一种采用甩盘反应釜制备甲基亚磷酸酯的系统的制作方法
本实用新型属于有机化学合成领域,涉及一种制备甲基亚磷酸酯的系统,尤其涉及一种以甩盘反应釜连续生成甲基亚磷酸酯的系统。
背景技术:
甲基亚磷酸酯是一种重要的化工原料中间体,可以广泛的应用于制备阻燃剂和除草剂。甲基亚磷酸酯结构如下所示:
CH3P(OR)n(OH)2-n
其中,基团R为C原子数在1~4的烷基,n为1或2。该类物质用途广泛,可以用于阻燃剂合成和农药合成等。
当R为甲基,n为2时,即为甲基亚膦酸二甲酯,分子式C3H9O3P,分子量为124.08,外观为无色液体,熔点小于-50℃,沸点为181℃(0.1005MPa),密度为1.16g/cm3。主要用于环氧树酯类的添加型阻燃剂、聚氨酯泡沫塑料、不饱和聚酯树酯和环氧树酯等高分子材料中,还可用作有机合成的中间体,稀有金属萃取剂等。
当R为乙基,n为2时,即为甲基亚膦酸二乙酯,分子式为C5H13O2P,分子量为136.13,外观为无色液体,沸点为124.5℃,闪点为26.5℃,密度为0.9g/cm3。甲基亚膦酸二乙酯是合成除草剂草铵膦的关键中间体。
当R为正丁基,n为1时,即为甲基亚磷酸单正丁酯,无色液体,主要用于塑料阻燃添加剂。
当R为正丁基,n为2时,即为甲基亚磷酸二丁酯,分子量为208.23,沸点为264.5℃,密度为0.973g/cm3。
CN 103319529A公开了一种甲基亚膦酸二乙酯的制备方法,其采用亚磷酸二乙酯、氯甲烷为原料,亚磷酸二乙酯先和氯甲烷反应,生成甲基磷酸二乙酯,甲基膦酸二乙酯再经过还原反应得到甲基亚膦酸二乙酯,其主要反应步骤是甲基化反应,还原反应两步。
CN 103374030A公开了一种草铵膦及其中间体的制备方法,其中甲基亚膦酸二乙酯的制备方法为三氯化磷和亚磷酸三乙酯反应得到氯代亚磷酸酯;镁和氯甲烷反应得到甲基氯化镁;氯代亚磷酸酯再和甲基氯化镁反应得到甲基亚膦酸二乙酯。
CN 105524109A公开了一种甲基亚磷酸酯的合成方法,其以五氧化二磷为起始原料先经硫化反应、氯化反应、水洗、蒸馏提纯后催化加氢得到氯代亚磷酸酯,再经格式反应得到甲基亚磷酸酯。该方法反应步骤多,工艺复杂,收率低。
CN 105131034A公开了一种甲基亚磷酸酯类化合物的合成纯化方法,具体为在惰性溶剂中,向含有甲基亚磷酸二烷酯和氯化镁混合物的溶液,低温下加入盐水,分离得到甲基亚磷酸二烷酯或甲基亚磷酸单烷酯。该方法主要作用在于提纯和以甲基亚磷酸二烷酯为原料合成甲基亚磷酸单烷酯。
CN 104892670A公开了一种草铵膦及其类似物的制备方法,其中设计了甲基亚磷酸酯的合成,其方法以甲基二氯化磷和醇为原料反应合成甲基亚磷酸酯,其特点在于所用的醇为C4以上的正醇或异醇,反应产物是甲基亚磷酸单酯。该方法采用间歇操作的方式,反应过程需要使用N,N-二甲基甲酰胺等作为缚酸剂,原子经济性差。
CN 106046051A公开了一种甲基亚磷酸二乙酯的制备方法,其采用甲基二氯化磷(MDP)为原料,在管式反应器中和乙醇气相反应,生成甲基亚膦酸二乙酯,其重点在于管式反应器。该方法在管式反应器中进行,但缺少脱酸、精馏等后处理工艺,导致产品转化率及含量较低。
CN 105949239A公开了一种甲基亚磷酸二烷酯的制备方法,采用pKa大于 10的叔胺作缚酸剂,甲基二氯化磷与醇在溶剂中反应,得到甲基亚磷酸二烷酯。该反应需要缚酸剂参与反应,且是在溶剂中进行反应,工艺复杂,对环保压力大;不能连续化生产,设备利用率低。
CN 106674275A公开了一种甲基次磷酸酯的制备工艺、以及相应的制备装置和制备方法,通过在-0.095MPa~-0.001MPa的负压条件下,以及反应温度为 35℃~90℃的条件下,在填料塔中进行甲基二氯化磷和醇ROH的反应制得甲基次磷酸酯。该方法虽然无需添加缚酸剂且可连续化生产,但填料塔对于原料负荷的操作范围较大,不易于反应控制。
国内还有一些针对亚磷酸二乙酯合成的研究,其基本路线多集中在以三氯化磷为原料,先和醇进行酯化反应得到氯代亚磷酸酯,接着再进行甲基化反应,得到甲基亚膦酸二乙酯。如“浙江工业大学学报”丁成荣发表的《甲基亚膦酸二乙酯合成工艺研究》、“精细化工中间体”第35卷第2期王宏亮发表的《甲基亚膦酸二乙酯的合成工艺研究》、“第十一届全国农药学科教学科研研讨会”论文集杜春华发表的《草铵膦及其中间体甲基亚膦酸二乙酯的合成工艺研究》都集中研究的这一合成路线。
此外还有报道采用反应釜向乙醇中滴加MDP,以三乙胺或吡啶或氨气为缚酸剂,但由于反应釜结构的限制,该反应需要在溶剂条件下进行,此外由于MDP 不稳定,易与加入的碱性物质在适宜条件下发生反应,一方面原料消耗大,原料成本增加,另一方面产品收率低,副反应增多,增加产品提纯成本。
