一种钯碳催化剂及其制备方法与应用与流程
本发明涉及一种钯碳催化剂及其制备方法与应用,属于多相催化技术领域。
背景技术:
燃油中有机含硫化合物是主要的大气污染源之一,化工原料中的含硫化合物则会造成贵金属加氢催化剂的中毒。随着原油重质化和劣质化趋势的加剧,石油中有机含硫化合物含量逐渐增高,但环保法规对燃油中硫含量的要求却日益严格,对油品的深度脱硫的要求越来越高。
在炼油厂中,油品中有机含硫化合物的脱除主要是通过加氢精制工艺中的加氢脱硫(HDS)过程实现的,即在高温高压以及催化剂作用下,将有机含硫化合物中硫原子还原为硫化氢,实现脱硫。传统的HDS催化剂为负载型Co-Mo、Ni-Mo或Ni-W双金属硫化物。以柴油馏分油为例,其深度和超深度加氢脱硫在反应物方面与常规的加氢脱硫有显著的差异。原油中的含硫化合物可分为非杂环与杂环两类。前者主要包括硫醇和硫醚类,易于脱除。杂环类主要包括噻吩及其烷基或苯基取代物。大分子的二苯并噻吩(DBT)及其烷基取代物如4,6-二甲基二苯并噻吩是柴油中最难脱除的含硫化合物。有关报道表明(Appl.Catal.B,2003,41:207-238),硫含量小于500ppm时,柴油馏分油中主要的含硫化合物是烷基取代的二苯并噻吩类化合物。因此柴油深度加氢脱硫针对的主要是这些大分子的芳香稠环含硫化合物。这对传统的硫化物催化剂提出了很大的挑战。原因是硫化物催化剂一般为层状结构,由于空间位阻效应,芳香稠环含硫化合物分子中的硫原子很难接近硫化物催化剂的活性中心进行反应。而当DBT类含硫化合物中的芳环加氢生成四氢二苯并噻吩或六氢二苯并噻吩后,分子结构发生扭曲,可以降低空间位阻,HDS活性显著提高。也就是说,要提高脱除DBT类稠环含硫化合物的效率,达到深度脱硫的目的,催化剂需要有良好的加氢活性。
负载型贵金属是一类重要的高活性加氢催化剂,因此在深度加氢脱硫领域受到广泛关注。在考察过的贵金属中,Pd和Pt在对DBT类含硫化合物的HDS反应中表现出很高的活性和最佳的耐硫性能(J.Catal.,2005,235:229-240;J.Catal.,2006,242:207-216)。二者各有特点:Pt有较高的C–S键断裂活性而Pd加氢活性较高但脱硫活性比Pt催化剂低(J.Catal.,2005,235:229-240;J.Catal.,2006,242:207-216)。制约贵金属催化剂应用的一个重要问题是贵金属在加氢脱硫反应条件下稳定性较差,易失活。硫中毒是造成贵金属催化剂失活的一个主要原因。硫主要通过以下两方面影响贵金属的催化性能:(1)硫在活性位上的强烈吸附;(2)反应生成的H2S吸附在贵金属活性组分表面,不仅降低了活性组分与载体相互作用,还会与活性组分反应生成迁移性更强但没有活性的金属硫化物,导致活性相的团聚,造成催化剂的永久失活(J.Catal.,1997,169:338-346)。
目前,提高贵金属耐硫性能和活性的一个常用的方法是提高催化剂载体的酸性。作为电子授体的贵金属活性中心的电荷会部分转移到作为电子受体的载体酸中心上,形成所谓的缺电子(electron-deficient)结构(Appl.Catal.A,1999,188:3-35)。这些缺电子的贵金属活性位与作为电子受体的硫之间结合力较弱,从而具有较高的耐硫性能(Catal.Today,2007,123:198-207)。缺电子贵金属对芳环有很高的加氢活性(Appl.Catal.A,1999,188:3-35),因此提高催化剂的酸性也是提高其HDS活性的一个有效途径。但是,提高载体的酸性会引发或加剧结焦和裂化等副反应,反而造成催化剂的失活(Catal.Today,2007,123:198-207)。因此,本领域亟需开发一种加氢脱硫催化剂以使在提高贵金属耐硫性能的同时不发生结焦和裂化等副反应,保持或提高催化剂的活性。
