使用杂交的靶核酸检测的制作方法
本国际PCT申请要求2014年8月1日提交的美国专利申请第14/450,144号和2014年8月6日提交的美国专利申请第14/453,396号的权益。
技术领域
本发明涉及从样品中检测靶基因组区域。
背景技术:
在下面的讨论中,将为了背景和介绍的目的描述某些文章和方法。本文所包含的任何内容不得被解释为对现有技术的“承认”。申请人明确地保留在适当时证明基于适用的法定条款本文提及的文章和方法不构成现有技术的权利。
遗传异常导致大量的病状,包括由染色体非整倍性引起的病状(例如,唐氏综合征(Down syndrome))、特定基因中的种系突变(例如,镰状细胞性贫血)和由体细胞突变引起的病状(例如,癌症)。用于确定遗传异常的诊断方法已成为用于鉴定特定疾病和病症以及提供关于疾病来源和治疗选择的宝贵信息的标准技术。
拷贝数变异(CNV)是对应于基因组(包括整个染色体)上已经缺失或重复的特定区域的基因组DNA改变。可以通过基因组重排如缺失、重复、倒位和易位引起CNV。CNV已经与多种形式的癌症(Cappuzzo F,Hirsch等人(2005)J Natl Cancer Inst.,97(9):643-55)、神经紊乱,包括孤独症(Sebat,J.等人(2007)Science 316(5823):445-9)和精神分裂症(St.Clair,D.,(2008).Schizophr Bull 35(1):9-12)相关。
因此,需要采用有效的可重现性测定和检测系统来筛选拷贝数变异的方法。
技术实现要素:
提供本发明内容以便以简化形式介绍将在以下详细描述中进一步描述的概念的选择。本发明内容并不旨在标识所要求保护主题的关键特征或必要特征,也不旨在用于限制所要求保护主题的范围。根据以下撰写的详细描述,包括在附图中图示的以及在所附权利要求中限定的那些方面,所要求保护主题的其他特征、细节、功用和优点将会是明显的。
本发明提供用于检测样品中的遗传特征,包括拷贝数变异(CNV)、插入、缺失、易位、多态性和突变的方法。本发明采用针对每个被探查基因座使用至少两个固定序列寡核苷酸并且经由连接使所述固定序列寡核苷酸直接或间接接合,从而从两个或更多个靶基因组区域探查基因座的技术。来自选定基因组区域内的不同基因座的连接产物包含核酸捕获区域,所述核酸捕获区域被设计为包括与固体支持物上的一个或多个捕获探针互补的区域。所述捕获区域包含一个或多个可检测标记,这些可检测标记将连接产物鉴定为源于特定靶基因组区域。对来自不同靶基因组区域的连接产物的鉴定通过使连接产物的捕获区域与固体支持物上的互补捕获探针结合来实现。
在一个具体实施方案中,本发明提供了一种用于提供胎儿非整倍性的统计可能性的测定方法,其包括:提供包含母体和胎儿细胞游离DNA的母体样品;使用包含捕获区域的序列特异性寡核苷酸从第一靶基因组区域探查一个或多个基因座;使用包含捕获区域的序列特异性寡核苷酸从第二靶基因组区域探查一个或多个基因座;经由与阵列的杂交来检测来自第一靶基因组区域和第二靶基因组区域的分离的选定基因座;量化分离的基因座的总计数以确定第一靶基因组区域和第二靶基因组区域的相对频率;使用序列特异性寡核苷酸从不同于第一靶基因组区域和第二靶基因组区域的至少一个靶基因组区域探查选定多态性基因座;检测分离的选定多态性基因座;量化分离的选定多态性基因座的总计数以计算母体样品中胎儿细胞游离DNA的百分比;计算母体样品中胎儿非整倍性的统计可能性,其中来自第一靶基因组区域的基因座的相对频率、来自第二靶基因组区域的基因座的相对频率和来自分离的选定多态性基因座的量化计数用于提供胎儿非整倍性的存在的统计可能性。
在其他具体实施方案中,本发明提供了确定胎儿非整倍性的存在或不存在的测定方法,其包括:提供包含母体和胎儿细胞游离DNA的母体样品;使用包含捕获区域的序列特异性寡核苷酸从第一靶基因组区域探查一个或多个基因座;使用包含捕获区域的序列特异性寡核苷酸从第二靶基因组区域探查一个或多个基因座;经由与阵列的杂交来检测来自第一靶基因组区域和第二靶基因组区域的基因座;量化所述基因座的总计数以确定第一靶基因组区域和第二靶基因组区域的相对频率;以及基于与分离的基因座的预期计数的偏差,使用来自第一靶基因组区域的基因座的相对频率和来自第二靶基因组区域的基因座的相对频率来确定母体样品中胎儿非整倍性的存在或不存在。在某些方面,使用由样品的代表性群体和优选包含来自相似母体年龄和/或孕龄的患者的样品的代表性群体测定的阈值水平来确定与预期计数的偏差。
在具体方面,从第一基因组区域和第二基因组区域探查基因座使用杂交,接着是连接。在更具体的方面,扩增步骤是在杂交和连接步骤之后进行。在其他具体方面,扩增是使用聚合酶链式反应的通用扩增。
在优选方面,使用具有捕获探针的单个固体载体;例如包含与指示不同靶基因组区域的多个捕获区域互补的捕获探针的阵列,来鉴定来自两个或更多个不同基因组区域的连接产物。将源于两个或更多个不同基因组区域的连接产物的混合物引入到阵列时,具有相同捕获区域的连接产物将与阵列上的互补捕获探针竞争性杂交,并且可以基于与捕获探针结合的检测标记的量估计来自每个基因组区域的连接产物的相对频率。以这种方式,可以确定靶基因组区域本身的相对频率。可通过鉴定对应于来自每个靶基因组区域的每个选定基因座的连接产物上的捕获区域与阵列上的特定已知位置的结合,或通过在源于靶基因组区域的连接产物结合之后估计来自阵列的总荧光量来确定每个靶基因组区域的相对频率。
在某些优选的实施方案中,捕获区域和捕获探针并不反映特定靶基因组区域核苷酸序列,而是用作鉴定特定靶基因组区域的替代物的“工程化”序列;因此对应于靶基因组区域的连接产物的核苷酸序列不需要直接确定。使用连接产物上的捕获区域允许连接产物与阵列上的捕获探针结合来指示作为连接产物来源的较大靶基因组区域,而无需为对应于靶基因组区域的实际核苷酸序列的连接产物部分测序。因为捕获区域和捕获探针是工程化序列,所以它们可以被认为是“通用”序列;即,这些捕获区域和捕获探针可结合许多不同的测定进行使用,唯一不同的是靶序列与捕获区域缔合。