技术实现要素:
针对现有技术无法连续化制备甲基亚磷酸酯,需要额外添加溶剂或缚酸剂等添加剂,产品收率低,副反应增多,产品提纯成本高等问题,本实用新型提供了一种采用甩盘反应釜制备甲基亚磷酸酯的系统。本实用新型以甩盘反应釜作为甲基二氯化磷和醇的反应器,并结合精馏塔等设备,通过混合合成、脱酸和精馏等工序,连续合成甲基亚磷酸酯产品,工艺收率高,操作简便,基本无三废产生,无需溶剂,绿色环保。
为达此目的,本实用新型采用以下技术方案:
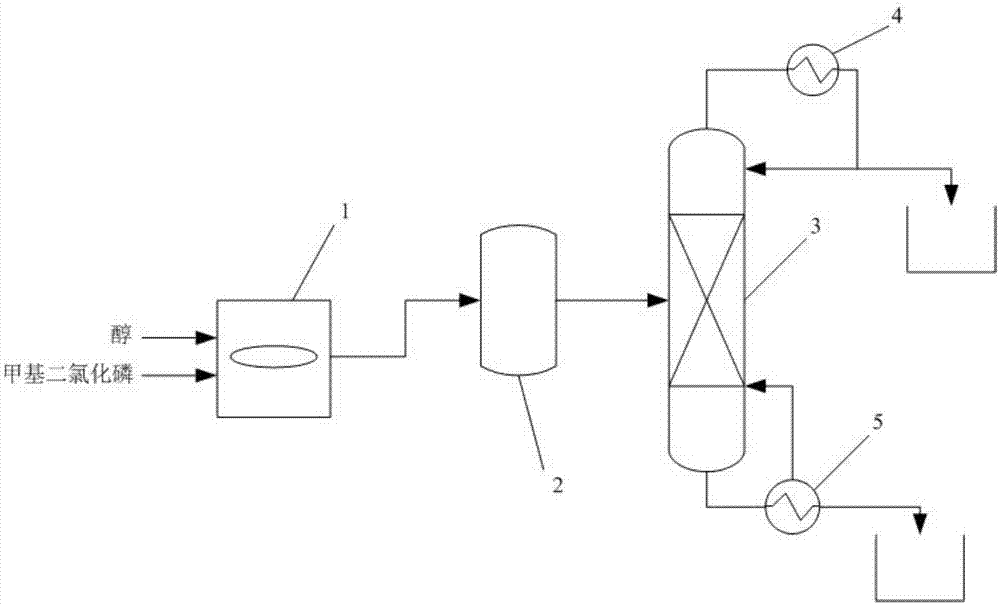
本实用新型提供了一种制备甲基亚磷酸酯的系统,所述系统包括依次连接的甩盘反应釜、脱副产单元和产物分离单元;其中,所述甩盘反应釜的物料出口与脱副产单元的物料入口相连。
本实用新型提供了一种连续的制备甲基亚磷酸酯的系统,以甲基二氯化磷和醇为原料,二者在甩盘反应釜中进行混合反应,反应原料甲基二氯化磷和醇先进入甩盘反应釜中的甩盘,通过甩盘被均匀地分布于反应釜内壁上,再进行连续混合反应。本实用新型可以使反应原料连续的进行均匀混合并反应,其较现有的常规反应釜、管式反应器以及填料塔等装置具结构简单,与后续脱酸及精馏装置匹配性好的特点。同时,其可有效防止物料受热不均局部形成沟流或无液流,在甩盘作用下在反应介质在器壁上均匀成膜,蒸发过程中尚未反应的甲基二氯化膦与乙醇继续反应,进一步提高了甲基亚磷酸酯产品的产率和生产效率。
本实用新型中,甲基二氯化磷和醇进行反应的过程中生成的氯化氢HCl等副产物需要经过后续脱副产单元和产物分离单元进行进一步的分离,以得到纯度较高的甲基亚磷酸酯产品。
以下作为本实用新型优选的技术方案,但不作为本实用新型提供的技术方案的限制,通过以下技术方案,可以更好地达到和实现本实用新型的技术目的和有益效果。
作为本实用新型优选的技术方案,所述甩盘反应釜在反应釜中内置甩盘,甩盘与液体物料入口相连。
本实用新型中,所述甩盘反应釜中内置的甩盘为本领域已有装置部件,故不再赘述。
作为本实用新型优选的技术方案,所述液体物料入口包括甲基二氯化磷入口和醇入口,甲基二氯化磷入口连接甲基二氯化磷输送管路;所述醇入口连接醇输送管路。
作为本实用新型优选的技术方案,所述脱副产单元包括脱酸装置;甩盘反应釜的物料出口与脱酸装置的物料入口相连;脱酸装置为反应釜、降膜蒸发器或塔式反应器中任意一种或至少两种的组合,所述组合典型但非限制性实例有:反应釜和降膜蒸发器的组合,降膜蒸发器和塔式反应器的组合,反应釜、降膜蒸发器和塔式反应器的组合等。
作为本实用新型优选的技术方案,所述脱酸装置包括依次连接的一级脱酸装置和二级脱酸装置;所述一级脱酸装置和二级脱酸装置均独立地为反应釜、降膜蒸发器或塔式反应器中任意一种或至少两种的组合,所述组合典型但非限制性实例有:反应釜和降膜蒸发器的组合,降膜蒸发器和塔式反应器的组合,反应釜、降膜蒸发器和塔式反应器的组合等。
本实用新型中,甲基二氯化磷和醇进行混合反应后,进入脱酸装置中继续进行反应并脱除副产氯化氢;更进一步的,甲基二氯化磷和醇进行混合反应后进入一级脱酸装置继续反应以及初步脱除副产氯化氢,再进入二级脱酸装置彻底脱除副产物氯化氢,脱除的氯化氢用水回收制备盐酸。
作为本实用新型优选的技术方案,所述产物分离单元包括至少一个精馏塔,但不限于一个精馏塔,可以2个、3个、4个或5个及以上,以使精馏塔交替使用,保证整个系统的连续运行;所述脱副产单元的产物出口与精馏塔的物料入口相连。
其中,脱副产单元包括产物出口与副产物出口,其产物出口与精馏塔的物料入口相连。
作为本实用新型优选的技术方案,所述精馏塔塔顶设冷凝器,冷凝器的冷凝液分两路,一路返回精馏塔,一路连接储罐。
作为本实用新型优选的技术方案,所述精馏塔塔底设再沸器,再沸器的出料分两路,一路返回精馏塔,一路连接储罐。
作为本实用新型优选的技术方案,所述系统包括依次连接的甩盘反应釜、一级脱酸装置、二级脱酸装置和精馏塔;
其中,所述甩盘反应釜的物料出口与一级脱酸装置的物料入口相连,所述甩盘反应釜在反应釜中内置甩盘,甩盘与液体物料入口相连;液体物料入口包括甲基二氯化磷入口和醇入口,所述甲基二氯化磷入口连接甲基二氯化磷输送管路,所述醇入口连接醇输送管路;二级脱酸装置的产物出口与精馏塔的物料入口相连,精馏塔塔顶设冷凝器,冷凝器的冷凝液分两路,一路返回精馏塔,一路连接储罐;精馏塔塔底设再沸器,再沸器的出料分两路,一路返回精馏塔,一路连接储罐。