技术实现要素:
本发明的目的之一在于提供一种钯碳催化剂的制备方法,以该方法制备得到的钯碳催化剂具有良好的活性、直接脱硫路径选择性强及稳定性好。
本发明的另一目的在于提供由所述制备方法制备得到的钯碳催化剂。
本发明的再一目的在于提供所述钯碳催化剂的应用。
为实现上述目的,一方面,本发明提供一种钯碳催化剂的制备方法,所述方法包括如下步骤:
(1)对活性碳原料进行酸氧化处理;
(2)使用碱金属氢氧化物处理步骤(1)酸氧化处理后的活性碳;
(3)以步骤(2)处理后得到的活性碳作为载体制备所述钯碳催化剂。
有别于传统的通过提高载体酸性提高贵金属活性组分活性和耐硫性能的方法,本发明研究发现由上述方法制备得到的钯碳催化剂具有很高的加氢脱硫活性和直接脱硫路径选择性,能够降低氢耗,具有良好的经济性,另外,由于所述钯碳催化剂载体活性碳消除表面强酸中心,抑制结焦等副反应,因此,本发明钯碳催化剂具有良好的活性、直接脱硫路径选择性及稳定性,具有广阔的应用前景。
本发明对步骤(1)中所采用的活性碳原料并不特别限定其来源及制备方法,其可商购或按现有技术制备得到。作为推荐的实施方式,本发明优选自果壳活性碳、椰壳活性炭和木质活性炭中的一种或多种。
在本发明一些具体实施方式中,所述步骤(1)可为:在所述活性碳原料中加入具有氧化性的酸性水溶液,回流煮沸,过滤并洗涤至中性,干燥制得酸氧化处理后的活性碳。在本发明一些具体实施方式中,所述硝酸水溶液的浓度为0.5~5mol/L,所述活性碳原料与该硝酸水溶液的质量体积比为1~5g:10~100ml。在本发明所述一些具体实施方式中,在该硝酸水溶液中回流煮沸1~8h,且在真空条件下于50~120℃烘干。
在一些具体实施方式中,本发明制备方法的步骤(2)中所述碱金属氢氧化物选自氢氧化锂、氢氧化钠、氢氧化钾、碳酸钠和碳酸钾中的一种或多种。优选地,在本发明的一些具体实施方法中,所述碱金属氢氧化物的水溶液为氢氧化钠水溶液和/或氢氧化钾水溶液。
在本发明一些具体实施方式中,步骤(2)可为:在步骤(1)酸氧化处理后的活性碳中加入所述碱金属氢氧化物的水溶液,搅拌,过滤并洗涤至中性,干燥制得碱处理后得到的活性碳。在本发明的一些具体实施方式中,所述碱金属氢氧化物的水溶液的浓度0.5~5mol/L,所述步骤(1)酸氧化处理后的活性碳与该碱金属氢氧化物的水溶液的质量体积比为1~5g:10~100ml。在本发明的一些具体实施方式中,该搅拌是在15~50℃处理2~50h,且该干燥是在真空条件下50~120℃烘干。
在本发明的的一些具体实施方式中,步骤(3)包括如下步骤:
(i)以步骤(2)处理后得到的活性碳作为载体制备负载型Pd催化剂前驱体;
(ii)以步骤(i)制得的负载型Pd催化剂前驱体制备所述钯碳催化剂。
本发明并不特别限定所述负载型Pd催化剂前驱体的制备方法,如可以采用传统的离子交换法、共沉淀法或浸渍法等制备所述负载型Pd催化剂前驱体。优选地,采用等体积浸渍法制备所述负载型Pd催化剂前驱体。在本发明的一些具体实施方式中,将所述负载型Pd催化剂前驱体置于氢气气氛下,采用程序升温还原的方法制备所述钯碳催化剂。在本发明的一些具体实施方式中,氢气气氛的压力为1~10MPa,所述程序升温是以1~10℃/min的速率升温至100~400℃,还原时间为1~24h。