在一个实施方案中,本发明的阵列包含全部具有基本上相同的序列的捕获探针。在另一个实施方案中,所用阵列包含两个至几个不同部件,其带有具有基本上相同的序列的捕获探针。这些阵列与本领域已知的通过与单独部件的互补性来鉴定单独序列的阵列形成对比,所述单独部件的每个部件包含不同于其他部件的核酸序列的阵列。使用阵列上的单独部件中的单个或有限数量的互补捕获探针序列可以简化产生该阵列所需的生物化学过程并且减少例如由连接产物上的捕获区域和捕获探针之间的结合亲和力差异所引起的检测频率上的潜在伪差。
在具体实施方案中,所述阵列包含用于检测来自两个或更多个不同靶基因组区域的单独连接产物、来自单个靶基因组区域的两个或更多个不同基因座或来自选定基因座的两个或更多个不同等位基因的两个或更多个不同捕获探针。即,不同序列的捕获探针与连接产物上对应于不同靶基因组区域的捕获区域、来自单个靶基因组区域的不同基因座或来自选定基因座的不同等位基因杂交。连接产物上的捕获区域与指示靶基因组区域或选定基因座,或指示作为连接产物来源的多态性等位基因的标记缔合。
因此,本发明的优选实施方案是使用捕获探针来鉴定连接产物,所述捕获探针与引入或引向连接产物的捕获区域互补,但仅通过与靶基因组区域互补的部件进行杂交无法鉴定连接产物所对应的靶基因组区域。在一些实施方案中,捕获探针与靶基因组区域部分互补并且与连接产物中的捕获区域部分互补。在一个优选实施方案中,用于鉴定对应于靶基因组区域的连接产物的捕获区域与靶基因组区域的任何部分都不互补。
优选地,在连接之前捕获区域作为其中一个固定序列寡核苷酸的一部分引入,但捕获区域可以在连接程序之后附连(例如,经由衔接子连接)至固定序列寡核苷酸的连接产物的一端或两端。
本发明的杂交测定形式的另一特征是,当使用捕获区域时靶基因组区域的量化可以通过量化与来自靶基因组区域内的基因座的连接产物缔合的标记来实现,而无需实际测定对应于靶基因组区域的连接产物的序列。以这种方式,可以估计来自靶基因组区域的连接产物的频率(并从而估计靶基因组区域本身的频率),而无需检测来自该靶基因组区域的基因座的实际核苷酸序列。
在许多优选实施方案中,与阵列上的捕获探针结合的标记的量化是估计由每个靶基因组区域产生的连接产物的水平或量所必需的唯一读数,连接产物的水平或量又可用于估计靶基因组区域的频率。
本发明方法的优点是连接产物可以与多个不同的可检测标记和/或捕获区域缔合。在不同的实验中使用不同的标记和/或捕获区域可以降低使用特定可检测标记、捕获区域或捕获探针的任何频率偏差。
在某些实施方案中,本发明提供了用于检测样品中第一靶基因组区域和第二靶基因组区域的频率的方法,其包括:将第一组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许第一固定序列寡核苷酸和第二固定序列寡核苷酸与来自第一靶基因组区域的基因座内的互补区域特异性杂交的条件下向样品中引入,其中第一组的第一固定序列寡核苷酸或第二固定序列寡核苷酸中的至少一个包含捕获区域和第一标记;将第二组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许第一固定序列寡核苷酸和第二固定序列寡核苷酸与来自第二靶基因组区域的基因座内的互补区域特异性杂交的条件下向样品中引入,其中第二组的第一固定序列寡核苷酸或第二固定序列寡核苷酸中的至少一个包含捕获区域和第二标记;连接杂交的固定序列寡核苷酸以产生与所述基因座互补的连接产物;向包含捕获探针的阵列引入连接产物,条件是允许阵列上的捕获探针与连接产物的捕获区域特异性杂交;检测第一标记和第二标记;并且量化第一标记和第二标记的相对频率以量化第一靶基因组区域和第二靶基因组区域的频率。在一些方面,第一组和第二组的连接产物的捕获区域不同。在其他方面中第一组和第二组的连接产物的捕获区域相同。
在一个优选方面,第一组和第二组的寡核苷酸的第一捕获区域和第二捕获区域相同。在其他方面,少量的捕获区域用于第一组和第二组两组的固定序列寡核苷酸中。在其他方面,对应于第一靶基因组区域的第一捕获区域不同于对应于第二基因组区域的第二捕获区域。
在其他优选实施方案中,除了探查第一靶基因组区域和第二靶基因组区域外,还探查了包含来自两个或更多个选定的多态性基因座的SNP的多态性序列。使用第三组固定序列寡核苷酸探查选定的多态性基因座以确定等位基因频率。等位基因频率用于例如计算母体血清样品中存在的胎儿核酸的百分比。
在某些实施方案中,第一固定序列寡核苷酸和第二固定序列寡核苷酸不与靶基因组区域内的基因座相邻杂交,而是在与基因座杂交的一组固定序列寡核苷酸之间具有间插区域或“空位”。可以例如使用聚合酶和dNTP来填补该间插区域,从而延伸一个固定序列寡核苷酸的末端,使得该末端变得与该组的另一个杂交寡核苷酸的末端相邻。在另一个实施方案中,可以使用在一组固定序列寡核苷酸之间结合并与所述一组固定序列寡核苷酸相邻的一个或多个“空位填补”或“桥接”寡核苷酸来填补间插区域。在后一种情况下,优选连接步骤会将所有寡核苷酸连接成包含单个捕获区域的单一连续连接产物,然后可以在阵列上对该连接产物进行检测。在另一个实施方案中,可以使用桥接寡核苷酸与dNTP和聚合酶的组合来填补介于固定序列寡核苷酸之间的间插空间。
在上述实施方案中,使用一组固定序列核酸,其包含设计成与每个选定基因座内的两个单独区域杂交(相邻或不相邻)的两个单独的固定序列寡核苷酸。然而,在一些实施方案中,一组固定序列寡核苷酸可以包含在任一末端具有与选定基因座互补的区域的单个探针。此单个探针与基因座杂交时,探针形成在基因座上可能会或可能不会相邻杂交的环状结构。.此类“环前(precircle)”探针也可以与探针末端之间的空位杂交,可通过一个或多个桥接寡核苷酸的杂交、通过使用聚合酶和dNTP或其组合延伸探针的一个末端来填补空位。
在某些方面,可检测标记与每一组的其中一个固定序列寡核苷酸中的捕获区域直接缔合(即共价或非共价结合)。在另一个实施方案中,在连接之后,例如以通用扩增来扩增连接产物,并且使可检测标记与用于扩增的引物中所含的捕获区域缔合。在其他具体方面,在单个容器中扩增来自第一靶基因组区域和第二靶基因组区域的分离的基因座和来自选定的多态性基因座的连接产物。在其他方面,可检测标记共价或非共价结合与连接产物、扩增子或其裂解产物上的互补序列杂交的寡核苷酸。此类经标记的寡核苷酸可以在将连接产物引入到捕获探针阵列之前或之后与连接产物杂交。
