳骨架的碳苷类抗生素,最初在1976年由takanoetal.分离自streptomycessp.k73,具有很强的抗菌与抗肿瘤活性,有希望成为抗菌抗肿瘤药物的前导化合物。
针对美达霉素的活性研究显示,当该类的小分子化合物与丝氨酸/苏氨酸蛋白激酶(akt/pkb)活性位点中的半胱氨酸残基结合后,能够不可逆地抑制丝氨酸/苏氨酸蛋白激酶,失去底物磷酸化能力。但是对于一些在活性位点中缺乏半胱氨酸残基的丝氨酸/苏氨酸蛋白激酶而言,美达霉素类化合物失去对其的抑制作用。此外美达霉素类的化合物还具有抑制细胞中哺乳动物雷帕霉素靶蛋白(mammaliantargetofrapamycin,mtor)信号通路,以及抑制加帽mrna翻译起始过程的活性。
在肿瘤细胞中,由于肿瘤细胞基因的突变引发一系列机制的变化,如pten活性的缺失,导致肿瘤细胞中激活的蛋白激酶akt的水平大幅升高。美达霉素通过与蛋白激酶相互作用、结合,阻止了生理状况下atp与蛋白激酶的结合,因此具有抑制该酶活性的能力,影响细胞周期的正常过程。但研究发现美达霉素没有特异性,在对肿瘤细胞发挥作用的同时也影响正常细胞的增殖分裂,作为临床开发应用毒性太大,因此无法成药。因此基于该类化合物的作用机制,不断开发研究得到一系列结构和功能与美达霉素相似的化合物。
技术实现要素:
本发明通过对放线菌进行研究发现了一种新的具有抗肿瘤活性的n-甲基-美达霉素类化合物。
一种n-甲基-美达霉素类化合物,命名为3’-n-甲基-美达霉素,结构式如式i:
本发明又提供了所述的n-甲基-美达霉素类化合物在制备治疗癌症的药物中的应用。
所述的癌症为黑色素瘤、急性髓系白血病、结肠癌、非小细胞肺癌、白血病、肝癌、肾癌、甲状腺癌、皮肤癌、胰腺癌、卵巢癌、乳腺癌、间皮瘤或者外周神经鞘膜瘤等。优选的,所述癌症为前列腺癌。
本发明还提供了所述的n-甲基-美达霉素类化合物的制备方法,包括以下步骤:
(1)发酵培养放线菌获得发酵产物;
(2)发酵产物过滤去除菌丝体获得粗提物;
(3)粗提物分离纯化获得所述n-甲基-美达霉素类化合物,
其中,所述放线菌为streptomycescavourensisstrainnrrl2740,购买自中国工业微生物菌种保藏管理中心。
发酵使用培养基由高氏一号液体培养基与海盐按10ml∶1g的比例配制。
步骤(3)中分离纯化方法如下:
(a)粗提物用乙酸乙酯萃取;
(b)萃取液浓缩后,使用硅胶柱色谱纯化,并用二氯甲烷体积占0%~20%的二氯甲烷-甲醇体系梯度洗脱;
(c)将包含目的化合物的洗脱产物合并后,使用凝胶柱色谱纯化,并用二氯甲烷体积占50%的二氯甲烷-甲醇体系洗脱;
(d)将包含目的化合物的洗脱产物合并后,使用高效制备液相色谱纯化获得所述n-甲基-美达霉素类化合物。
步骤(a)中萃取方法为:用与粗提物等体积的乙酸乙酯萃取3次。
步骤(b)中硅胶柱色谱纯化使用的填料为硅胶。
步骤(c)中凝胶柱色谱纯化使用的填料为羟丙基葡聚糖凝胶。
步骤(d)中高效制备液相色谱纯化使用的填料为十八烷基键合硅胶,流动相为体积浓度20%-100%的甲醇-水溶液,且流动相中还添加体积浓度0.5‰的三氟乙酸,以梯度洗脱。
本发明n-甲基-美达霉素类化合物为从一种放线菌中分离纯化获得,经鉴定为一种n-甲基-美达霉素类化合物,具体命名为3’-n-甲基-美达霉素。经实验证明该化合物对人前列腺癌细胞具有较强的抑制作用,ic50值达到0.51255μm。本发明化合物在制备治疗癌症的药物方面具有良好的应用前景。
附图说明
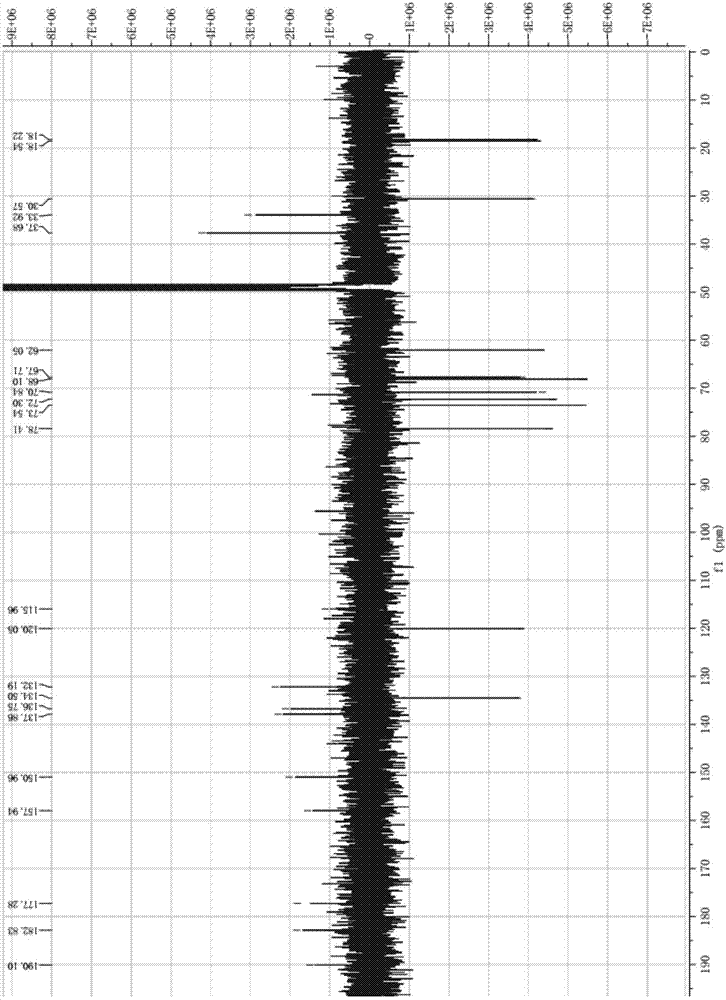
图1为本发明化合物的1h-nmr图谱。
图2为本发明化合物的13c-nmr图谱。
图3为本发明化合物的结构图。
具体实施方式
菌种来源