另一方面,本发明提供一种钯碳催化剂,其是由前述制备方法制备得到的。
本发明对所述钯碳催化剂中活性金属Pd的负载量不作限定,本领域技术人员可根据实际应用需要制备Pd质量含量不同的钯碳催化剂。本发明所述质量含量应当理解为特定组分的质量占催化剂总质量的含量比例,在本发明的一具体实施方式中,以钯碳催化剂的总质量为100%计,该钯碳催化剂含有质量分数为0.1%~10%的活性组分Pd。本发明Pd活性组分特指金属Pd,而不是钯的化合物。在本发明的一些具体实施方式中,所述钯碳催化剂还含有质量分数为1%~10%的碱金属。
再一方面,本发明还提供所述钯碳催化剂在加氢脱硫反应中的应用。如前所述,本发明的钯碳催化剂在加氢脱硫反应中具有良好的活性、直接脱硫路径选择性强及稳定性好等优势。在本发明的一些具体实施方式中,所述加氢脱硫反应的原料选自汽油、煤油、柴油馏分油或化工原料中噻吩类芳香环含硫化物中的一种或多种,优选地,所述噻吩类芳香环含硫化物为二苯并噻吩。在本发明的一些具体实施方式中,所述加氢脱硫反应在固定床反应器中进行;优选地,所述加氢脱硫反应的条件包括:反应温度200~400℃,压力1.0~10.0MPa,氢油体积比不超过10000Nm3/m3,重时空速0.1~100小时-1。
再一方面,本发明还可提供所述钯碳催化剂在以二苯并噻吩衍生物制备相应联苯衍生物中的应用。本发明所述二苯并噻吩衍生物通常情况下可含有一些对本发明钯碳催化剂不反应的官能团,但这并非限制要求,本领域技术人员可根据实际需要也可选择一些与二苯并噻吩脱硫一起被氢化的官能团以实现相应目的。
综上可知,本发明主要提供了一种钯碳催化剂及其制备方法,由该方法制备得到的钯碳催化剂具有很高的加氢脱硫活性和直接脱硫路径选择性,能够降低氢耗,具有良好的经济性。更为重要的是,所述钯碳催化剂稳定性显著优于相应的以酸性活性炭作载体的钯碳催化剂,在深度脱硫领域展现出良好的应用前景。本发明钯碳催化剂增加了Pd活性组分的电子密度,维持其金属状态,另一方面消除活性炭载体表面强酸中心,抑制结焦等副反应,从而使得催化剂有良好的活性、直接脱硫路径选择性及稳定性。
附图说明
图1是以果壳活性炭(AC)、硝酸处理的果壳活性炭(HC)、NaOH处理的HC(NaC)和KOH处理的HC(KC)作载体的Pd催化剂在Pd 3d区域的XPS谱图。
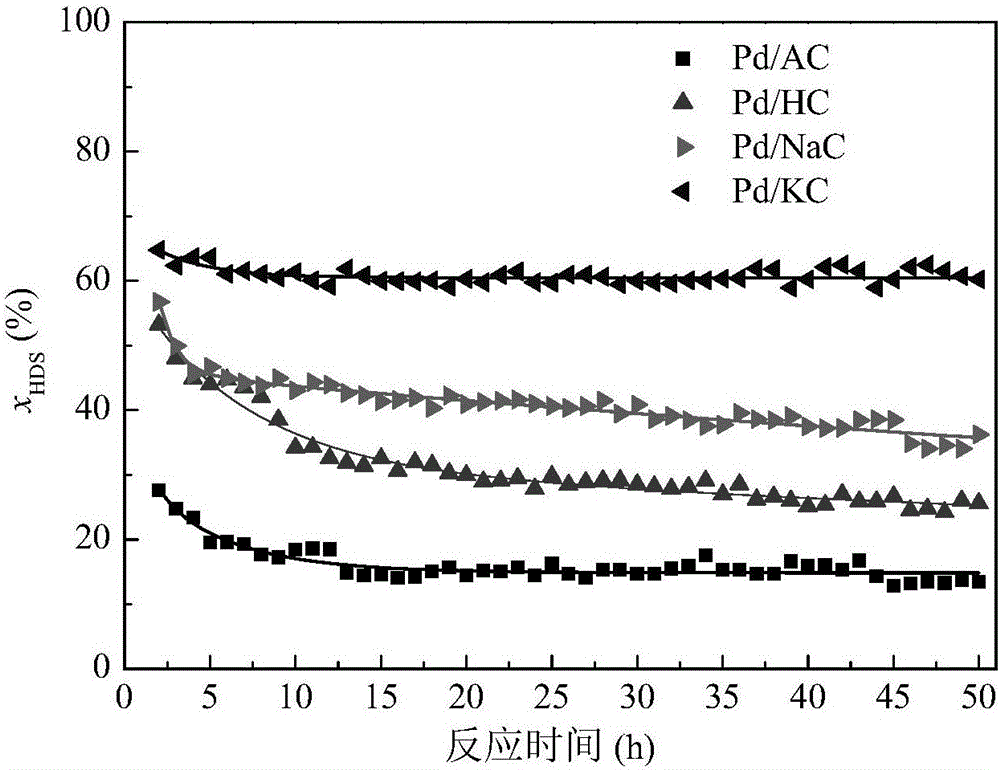
图2是DBT在Pd/AC、Pd/HC、Pd/NaC和Pd/KC上进行加氢脱硫反应时脱硫率(xHDS)随反应时间的变化。
图3是DBT在Pd/AC、Pd/HC、Pd/NaC和Pd/KC上进行加氢脱硫反应时联苯(BP)选择性。
具体实施方式
为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现结合具体实施例对本发明的技术方案进行以下详细说明,应理解这些实例仅用于说明本发明而不用于限制本发明的范围。实施例中,各原始试剂材料均可商购获得,未注明具体条件的实验方法为所属领域熟知的常规方法和常规条件,或按照仪器制造商所建议的条件。
对比例1
制备果壳活性炭载体。