在本发明的其他实施方案中,对应于不同靶基因组区域的连接产物上的捕获区域包含不同序列,并且基于与不同捕获区域缔合的不同可检测标记的使用来确定至少第一靶基因组区域和第二靶基因组区域的对比频率。
在某些方面,通过来自样品中的靶基因组区域的被结合连接产物的被结合可检测标记的预期比率的变化来检测拷贝数变异。在某些具体方面,通过来自第一选定基因座的第一组连接产物与来自第二选定基因座的第二组连接产物相比升高或降低的杂交水平来检测拷贝数变异。
样品中基因座的相对频率不但可用于确定小靶基因组区域的拷贝数变异,而且连同和/或相比于其他基因座,基因座的相对频率可用于确定包括部分或整个染色体在内的较大靶基因组区域的拷贝数变异。
在本发明的另一个一般方面,提供了一种用于检测样品中第一靶基因组区域和第二靶基因组区域的频率的方法,其包括将第一组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许第一固定序列寡核苷酸和第二固定序列寡核苷酸与第一靶基因组区域中的基因座内的互补区域特异性杂交的条件下向样品中引入以产生第一杂交的固定序列寡核苷酸;将第二组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许第一固定序列寡核苷酸和第二固定序列寡核苷酸与互补区域特异性杂交的条件下向样品中引入以产生第二杂交的固定序列寡核苷酸;以及连接每一组的杂交的固定序列寡核苷酸以产生与第一靶基因组区域和第二靶基因组区域中的基因座互补的连接产物。每一组的至少一个固定序列寡核苷酸包含捕获区域(其包含与阵列上的捕获探针互补的序列)和可检测标记的结合区域。将连接产物引入到包含与连接产物的捕获区域互补的捕获探针的阵列中,条件是允许捕获探针与连接产物的捕获区域特异性杂交。向阵列中引入第一可检测标记,条件是允许可检测标记与来自第一靶基因组区域的基因座的连接产物上的捕获区域的结合区域特异性杂交,,并且向阵列中引入第二可检测标记,条件是允许可检测标记与来自第二靶基因组区域的基因座的连接产物上的捕获区域的结合区域特异性杂交。检测并量化第一可检测标记和第二可检测标记以提供样品中第一靶基因组区域和第二靶基因组区域的相对频率。如以上关于一个实施方案所讨论的,除探查第一靶基因组区域和第二靶基因组区域外,此实施方案的各个方面探查包含来自两个或更多个选定的多态性基因座的SNP的多态性序列。使用第三组固定序列寡核苷酸探查选定的多态性基因座以确定等位基因频率。等位基因频率用于例如计算母体血清样品中存在的胎儿核酸的百分比。还如以上所讨论的,在此实施方案的一些方面,除所述两个固定序列寡核苷酸外还可采用“空位填补”或“桥接”寡核苷酸,并且在此实施方案的一些方面,固定序列寡核苷酸或桥接寡核苷酸具等位基因特异性,如下文所详述。
在具体方面,本发明的测定提供从分离的选定多态性基因座中鉴定低频等位基因,其中母体DNA为纯合的并且非母体DNA为杂合的;计算来自分离的选定多态性基因座的低频等位基因的总和;以及使用来自分离的选定多态性基因座的低频等位基因的总和来计算第一靶基因组区域和第二靶基因组区域的靶基因组区域频率的统计显著性差异,从而计算母体样品中胎儿非整倍性的统计可能性,并且其中染色体频率的统计显著性差异提供了胎儿非整倍性的存在的统计可能性。
在本发明的另一个一般方面,提供了一种用于检测样品中第一靶基因组区域和第二靶基因组区域的频率的方法,所述方法包括提供样品;将第一组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许固定序列寡核苷酸与第一靶基因组区域的第一基因座内的互补区域特异性杂交的条件下向样品中引入以产生第一杂交的固定序列寡核苷酸;以及将第二组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许固定序列寡核苷酸与第二靶基因组区域的第二基因座内的互补区域特异性杂交的条件下向样品中引入以产生第二杂交的固定序列寡核苷酸。第一组固定序列寡核苷酸中的至少一个固定序列寡核苷酸包含捕获区域和与第一可检测标记互补的标记结合区域,并且第二组固定序列寡核苷酸中的至少一个固定序列寡核苷酸包含与第一组基本上相同的捕获区域和与第二可检测标记互补的标记结合区域。连接第一组和第二组的固定序列寡核苷酸以产生包含分别与第一基因座和第二基因座互补的区域并且标记有第一可检测标记和第二可检测标记的第一连接产物和第二连接产物。将第一连接产物和第二连接产物向包含捕获探针的阵列中引入,条件是允许捕获探针与连接产物的捕获区域特异性杂交。检测并量化第一标记和第二标记以确定第一标记和第二标记的相对频率,从而量化样品中第一靶基因组区域和第二靶基因组区域的相对频率。
可以在将连接产物引入到所述阵列之前将可检测标记引入到连接产物中。可选地,可以在连接产物杂交之后将可检测标记引入到所述阵列中。
同样,在某些实施方案中,所述方法还采用与感兴趣的序列杂交的至少一个固定序列寡核苷酸的延伸。即,在一些实施方案中,与一个或多个基因座杂交的固定序列寡核苷酸可能不会相邻杂交,从而留下“空位”或“间插”区。可以例如使用聚合酶和dNTP填补该间插区域,从而延伸一个固定序列寡核苷酸的末端,使得该末端与该组的另一个杂交寡核苷酸的末端相邻。在另一个实施方案中,可以使用在一组固定序列寡核苷酸之间和相邻处结合的一个或多个“空位填补”或“桥接”寡核苷酸填补间插区域。在后一种情况下,优选连接步骤会将所有寡核苷酸连接成包含单个捕获区域的单一连续连接产物,然后可以在阵列上对该连接产物进行检测。同样,可以使用桥接或空位填补寡核苷酸与dNTP和聚合酶的组合来填补空位。另外在一些实施方案中,环前、扣锁(padlock)或分子倒置探针(molecular inversion probe)可用于代替一组中的两个固定序列寡核苷酸。
当使用空位填补或桥接寡核苷酸时,桥接寡核苷酸通常很短,长度优选介于2-30个核苷酸并且更优选介于3-28个核苷酸。在一个方面,可以设计桥接寡核苷酸以在多个或所有位置提供简并性,例如,桥接寡核苷酸可以是带有各种序列变异的完全或部分随机物(randomer)以便在即使基因座在一个或多个位置含有多态性核苷酸时也确保对基因座的检测。可以基于可存在于基因座中的预测多态性来设计桥接寡核苷酸的简并性。可选地,在另一个方面,反应中使用的桥接寡核苷酸的混合物可以提供基于可存在于所述基因座区域中的预测多态性而特异性靶向一个或多个位置的有限简并性。在另一个方面,反应中使用的桥接寡核苷酸的混合物可以提供每个内部位置的简并性,其中与固定序列寡核苷酸连接位点相邻的核苷酸保持不变。有利的是,使用简并性桥接寡核苷酸避免了在采用本发明的检测方法之前预先确定选定基因座的母体和胎儿多态性含量的需求。