上述制备甲基亚磷酸酯的系统的制备方法包括以下步骤:
原料甲基二氯化磷和醇在甩盘反应釜中进行反应,反应后物料依次经脱酸和精馏,得到甲基亚磷酸酯产品。
更为具体的,上述制备甲基亚磷酸酯的系统的制备方法包括以下步骤:
(a)原料甲基二氯化磷和醇按摩尔比为1:(0.5~10)在甩盘反应釜中进行反应,反应温度为10℃~40℃,反应压力为-0.05MPa~-0.095MPa,甩盘反应釜中甩盘的转动速率为10r/min~200r/min,得到反应后物料;
(b)步骤(a)所述反应后物料依次进入一级脱酸装置和二级脱酸装置中在温度为40℃~80℃,压力为-0.05MPa~-0.095MPa的条件下进行脱酸,脱除副产物氯化氢;
(c)步骤(b)脱酸后的物料进入精馏塔中进行精馏,精馏的温度为80℃~160℃,压力为-0.05MPa~-0.095MPa,得到甲基亚磷酸酯产品。
与现有技术相比,本实用新型具有以下有益效果:
(1)本实用新型所述系统是连续化的生产系统,以甲基二氯化磷和醇为原料,先在甩盘反应釜中混合反应,再经脱酸和精馏得到甲基亚膦酸酯,连续化反应,简化了工艺操作过程,极大地提高了设备的有效利用率,降低了人工成本,节省了生产成本;
(2)本实用新型采用甩盘反应釜,相对管式反应器、常规反应釜以及填料塔等反应装置而言,反应更充分,可以极大地提高产品的收率,使甲基亚膦酸酯的收到达到90%以上,并且反应条件温和,仅在低温条件下就可以进行;
(3)本实用新型在反应过中无需溶剂和缚酸剂等添加剂,减少了后续蒸馏等过程,降低了能耗,节约了资源,反应过程中极少产生废物,处理方便,对环境友好。
附图说明
图1是本实用新型实施例1中所述制备甲基亚磷酸酯的系统的结构示意图;
图2是本实用新型实施例2中所述制备甲基亚磷酸酯的系统的结构示意图;
其中,1-甩盘反应釜,2-脱酸装置,21-一级脱酸装置,22-二级脱酸装置,3-精馏塔,4-冷凝器,5-再沸器。
具体实施方式
为更好地说明本实用新型,便于理解本实用新型的技术方案,下面对本实用新型进一步详细说明。但下述的实施例仅仅是本实用新型的简易例子,并不代表或限制本实用新型的权利保护范围,本实用新型保护范围以权利要求书为准。
本实用新型具体实施方式部分提供了一种采用甩盘反应釜制备甲基亚磷酸酯的系统及制备方法,所述系统包括依次连接的甩盘反应釜1、脱副产单元和产物分离单元;其中,所述甩盘反应釜1物料出口与脱副产单元的物料入口相连。
制备方法包括以下步骤:
原料甲基二氯化磷和醇在甩盘反应釜1中进行反应,反应后物料依次经脱酸和精馏,得到甲基亚磷酸酯产品。
以下为本实用新型典型但非限制性实施例:
实施例1:
本实施例提供了一种制备甲基亚磷酸酯的系统,如图1所示,所述系统包括依次连接的甩盘反应釜1、脱酸装置2和精馏塔3;
其中,所述甩盘反应釜1的物料出口与脱酸装置2的物料入口相连,甩盘反应釜1在反应釜中内置甩盘,甩盘与液体物料入口相连,液体物料入口包括甲基二氯化磷入口和醇入口,甲基二氯化磷入口连接甲基二氯化磷输送管路,醇入口连接醇输送管路;脱酸装置2为反应釜,脱酸装置2的产物出口精馏塔3 的物料入口相连,精馏塔3塔顶设冷凝器4,冷凝器4的冷凝液分两路,一路返回精馏塔3,一路连接储罐;精馏塔3塔底设再沸器5,再沸器5的出料分两路,一路返回精馏塔3,一路连接储罐。
实施例2:
本实施例提供了一种制备甲基亚磷酸酯的系统,如图2所示,所述系统结构参照实施例1,区别仅在于:脱酸装置2包括依次连接的一级脱酸装置21和二级脱酸装置22,一级脱酸装置21为降膜蒸发器,二级脱酸装置22为塔式反应器。
实施例3:
本实施例提供了一种制备甲基亚磷酸酯的系统,所述系统结构参照实施例 1,区别仅在于:脱酸装置2包括依次连接的一级脱酸装置21和二级脱酸装置 22,一级脱酸装置21为反应釜,二级脱酸装置22为降膜蒸发器。
实施例4:
本实施例提供了一种制备甲基亚磷酸酯的系统,所述系统结构参照实施例 1,区别仅在于:脱酸装置2包括依次连接的一级脱酸装置21和二级脱酸装置 22,一级脱酸装置21和二级脱酸装置22均为降膜蒸发器。
实施例5:
本实施例提供了一种制备甲基亚磷酸酯的系统,所述系统结构参照实施例 1,区别仅在于:脱酸装置2包括依次连接的一级脱酸装置21和二级脱酸装置 22,一级脱酸装置21和二级脱酸装置22均为塔式反应器。
实施例6:
本实施例提供了一种甲基亚膦酸二乙酯的制备方法,所述方法采用实施例2 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,乙醇(纯度99wt%) 按760kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为25℃~35℃,反应压力≤-0.08MPa,甩盘反应釜1中甩盘的转速为10~20r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度35℃~65℃,一级脱酸装置21的压力为负压,真空度≤-0.085MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置 22的进料流量0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为 0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.085MPa
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.07Mpa~-0.095MPa,塔底温度≤165℃,精馏塔进料流量 0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到165℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸二乙酯产品。
本实施例中,精馏后甲基亚膦酸二乙酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为94%。
实施例7:
本实施例提供了一种甲基亚膦酸单乙酯的制备方法,所述方法采用实施例3 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,乙醇(纯度99wt%) 按370kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为30℃~40℃,反应压力≤-0.06MPa,甩盘反应釜1中甩盘的转速为30~40r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度40℃~65℃,一级脱酸装置21的压力≤-0.07MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.08Mpa~-0.095MPa,温度≤150℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到150℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸单乙酯产品。
本实施例中,精馏后甲基亚膦酸单乙酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为92%。
实施例8:
本实施例提供了一种甲基亚膦酸二甲酯的制备方法,所述方法采用实施例4 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,甲醇(纯度99wt%) 按570kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为20~30℃,反应压力≤-0.05MPa,甩盘反应釜1中甩盘的转速为20~30r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度30℃~50℃,一级脱酸装置21的压力≤-0.06MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度50℃~70℃,二级脱酸装置22的压力为负压,真空度≤-0.07MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.06Mpa~-0.085MPa,温度≤155℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到155℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸二甲酯产品。
本实施例中,精馏后甲基亚膦酸二甲酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为95%。
实施例9:
本实施例提供了一种甲基亚膦酸单甲酯的制备方法,所述方法采用实施例4 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,甲醇(纯度99wt%) 按265kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为30℃~40℃,反应压力≤-0.06MPa,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度40℃~65℃,一级脱酸装置21的压力≤-0.07MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.07Mpa~-0.095MPa,温度≤150℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到150℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸单甲酯产品。
本实施例中,精馏后甲基亚膦酸单甲酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为93%。
实施例10:
本实施例提供了一种甲基亚膦酸二丙酯的制备方法,所述方法采用实施例4 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,丙醇(纯度99wt%) 按990kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为15℃~25℃,反应压力≤-0.07MPa,甩盘反应釜1中甩盘的转速为60~70r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度25℃~60℃,一级脱酸装置21的压力≤-0.08MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.05Mpa~-0.07MPa,温度≤175℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到175℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸单甲酯产品。
本实施例中,精馏后甲基亚膦酸二丙酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为93%。
实施例11:
本实施例提供了一种甲基亚膦酸单丙酯的制备方法,所述方法采用实施例5 中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,丙醇(纯度99wt%) 按540kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为10℃~20℃,反应压力≤-0.06MPa,甩盘反应釜1中甩盘的转速为90~100r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度20℃~50℃,一级脱酸装置21的压力≤-0.07MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度50℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.07Mpa~-0.095MPa,温度≤165℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到165℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸单丙酯产品。
本实施例中,精馏后甲基亚膦酸单丙酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为95%。
实施例12:
本实施例提供了一种甲基亚膦酸二正丁酯的制备方法,所述方法采用实施例2中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,正丁醇(纯度99wt%) 按1170kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为20℃~30℃,反应压力≤-0.07MPa,甩盘反应釜1中甩盘的转速为120~130r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度30℃~65℃,一级脱酸装置21的压力≤-0.08MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.07Mpa~-0.095MPa,温度≤175℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到175℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸二正丁酯产品。