称取1克市售果壳炭,加入到20mL去离子水中,回流状态下煮沸0.5小时去除杂质。过滤后将所得固体用去离子水充分洗涤,然后在真空烘箱中于95℃烘干,制得的载体记作AC。AC的比表面积如下表1所示,由表1可见,AC具有较大的比表面积(560m2/g)。分别用质量滴定(Carbon,1990,28:675-82)和Boehm滴定(Appl.Surf.Sci.,2008,254:7035-41;Appl.Surf.Sci.,2012,258:8247-52)的方法测定活性炭载体的零电荷点(pHPZC)以及表面含氧官能团分布,结果列于表1。AC的pHPZC值接近于中性(7.62),主要的含氧官能团为酸性的羰基含氧官能团以及少量的酚羟基酸性官能团和碱性含氧官能团。
对比例2
稀硝酸处理果壳活性炭载体。
称取1.0克AC,加到15mL的7.5mol/L硝酸水溶液中,回流状态下煮沸6小时。过滤后将所得固体用去离子水充分洗涤至液体为中性,然后在真空烘箱中于95℃烘干,制得的载体记作HC。HC的比表面积如下表1所示,由表1可见,HC的比表面积(489m2/g)略低于AC(560m2/g)。HC的pHPZC值呈酸性(2.55)。HNO3处理产生了大量的强酸性的羧基含氧官能团以及少量的内酯基。酚羟基的含量较AC有所增加,但羰基含量有所降低。在HC中没有检测到碱性含氧官能团。
实施例1
用NaOH处理HC载体。
称取1.0克HC,加到20mL的NaOH水溶液(1mol/L)中,室温下搅拌48小时。过滤后将所得固体用去离子水充分洗涤至液体为中性,然后在真空烘箱中于95℃烘干,制得的载体记作NaC。采用电感耦合等离子体原子发射光谱测得Na的质量含量为3.1%。NaC的比表面积如下表1所示,由表1可见,NaC的比表面积(284m2/g)显著低于AC(560m2/g)。NaC的pHPZC值呈碱性(9.61)。NaOH处理将HC中大量的酸性含氧官能团转化为碱性的含氧官能团。几乎完全消除了羧基和内酯基官能团,酚羟基降低了一半左右。但羰基含氧官能团含量明显增加。NaC中总的碱性含氧官能团和酸性含氧官能团含量相当。
实施例2
用KOH处理HC载体。
称取1.0克HC,加到20mL的KOH水溶液(1mol/L)中,室温下搅拌48小时。过滤后将所得固体用去离子水充分洗涤至液体为中性,然后在真空烘箱中于95℃烘干,制得的载体记作KC。采用电感耦合等离子体原子发射光谱测得K的质量含量为4.1%。KC的比表面积如下表1所示,由表1可见,KC的比表面积(234m2/g)显著低于AC(560m2/g)。KC的pHPZC值呈碱性(9.49)。KC中仍含有少量的羧基。与HC相比,内酯基和羰基含量都有增加,而酚羟基数量显著降低。KC中总的碱性含氧官能团和酸性含氧官能团含量相当。
表1活性炭载体比表面积(Sg)、pHPZC及含氧官能团分布
实施例3
负载型Pd催化剂的制备
采用等体积浸渍的方法制备负载型Pd催化剂前驱体:首先将计量的PdCl2溶于计量的稀HCl溶液(0.4mol/L)中,室温下分别浸渍到前述制备的活性炭载体(AC、HC、NaC或KC)上。室温下静置8小时后于真空烘箱95℃干燥制得催化剂前驱体。
将0.05克制得的催化剂前驱体压片成型并破碎至20~40目,置于内径8mm的固定床反应器中,用程序升温还原的方法制备负载型的Pd催化剂。具体条件如下:在总压1.0MPa的氢气气氛中以10℃/min的升温速率从室温升到300℃,气体流量为75NmL/min,保持1小时后自然降温至反应温度,制得负载型Pd催化剂。催化剂中Pd的质量含量为0.5%。