在另一个方面,桥接寡核苷酸长度长于10个核苷酸并且优选长度长于18-30个核苷酸。在一个优选方面,存在与每个选定基因座互补,设计为在选定基因座与第一固定序列寡核苷酸和第二固定序列寡核苷酸互补的区域之间杂交的单个桥接寡核苷酸。在另一方面,设计两个或更多个桥接寡核苷酸以在每个选定基因座处的固定序列寡核苷酸之间杂交,并且优选桥接寡核苷酸与第一固定序列寡核苷酸和第二固定序列寡核苷酸相邻杂交。
在存在两个桥接寡核苷酸的情况下,每个选定基因座发生三个连接事件:第一固定寡核苷酸和第一桥接寡核苷酸之间的连接,第一桥接寡核苷酸和第二桥接寡核苷酸之间的连接,及第二桥接寡核苷酸和第二固定序列寡核苷酸之间的连接。在另一方面,在桥接寡核苷酸之间和/或在桥接寡核苷酸和固定序列寡核苷酸之间存在空位。可以通过延伸—例如通过使用聚合酶和dNTP—在连接之前填补这些空位。
在本发明的一个方面,在将桥接寡核苷酸引入到样品中之前将第一固定序列寡核苷酸和第二固定序列寡核苷酸引入到样品中并且与基因座的互补部分特异性杂交。另一方面,在将第一组和第二组固定序列寡核苷酸引入到样品中的同时将桥接寡核苷酸引入到样品中。
在本发明的另一个一般方面,提供了一种用于确定混合样品中非整倍性的存在或不存在的方法,所述方法包括提供混合样品;将第一组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许固定序列寡核苷酸与第一染色体上的第一基因座特异性杂交的条件下向混合样品中引入以产生第一杂交的固定序列寡核苷酸,其中第一组的固定序列寡核苷酸包含第一捕获区域和第一标记结合区域;将第二组的第一固定序列寡核苷酸和第二固定序列寡核苷酸在允许固定序列寡核苷酸与第二染色体上的第二基因座特异性杂交的条件下向混合样品中引入以产生第二杂交的固定序列寡核苷酸,其中第二组的固定序列寡核苷酸包含第二捕获区域和第二标记结合区域;连接杂交的寡核苷酸以产生与所述基因座互补的连接产物;向包含捕获探针的阵列中引入连接产物,条件是允许捕获探针与连接产物的第一捕获区域和第二捕获区域特异性杂交;向阵列中引入第一经标记的寡核苷酸,条件是允许第一经标记的寡核苷酸的靶识别区域与第一标记结合区域特异性杂交;向阵列中引入第二经标记的寡核苷酸,条件是允许第二经标记的寡核苷酸的靶识别区域与第二标记结合区域特异性杂交;检测第一标记和第二标记;量化第一标记和第二标记的相对频率,从而量化混合样品中第一染色体和第二染色体(或基因组区域)的相对频率,其中阵列上所述标记的相对频率的统计显著性差异指示混合样品中染色体非整倍性的存在或不存在。
在替代性实施方案中,可基于对混合样品中第一染色体和第二染色体相对频率的观测偏差,使用“阈值”水平来确定胎儿非整倍性的存在或不存在。该阈值可以,例如使用如美国申请2012/0149583、2013/0324420、2013/0029852和美国专利第8,532,936号中描述的那些技术来确定。在某些方面,使用由样品代表性群体和优选包含来自相似特征,诸如先前风险状况、母体年龄和/或孕龄的患者的样品的代表性群体测定的阈值水平来确定与预期计数的偏差。
在本发明的优选方面,在添加固定序列寡核苷酸之前、期间或之后使样品DNA与固体载体结合。在本发明的优选方面,所述测定采用在产生连接产物之前,例如通过洗涤或核酸外切酶消化而去除未杂交的寡核苷酸的步骤。在其他优选方面,在连接之后但是在进一步处理和/或引入到阵列中进行检测之前分离连接产物。在其他优选实施方案中,优选使用通用引物扩增连接产物,以形成扩增子。在其他优选实施方案中,扩增子随后在与阵列杂交之前被裂解形成裂解的扩增子。在涉及连接产物的裂解的实施方案中,优选在将捕获区域部分引入到阵列中之前将包含捕获区域的裂解区域与裂解产物的其余部分分开。
在某些方面,使用本领域的常规技术分离样品DNA、连接产物和/或扩增产物。例如,可以通过附连到固体底物,接着通过分离步骤例如洗涤或核酸酶消化来分离杂交复合物(例如,与靶基因座结合的固定序列寡核苷酸)、连接产物和/或扩增产物。在具体实例中,可以使用与磁珠附连来将它们分离。在其他具体实例中,可以使用与带结合伴侣的底物的附连将它们分离,例如将寡核苷酸生物素化并且底物包含亲和素或链霉亲和素。在使用环前探针的各个方面,可通过核酸酶破坏非环化探针来分离杂交复合物和/或连接产物。
在此实施方案的一些方面,第一捕获区域和第二捕获区域具有相同的核苷酸序列。在此实施方案的其他方面,第一捕获区域和第二捕获区域具有不同的核苷酸序列。也如以上关于其他实施方案所讨论的,除了探查第一靶基因组区域和第二靶基因组区域外,此实施方案的各个方面探查包含来自两个或更多个选定的多态性基因座的SNP的多态性序列。例如,使用第三组固定序列寡核苷酸探查选定的多态性基因座以确定等位基因频率。等位基因频率用于例如计算母体血清样品中存在的胎儿核酸的百分比。
如以上所讨论的,在此实施方案的一些方面,除所述两个固定序列寡核苷酸以外,还可以采用延伸连接和/或“桥接”寡核苷酸。因此,本发明提供了一种用于确定胎儿非整倍性的可能性的方法,其包括以下步骤:提供包含母体和胎儿细胞游离DNA的母体样品;将与母体样品中的第一靶基因组区域内的基因座互补的第一组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;将与母体样品中的第二靶基因组区域内的基因座互补的第二组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;将与母体样品中不同于第一靶基因组区域的靶基因组区域内的一组多态性基因座互补的第三组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与选定的多态性基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;将桥接寡核苷酸在允许桥接寡核苷酸与固定序列寡核苷酸之间的基因座内的互补区域特异性杂交的条件下向母体样品中引入;连接杂交的第一固定序列寡核苷酸和第二固定序列寡核苷酸和桥接寡核苷酸以产生与基因座互补的连接产物;分离连接产物;使用通用引物位点扩增分离的连接产物;将扩增的连接产物施加到阵列上,其中所述阵列包含与连接产物上的捕获区域互补的捕获探针;量化来自选定的多态性基因座的每个等位基因的相对频率以确定样品中的胎儿细胞游离DNA百分比;量化来自第一靶基因组区域的基因座的相对频率和来自第二靶基因组区域的基因座的相对频率;以及使用来自第一靶基因组区域和第二靶基因组区域的基因座的相对频率和胎儿细胞游离DNA百分比计算胎儿非整倍性存在或不存在的可能性以确定胎儿非整倍性存在或不存在的可能性。