链霉菌streptomycescavourensisstrainnrrl2740:购买自位于北京市朝阳区酒仙桥中路24号院6号楼的中国工业微生物菌种保藏管理中心(cicc),订购网址:http://www.china-cicc.org/。
培养基
高氏一号液体培养基:以发酵培养基1l计,可溶性淀粉20g,kno31g,k2hpo40.5g,mgso4·7h2o0.5g,nacl0.5g,feso4·7h2o0.01g,加水至1l,调ph7.2。
实施例1
1、种子液
将链霉菌streptomycescavourensisstrainnrrl2740接种于含有250ml高氏一号液体培养基的500ml锥形瓶中,在摇床中28℃下以180rpm振荡培养3天,获得种子液。
2、发酵
以每瓶接种12ml的量将上述种子液接种到培养基中(每瓶中培养基为250ml高氏一号液体培养基中加入25g海盐,然后经高压湿热灭菌所得),在摇床中28℃下以180rpm振荡培养培养7天后终止发酵。
3、粗提
每瓶固体发酵物用ea(乙酸乙酯)约200ml浸泡过夜,三层纱布过滤除去菌丝体,收集滤液,减压浓缩得粗提物(油状浸膏)。
4、化合物分离
(a)将上述粗提物用等体积乙酸乙酯萃取3次,所得萃取液浓缩;
(b)萃取液浓缩后,使用硅胶柱色谱纯化,并用二氯甲烷体积占0%~20%的二氯甲烷-甲醇体系梯度洗脱,每1/4柱体积为一个馏分,薄层色谱(tlc)分析合并含有目标化合物的馏分;
(c)将包含目的化合物的洗脱产物合并后,使用凝胶柱色谱纯化,并用二氯甲烷体积占50%的二氯甲烷-甲醇体系洗脱,每1/4柱体积为一个馏分,薄层色谱(tlc)分析合并含有目标化合物的馏分;
(d)将包含目的化合物的洗脱产物合并后,使用高效制备液相色谱纯化(agilentpursuitc-18(10μm,21.2×250mm)色谱柱,填料为十八烷基键合硅胶,检测波长254nm),流动相为体积浓度20%-100%的甲醇-水溶液,且流动相中还添加体积浓度0.5‰的三氟乙酸,以梯度洗脱,收集所述n-甲基-美达霉素类化合物所在的色谱峰,然后回收溶剂获得所述n-甲基-美达霉素类化合物,呈红棕色结晶状。
实施例2
将实施例1分离纯化到的化合物进行结构鉴定。
该化合物的核磁共振数据如表1所示,核磁共振参数1h500mhz,13c125.7mhz。
表1
再结合质谱数据(图1和图2),得出该化合物的分子式为c23h25no8([m+h]+444.1659),结构式如图3所示,鉴定该化合物命名为3’-n-甲基-美达霉素。
实施例3
采用sbr方法检测实施例1纯化得到的化合物对人前列腺癌细胞株pc3细胞的增殖抑制作用。
取对数生长期的细胞,配置成5×104个/ml,以100μl/孔铺于96孔培养板,co2培养箱中培养24小时,取出培养板后于每孔中加入不同浓度的待测样品,每个浓度设3个复孔,加药完成后,置于co2培养箱中继续培养72小时后取出培养板,弃去培养液,每孔加入100μl4℃冰箱预冷的10%的三氯醋酸(tca)固定,静置5分钟后,再将培养板移至4℃冰箱过夜。倒掉固定液,每孔用去离子水洗涤5遍,甩干,空气干燥。每孔加入70μlsrb溶液,室温放置20分钟,去上清液,用1%醋酸洗涤5次,空气干燥。结合的srb用100μl/孔10mmol/ltris碱液(ph=10.5)振荡溶解。置于酶标仪中测定各孔光吸收,测定波长为515nm。
根据各孔od值计算药物对细胞增殖抑制率:抑制率=[1-(od515给药孔/od515对照孔)]×100%,根据各浓度抑制率计算半数抑制浓度ic50。结果如表2所示,本发明化合物对人前列腺癌细胞株pc3细胞的ic50值为0.51255μm,说明具有较强的抑制作用。
表2
- 还没有人留言评论。精彩留言会获得点赞!