以AC、HC、NaC和KC作载体制得的Pd/AC、Pd/HC、Pd/KC和Pd/NaC催化剂在Pd 3d区域的XPS谱图如图1所示,由图1可以看出,Pd/HC和Pd/AC中同时含有金属态的Pd(记作Pd0)和缺电子状态的Pd(记作Pdδ+),而Pd/KC和Pd/NaC中主要活性组分为Pd0。碱金属助剂起到了提高Pd活性组分电子密度,维持其金属状态的作用。
实施例4
以实施例3中所述的Pd/AC、Pd/HC、Pd/KC和Pd/NaC作催化剂,用质量分数0.8%的二苯并噻吩的十氢萘溶液为模拟油品在固定床反应器中进行了加氢脱硫的试验。按实施例3所述的方法制备好催化剂并将床层温度调整至反应温度后(300℃),将氢气压力增加至5.0MPa,然后用高压计量泵向反应器中输送模拟油品,在反应器出口处经气液分离器分离出液体用于产物分析。其他反应条件:重时空速(WHSV)为54小时-1,H2/油品体积比为1500Nm3/m3。原料和产物用Agilent 6890气相色谱分析。定义脱硫率xHDS为:
xHDS=(C0-CDBT-Ci)/C0×100% (1)
其中,C0和CDBT分别为原料油品和产品中DBT的含量,Ci为产品中DBT的含硫中间体如四氢二苯并噻吩和六氢二苯并噻吩的含量。反应结果示于图2。由图2可以看出,Pd/NaC和Pd/KC的活性和稳定性均显著优于Pd/AC和Pd/HC。其中,Pd/KC表现出最高的活性和优良的稳定性,在50小时内未见明显失活。
DBT类含硫化合物的HDS反应网络复杂,可以看作由直接脱硫(DDS)和加氢两条平行反应路径构成。联苯(BP)是DDS反应路径的唯一产物。在有机含硫化合物存在的情况下,BP很难进一步加氢生成苯基环己烷(Appl.Catal.A,2008,344:175-182),因此可以用BP的选择性(SBP)作为催化剂DDS路径选择性的指标。由图3可以看出,在Pd/HC催化剂上,BP选择性为53%,则加氢路径的选择性为47%,说明直接脱硫路径和加氢路径并重。在Pd/AC催化剂上,BP选择性(63%)略高于在Pd/HC上的值(53%)。但是在Pd/NaC和Pd/KC催化剂上,BP选择性显著增加,均大于95%,DBT几乎完全通过DDS路径脱硫,因此催化剂氢耗较低,具有较好的经济性。
一般认为DBT主要通过氢解的方式脱硫生成BP(Top.Catal.,2011,54:290-298),而氢解反应主要由金属活性中心催化(J.Phys.Chem.,1983,87:2284-2287)。由实施例3可见,Pd主要以Pd0的形式存在于Pd/KC和Pd/NaC中,说明HC经KOH和NaOH处理引入碱金属助剂后,有利于提高Pd的电子密度,维持其金属状态。另外,由对比例2及实施例1~2可以看出,HC经KOH或NaOH碱处理后会消除强酸中心,抑制结焦等副反应,从而保障了催化剂的优良稳定性。碱金属的助催化作用使得Pd/KC和Pd/NaC催化剂表现出良好的活性、直接脱硫路径选择性及稳定性。
本发明经过上述的描述,已清楚地公开了本发明催化剂组成和制备条件。但是,本领域内的技术人员十分清楚,对本发明可以进行一些修改和改进。所以,只要不离开本发明的精神,对本发明所进行的任何修改和改进都应在本发明的范围内。本发明的范围在附属的权利要求书中提出。
- 还没有人留言评论。精彩留言会获得点赞!