本发明还提供了一种用于确定胎儿非整倍性的可能性的方法,其包括以下步骤:提供包含母体和胎儿细胞游离DNA的母体样品;将与母体样品中的第一靶基因组区域内的基因座互补的第一组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;将与母体样品中的第二靶基因组区域内的基因座互补的第二组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;将与母体样品中不同于第一靶基因组区域的靶基因组区域内的一组多态性基因座互补的第三组的两个固定序列寡核苷酸在允许每个固定序列寡核苷酸的互补区域与选定的多态性基因座特异性杂交的条件下引入,其中每一组的两个固定序列寡核苷酸中的至少一个包含通用引物位点和捕获区域;使用dNTP和聚合酶延伸至少一个杂交的固定序列寡核苷酸以产生相邻杂交的寡核苷酸;连接相邻杂交的寡核苷酸以产生与基因座互补的连接产物;分离连接产物;使用通用引物位点扩增分离的连接产物;将扩增的连接产物施加到阵列上,其中所述阵列包含与连接产物上的捕获区域互补的捕获探针;量化来自选定的多态性基因座的每个等位基因的相对频率以确定样品中的胎儿细胞游离DNA百分比;量化来自第一靶基因组区域的基因座的相对频率和来自第二靶基因组区域的基因座的相对频率;以及使用来自第一靶基因组区域和第二靶基因组区域的基因座的相对频率和胎儿细胞游离DNA百分比计算胎儿非整倍性存在或不存在的可能性以确定胎儿非整倍性存在或不存在的可能性。
在某些方面,本发明还包括比较来自第一靶基因组区域和第二靶基因组区域的基因座的相对频率并且基于胎儿细胞游离DNA百分比调节来自第一靶基因组区域和第二靶基因组区域的基因座的相对频率以确定胎儿非整倍性存在或不存在的可能性。在具体方面,对每个靶基因组区域的每个选定基因座的相对频率求和并且比较每条染色体的总和以计算靶基因组区域比率。
可以通过检测一个或多个非母体贡献的基因座的水平检测样品的胎儿细胞游离DNA百分比,例如Y染色体上的非母体基因座和/或非母体基因座为常染色体基因座。在优选方面,非母体基因座与母体基因座相比包含一个或多个遗传变异,例如SNP或甲基化差异。
在某些实施方案中,裂解连接产物(例如,使用酶裂解机制诸如限制性内切核酸酶)以减小连接产物的尺寸,同时留下可用于检测的捕获区域和标记结合区域。在某些方面,裂解在通用扩增之后进行。
在优选实施方案中,杂交后从未结合的固定序列寡核苷酸分离基因座和与基因座杂交的固定序列寡核苷酸以去除反应中过量的未结合的寡核苷酸;例如,通过洗涤步骤或酶降解未结合非寡核苷酸。
所述方法中所用的第一组和第二组固定序列寡核苷酸优选包含—除至少一个捕获区域以外—可用于扩增连接产物的通用引物区域。可选地,可以在连接之后,例如通过引入包含通用引物序列的衔接子,向连接产物的末端添加通用引物序列。
在某些方面,本发明的固定序列寡核苷酸包含一个或多个指示物。除捕获区域以外,这些指示物也可用作鉴定基因座或基因座的特定等位基因的替代序列。具体而言,这些指示物可用作检测连接产物或其扩增子与阵列的杂交的替代鉴定序列。在具体方法中,每一组固定序列寡核苷酸中的第一固定序列寡核苷酸或第二固定序列寡核苷酸包含使特定等位基因与固定序列寡核苷酸缔合的等位基因指示物。
在某些具体方面,对来自每个靶基因组区域的至少50个基因座,更优选来自靶基因组区域的50-100个基因座,更优选100-200个基因座,更优选200-500个基因座,更优选500-1000个基因座,优选1000-2000个基因座,优选2000-5000个基因座和优选5000-10,000个基因座或之间的任何介入范围进行所述方法。在某些方面,除靶基因组区域以外,还探查至少50个选定的多态性基因座。更优选地,探查50-100个选定的多态性基因座,更优选地探查100-200个选定的多态性基因座,更优选地探查200-500个选定的多态性基因座,更优选地探查500-1000个选定的多态性基因座,探查1000-2000个选定的多态性基因座,探查2000-5000个选定的多态性基因座,及探查5000-10,000个选定的多态性基因座,包括所有介入范围。
在其他方面,估计所述测定方法检测对应于靶基因组区域内的每个基因座的至少5个捕获区域,更优选对应于靶基因组区域内的每个基因座的至少10个捕获区域,更优选对应于靶基因组区域内的每个基因座的至少20个捕获区域,优选对应于靶基因组区域内的每个基因座的至少50个捕获区域,更优选对应于靶基因组区域内的每个基因座的至少100个捕获区域,更优选对应于靶基因组区域内的每个基因座的至少200个捕获区域。在一些实施方案中,对于每份样品检测对应于靶基因组区域内的每个基因座的不超过5000个捕获区域。在其他实施方案中,对于每份样品检测对应于靶基因组区域内的每个基因座的不超过2000个捕获区域。
下面更详细地描述本发明的这些方面和其他特征和优点。
附图简述
图1是描述本发明的一个一般方面的流程图。
图2示出利用基因座杂交检测的本发明方法的一个实施方案。
图3示出利用基因座杂交检测的本发明方法的一个替代性实施方案。
图4示出利用基因座杂交检测的本发明方法的另一个替代性实施方案。
图5示出利用基因座杂交检测的本发明方法的另一个替代性实施方案。
图6示出利用桥接寡核苷酸结合固定序列寡核苷酸及基因座杂交检测的本发明方法的另一个替代性实施方案。
图7示出利用基因座杂交检测来检测多态性的本发明方法的另一个替代性实施方案。
图8示出利用基因座杂交检测来检测多态性的本发明方法的另一个替代性实施方案。