本实施例中,精馏后甲基亚膦酸二正丁酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为91%。
实施例13:
本实施例提供了一种甲基亚膦酸二异丁酯的制备方法,所述方法采用实施例2中系统进行制备,具体包括以下步骤:
(a)原料甲基二氯化磷(纯度98wt%)按1000kg/h,异丁醇(纯度99wt%) 按1150kg/h通入甩盘反应釜1中,进行混合反应,控制反应温度为20℃~30℃,反应压力≤-0.07MPa,甩盘反应釜1中甩盘的转速为190~200r/min,得到反应后物料;
(b)步骤(a)中反应后物料先进入一级脱酸装置21中继续进行反应以及初步脱除副产氯化氢,一级脱酸装置21的进料流量0.4m3/h~4m3/h,回流量 0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,一级脱酸装置21的温度30℃~65℃,一级脱酸装置21的压力≤-0.08MPa;一级脱酸装置21脱酸后的物料进入二级脱酸装置22中进行脱酸,彻底脱除副产氯化氢,二级脱酸装置22的进料流量 0.4m3/h~4m3/h,回流量为0.4m3/h~1.5m3/h,出料流量为0.4m3/h~4m3/h,二级脱酸装置22的温度60℃~80℃,二级脱酸装置22的压力≤-0.08MPa;
(c)步骤(b)脱酸后的物料进入精馏塔3中进行精馏,控制精馏塔3塔顶压力为-0.07Mpa~-0.095MPa,温度≤175℃,精馏塔进料流量0.4m3/h~4m3/h,出料流量0.4m3/h~4m3/h,等精馏塔3温度达到175℃后,切换精馏塔3,原先精馏塔3停止进料,出一次副产物高沸,最终得到甲基亚膦酸二异丁酯产品。
本实施例中,精馏后甲基亚膦酸二异丁酯产品的纯度可达98wt%以上,产品收率以甲基二氯化磷计约为90%。
对比例1:
本对比例提供了一种甲基亚膦酸二乙酯的制备系统及方法,所述系统的结构参照实施例2中结构,区别仅在于:所用反应装置为管式反应器。
所述制备方法参照实施例6中方法。
本对比例中,精馏后甲基亚膦酸二乙酯产品的纯度仅为94%,产品收率以甲基二氯化磷计仅为80%。
对比例2:
本对比例提供了一种甲基亚膦酸二乙酯的制备系统及方法,所述系统的结构参照实施例2中结构,区别仅在于:所用反应装置为常规反应釜。
所述制备方法参照实施例6中方法。
本对比例中,精馏后甲基亚膦酸二乙酯产品的纯度仅为93%,产品收率以甲基二氯化磷计仅为75%。
对比例3:
本对比例提供了一种甲基亚膦酸二乙酯的制备系统及方法,所述系统的结构参照实施例2中结构,区别仅在于:所用反应装置为填料塔。
所述制备方法参照实施例6中方法。
本对比例中,精馏后甲基亚膦酸二乙酯产品的纯度仅为94%,产品收率以甲基二氯化磷计仅为78%。
综合上述实施例和对比例可以看出,本实用新型所述系统和方法是连续化的生产系统及方法,以甲基二氯化磷和醇为原料,先在甩盘反应釜中混合反应,再经脱酸和精馏得到甲基亚膦酸酯,连续化反应,简化了工艺操作过程,极大地提高了设备的有效利用率,降低了人工成本,节省了生产成本;
同时,本实用新型采用甩盘反应釜,相对管式反应器、常规反应釜以及填料塔等反应装置而言,反应更充分,可以极大地提高产品的收率,使甲基亚膦酸酯的收率达到90%以上,并且反应条件温和,仅在低温条件下就可以进行;
并且,本实用新型在反应过中无需溶剂和缚酸剂等添加剂,减少了后续蒸馏等过程,降低了能耗,节约了资源,反应过程中极少产生废物,处理方便,对环境友好。
申请人申明,本实用新型通过上述实施例来说明本实用新型的详细应用方法,但本实用新型并不局限于上述详细应用方法,即不意味着本实用新型必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本实用新型的任何改进,对本实用新型产品原料的等效替换及辅助成分的添加、具体操作条件和方式的选择等,均落在本实用新型的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!