图9示出利用核酸区域的杂交检测来检测多态性的本发明方法的另一个替代性实施方案。
图10示出利用具有桥接寡核苷酸的核酸区域的杂交检测和双重裂解的本发明方法。
图11示出利用具有桥接寡核苷酸的核酸区域的杂交检测和双重裂解来检测多态性的本发明方法。
图12示出利用由单个裂解事件产生的核酸区域的杂交检测并且采用差异性标记的通用引物的本发明方法。
图13示出图12中所说明的方法的替代方法,其也利用由单个裂解事件产生的核酸区域的杂交检测并且采用差异性标记的通用引物。
图14示出对于阵列和下一代测序而言样品间的测定可变性分布。
定义
本文使用的术语旨在具有如本领域的普通技术人员理解的简单和普通含义。下列定义旨在帮助读者理解本发明,但是并非意味着改变或以其他方式限制这样的术语的含义,除非特别地指出。
术语“等位基因指示物”通常是指对应于特定SNP的一系列核苷酸。等位基因指示物可以含有允许检测一个或更多个碱基的缺失、取代或插入的另外的核苷酸。可以将等位基因指示物与任何其他指示物组合以便产生提供用于两种性能(例如,样品-鉴定指示物、等位基因-基因座指示物)的信息的一个指示物。
“阵列”是指具有表面,优选但不仅限于平面或基本上为平面的表面的固相载体,其携带含有核酸的位点阵列,使得阵列的每个位点均包含基本上相同或相同拷贝的寡核苷酸或多核苷酸,且被空间限定并不与阵列的其他成员位点重叠;即,位点是空间上离散的。阵列或微阵列也可以包括具有表面的非平面可探查结构如珠粒或孔。阵列的寡核苷酸或多核苷酸可以共价地结合到固体载体上,或可以非共价地结合。常规的微阵列技术综述于例如Schena编辑的Microarrays:A Practical Approach,IRL Press,Oxford(2000)中。“阵列分析”、“通过阵列的分析”或“通过微阵列的分析”是指使用阵列对一种或多种生物分子的分析,例如,序列分析。术语阵列是指任何形式的阵列化固体底物,包括微阵列、阵列化珠粒、孔内的分子阵列或“液体”阵列。
术语“结合对”意指使用共价和/或非共价结合彼此特异性地结合的任何两个分子,且其可以用于例如遗传物质与底物的附连。实例包括但不限于配体及其蛋白质结合伴侣,例如,生物素和亲和素、生物素和链霉亲和素、抗体及其特定表位,等等。
术语“染色体异常”是指全部或部分染色体的任何遗传变异。遗传变异可以包括但不限于任何拷贝数变异如重复或缺失、易位、倒位和突变。
术语“互补的”或“互补性”用于指通过碱基配对规则相关的核酸分子(即,核苷酸序列)。通常,互补核苷酸是A与T(或A与U)或C与G。两条单链RNA或DNA分子在最佳比对且具有适当的核苷酸插入或缺失的一条链的核苷酸以至少约90%至约95%互补性配对,且更优选地以约98%至约100%互补性配对,和甚至更优选地以100%互补性配对时,被认为是基本上互补的。可选地,当RNA或DNA链将在选择性杂交条件下与其补体杂交时,存在实质互补性。选择性杂交条件包括但不限于严格的杂交条件。严格的杂交条件通常将包括低于约1M,更通常地低于约500mM和优选地低于约200mM的盐浓度。杂交温度通常比解链温度(Tm)低至少约2℃至约6℃。
如本文中所用,术语“诊断工具”是指,为了在患者样品上实施诊断测试或检测系统,如,例如在系统中,联合使用的本发明的任何组合物或系统。
术语“杂交”通常意指反应,通过其进行核酸互补链的配对。DNA通常是双链的,且当链分开时,它们将在适当条件下再次杂交。可以在DNA-DNA、DNA-RNA或RNA-RNA之间形成杂交体。可以在短链和含有与该短链互补的区域的长链之间形成杂交体。可以形成不完全的杂交体,但是杂交体越不完全,它们将越不稳定(且越不太可能形成)。
如本文中所用,术语“连接酶”通常是指一类酶,DNA连接酶(通常为T4DNA连接酶),其可以将DNA片段连接在一起。片段必须具有匹配的末端–两者都为平末端或具有相互匹配的粘性末端–且反应需要ATP。“连接”是两个DNA片段连接在一起的过程。
如本文中所用,术语“基因座”和“多个基因座”是指基因组中已知位置的核酸区域。
如本文中所用,术语“母体样品”是指从怀孕的哺乳动物获得的任何样品,其包含胎儿和母体细胞游离DNA。优选地,用于本发明的母体样品通过相对非侵入的方式,例如,用于从受试者提取外周样品的静脉切开术或其他标准技术来获得。
如本文中所用,术语“寡核苷酸”是指天然的或修饰的核酸单体的直链寡聚物,所述天然的或修饰的核酸单体包括脱氧核糖核苷酸、核糖核苷酸、其异头物形式、肽核酸单体(PNA)、锁定核酸单体(LNA)等等,或其组合,所述直链寡聚物能够经由单体与单体相互作用的常规模式,如Watson-Crick类型的碱基配对、碱基堆积、Hoogsteen或反向Hoogsteen类型(reverse Hoogsteen)的碱基配对等等,特异性地结合单链多核苷酸。通常,通过磷酸二酯键或其类似物连接单体以形成大小范围是从几个单体单元例如,8-12个到几十个单体单元,例如,100-200个或更多个的寡核苷酸。可以通过Beaucage和Carruthers(Tetrahedron Lett.,22:1859-1862(1981))描述的亚磷酰胺法,或通过根据Matteucci等人(J.Am.Chem.Soc.,103:3185(1981))的三酯法,两者都通过引用的方式并入本文,或通过其他化学方法如使用商业自动化寡核苷酸合成器制备合适的核酸分子。
如本文中所用,“核苷酸”是指碱基-糖-磷酸组合。核苷酸是核酸序列(DNA和RNA)的单体单元。术语核苷酸包括核糖核苷三磷酸ATP、UTP、CTG、GTP和脱氧核糖核苷三磷酸如dATP、dCTP、dITP、dUTP、dGTP、dTTP,或其衍生物。此类衍生物包括,例如,[αS]dATP、7-去氮-dGTP和7-去氮-dATP,和对含有它们的核酸分子赋予核酸酶抗性的核苷酸衍生物。如本文中所用的术语核苷酸也指双脱氧核苷三磷酸(ddNTP)及其衍生物。说明的双脱氧核苷三磷酸的实例包括,但不限于ddATP、ddCTP、ddGTP、ddITP和ddTTP。
如本文中所用,术语“聚合酶”是指使用另一条链作为模板,将单个核苷酸连接在一起形成长链的酶。存在两种一般类型的聚合酶-合成DNA的DNA聚合酶和合成RNA的RNA聚合酶。在这两种类别中,聚合酶存在许多亚型,这取决于何种类型的核酸可以起到模板的功能和形成何种类型的核酸。
如本文中所用,“聚合酶链式反应”或“PCR”是指用于在体外,甚至在过量的非特异性DNA的存在下,复制靶DNA的特异性片段的技术。将引物添加到靶DNA上,其中引物利用核苷酸并且通常使用Taq聚合酶等来引发靶DNA的拷贝。通过循环温度,重复地使靶DNA变性和拷贝。即使与其他随机DNA混合时,靶DNA的单个拷贝也可以扩增获得数十亿的复制物。聚合酶链式反应可以用于检测和测量非常少量的DNA和产生定制的DNA片段。在某些情况下,线性扩增方法可以用作PCR的替代法。
如本文中所用,术语“多态性”是指可以表示该特定基因座的基因座内的任何遗传改变或变异,包括但不限于单核苷酸多态性(SNP)、甲基化差异、短串联重复(STR)等等。
通常,“引物”是在如聚合酶链式反应的合成步骤中或在引物延伸技术中用于例如引起DNA延伸、连接和/或合成的寡核苷酸。引物也可以用于杂交技术,作为提供核酸区域与捕获寡核苷酸的互补性的手段用于检测特定的核酸区域。
如本文中所用术语“研究工具”是指用于实质上学术或商业的科学探究的本发明的任何组合物或系统,科学探究包括药物治疗剂和/或生物治疗剂的开发。本发明的研究工具并不旨在是治疗性的或经监管部门批准;而是,本发明的研究工具旨在促进研究且帮助这样的开发活动,包括旨在产生支持注册申报的信息而进行的任何活动。
术语“样品”是指包含生物体的所有或一部分遗传信息的任何样品,生物体包括但不限于病毒、细菌、真菌、植物和动物,尤其是哺乳动物。遗传样品内可以被探查的遗传信息包括基因组DNA(编码区和非编码区)、线粒体DNA、RNA和源自这些中的每一种的核酸产物。此类核酸产物包括由mRNA产生的cDNA或为了增加用于分析的材料预扩增的产物。
术语“靶基因组区域”是指一条染色体或多条染色体的全部或一部分,包括完整染色体、亚染色体区域、成组基因座和单基因座。
发明详述
除非另外指出,否则本文描述的技术的实践可以采用有机化学、聚合物技术、分子生物学(包括重组技术)、细胞生物学、生物化学和阵列技术的常规技术和描述,这些都在本领域的技术人员的技术范围内。此类常规技术包括聚合物阵列合成、多核苷酸的杂交和连接及使用标记的杂交检测。合适技术的具体说明可以参考本文的实例。然而,当然也可以使用其他等效的常规程序。可以在标准实验室手册中找到此类常规技术和描述,如Green等人编辑(1999),Genome Analysis:A Laboratory Manual Series(第I-IV卷);Weiner、Gabriel、Stephens编辑(2007),Genetic Variation:A Laboratory Manual;Dieffenbach、Dveksler编辑(2003),PCR Primer:A Laboratory Manual;Bowtell和Sambrook(2003),DNA Microarrays:A Molecular Cloning Manual;Mount(2004),Bioinformatics:Sequence and Genome Analysis;Sambrook和Russell(2006),Condensed Protocols from Molecular Cloning:A Laboratory Manual;及Sambrook和Russell(2002),Molecular Cloning:A Laboratory Manual(全部来自Cold Spring Harbor Laboratory Press);Stryer,L.(1995)Biochemistry(第4版)W.H.Freeman,New York N.Y.;Gait,“Oligonucleotide Synthesis:A Practical Approach”1984,IRL Press,London;Nelson和Cox(2000),Lehninger,Principles of Biochemistry第3版,W.H.Freeman Pub.,New York,N.Y.;以及Berg等人(2002)Biochemistry,第5版,W.H.Freeman Pub.,New York,N.Y.,其全部通过引用整体并入本文用于所有目的。
注意,除非上下文另外明确指出,否则如本文和所附权利要求中所用,“一种”、“一个”和“所述(该)”包含复数指代物。因此,例如,提及“一个等位基因”是指具有多种序列变异的等位基因的一个或多个拷贝,并且提及“检测系统”包括对本领域的技术人员已知的等效步骤和方法的提及,等等。
除非另有定义,否则本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常所理解的相同含义。本文提到的所有出版物为了所有目的,包括描述和公开可以用于目前描述的发明中或联合目前描述的发明使用的设备、试剂、技术和方法的目的,通过引用整体并入。
在提供值的范围时,应理解,该范围上下限之间每个居中值和该规定范围中的任何其他规定或居中值涵盖在本发明内。这些较小范围的上下限可以独立地被包括在较小范围内,且也被包括在本发明内,受制于规定范围中任何明确排除的限值。在规定范围包括一个或两个限值时,排除那些被包括的限值中的两个的范围也被包括在本发明内。
在下面的描述中,阐述了许多具体细节,以便提供对本发明更透彻的理解。然而,本领域的技术人员将显而易见,可以在没有这些具体细节中的一个或多个的情况下,实践本发明。在其他情况下,为了避免使本发明含糊,没有描述公知的特征和本领域的技术人员公知的程序。
发明概述
本发明提供了鉴定遗传样品中核酸区域(包括基因座、多组基因座和较大靶基因组区域,例如,染色体)的拷贝数变异,包括插入、缺失、易位、突变和多态性的测定方法。一方面,所述测定方法使用定向连接测定,接着检测附连到阵列的经标记的寡核苷酸来探查来自样品中的两个或更多个靶基因组区域的基因座。对经标记的寡核苷酸的量化允许基于来自样品中的靶基因组区域的受检基因座的量之间的比较(例如,单条染色体的两个或更多个部分之间的比较或两条或更多条不同染色体之间的比较)或通过与来自相同或不同样品的参考染色体的比较来确定特定靶基因组区域的非典型拷贝数。
在一些实施方案中,所述方法采用对样品中的靶基因组区域的定向分析,其使用与两个或更多个靶基因组区域内的基因座选择性杂交的多组固定序列寡核苷酸。固定序列寡核苷酸直接或间接连接以产生连接产物。对应于与第一靶基因组区域缔合的基因座的连接产物与第一可检测标记缔合并且对应于与第二靶基因组区域缔合的基因座的连接产物与第二可检测标记缔合。如果第一可检测标记和第二可检测标记经量化,则可以确定第一靶基因组区域和第二靶基因组区域中的每一个的相对频率。在本发明的某些方面,所述方法采用用于鉴定两个不同靶基因组区域的两种不同标记。在本发明的其他方面,所述方法采用对应于三个不同靶基因组区域的三种不同标记,等等。
通过杂交,尤其是通过与和连接产物中存在的捕获区域互补的捕获探针阵列的杂交来检测连接产物。在某些实施方案中,使用包括具有相同或基本上相似的捕获探针的部件的“通用阵列”来检测连接产物。在某些其他实施方案中,所述阵列包括两组或更多组具有常见序列的多个部件,其中每一组具有不同序列,例如阵列上多达几百个部件具有基本上相同的序列的阵列。在任一种情况下,阵列上的捕获探针与连接产物的捕获区域互补而不是与基因座或其补体的序列互补。这些阵列可用于探查任何靶基因组区域的任何基因座,而与基因座的序列无关。
优选将捕获区域引入到连接产物的用于探查样品中的基因座的固定序列寡核苷酸中。在一些优选实施方案中,在使用的所有固定序列寡核苷酸中捕获区域相同,使得来自所有基因座的连接产物或扩增子或其裂解产物与阵列上相同序列的捕获探针竞争性杂交。在其他实施方案中,所述阵列由许多不同的捕获探针组成,并且来自不同基因座的多组固定序列寡核苷酸包含不同的捕获区域。
图1是示出本发明的示例性方法的流程图100。在步骤102中,提供样品。在步骤104中,将包含标记结合区域的多组固定序列寡核苷酸在允许固定序列寡核苷酸与靶基因组区域内的基因座杂交的条件下引入到样品中,并且在步骤106中使寡核苷酸与靶基因组区域杂交。在步骤108中,连接来自每一组的杂交的固定序列寡核苷酸以产生连接产物,然后在步骤110中扩增连接产物以产生与连接产物互补的扩增子。在步骤112中,将扩增子引入到杂交阵列中并允许其与阵列上的捕获探针竞争性杂交。在步骤114中,将一组经标记的寡核苷酸引入到扩增子中并允许其与扩增子上的互补序列杂交。任选地,使经标记的寡核苷酸与阵列上的捕获探针连接(未示出)。在步骤116中,检测标记。
每一组的每个固定序列寡核苷酸均包含与选定基因座互补的区域(如图2中更详细地描述)。每一组的至少一个固定序列寡核苷酸还包含捕获区域,对于用于探查两个或更多个靶基因组区域的所有组的固定序列寡核苷酸而言捕获区域可以相同,对于用于探查两个或更多个靶基因组区域的成对的多组固定序列寡核苷酸而言捕获区域可以相同,或对于单独靶基因组区域的多组固定序列寡核苷酸间捕获区域可以不同。另外,根据该实施方案,一组的一个固定序列寡核苷酸包含可检测标记或用于使固定序列寡核苷酸与可检测标记缔合的标记结合区域。在具体实施方案中,标记结合区域可为与经标记的寡核苷酸互补的区域,所述标记的寡核苷酸与可检测标记缔合。
在一些实施方案中,包含捕获区域的每一组的固定序列寡核苷酸将不包含标记或标记结合区域;即,该组的另一个固定序列寡核苷酸包含标记或标记结合区域(参见例如图2、3和5-8中说明的示例性实施方案);在其他实施方案中,包含捕获区域的每一组的固定序列寡核苷酸还将包含标记或标记结合区域(参见例如,图4中示出的示例性实施方案)。
在某些具体方面,第一组固定序列寡核苷酸与第一靶基因组区域内的基因座杂交,而第二组固定序列寡核苷酸与第二靶基因组区域内的基因座杂交。连接产生连接产物之后,任选地使用通用引物扩增连接产物,然后与阵列杂交。在其他实施方案中,裂解扩增产物(例如,使用限制性内切核酸酶)并且将扩增产物包含捕获区域的部分引入到阵列中进行杂交和检测。在优选实施方案中,用于探查靶基因组区域的每一组固定序列寡核苷酸均含相同标记或标记结合区域。即,第一组的所有固定序列寡核苷酸均与第一标记缔合,第二组的所有固定序列寡核苷酸均与第二标记缔合,并且第三组的所有固定序列寡核苷酸均与第三标记缔合。
如果直接标记固定序列寡核苷酸,则连接产物、扩增或其裂解产物可以与阵列上的捕获探针杂交并通过来自标记的读数检测。如果固定序列寡核苷酸改为含有与经标记的寡核苷酸互补的标记结合区域(“标记结合序列”),则必须在检测之前向连接产物或扩增子添加经标记的寡核苷酸。在任一种情形下,然后都检测并量化标记并且确定每种标记的相对频率。量化每种标记允许量化每个靶基因组区域。
在一些实施方案中,所有组的第一固定序列寡核苷酸和第二固定序列寡核苷酸含有基本上相同的捕获区域。在这些实施方案中,因为来自所有靶基因组区域的所有基因座的连接产物共有与阵列上的捕获探针互补的相同捕获区域,所以来自每个靶基因组区域的连接产物竞争与通用阵列上的捕获探针杂交。
在其他实施方案中,阵列上的捕获探针包含与不同捕获区域互补的多个序列,并且该阵列包括含有这些不同捕获探针的部件。在一些此类实施方案中,每个捕获探针可与唯一的捕获区域杂交。在其他此类实施方案中,一个以上代表来自不同基因组区域的基因座的捕获探针可与单个捕获区域杂交。
在其他实施方案中,根据所述测定,来自相同或不同靶基因组区域的不同基因座可配置成相互竞争性杂交并且因此将包含相同捕获区域,而来自相同或不同靶基因组区域的其他基因座可配置成相互竞争性杂交。
靶基因组区域可为大基因组区域,如整个染色体,或者可为较小基因组区域如单条染色体的亚区或不同染色体上的亚区,甚至小到单个基因座。因此,本发明可用于检测基因组变异如非整倍性或局部非整倍性,以及突变、SNP、重排、插入和缺失。在比较整个染色体的情况下,第一靶基因组区域可以是,例如染色体21,并且待用第一组固定序列寡核苷酸探查的所有基因座将来自染色体21。
在连接测定中,如果固定序列寡核苷酸结合选定基因座内的紧邻区域,则可连接固定寡核苷酸以产生与靶基因组区域特异性标记缔合的连接产物。在固定序列寡核苷酸不结合基因组区域内的紧邻区域—即,在杂交的固定序列寡核苷酸之间存在空位的情况下—可以使用引物延伸和/或一个或多个桥接寡核苷酸闭合空位。寡核苷酸一旦直接地或在延伸操作或引入桥接寡核苷酸之后连续杂交,然后就可将其连接以产生与靶基因组区域特异性标记缔合的连接产物。
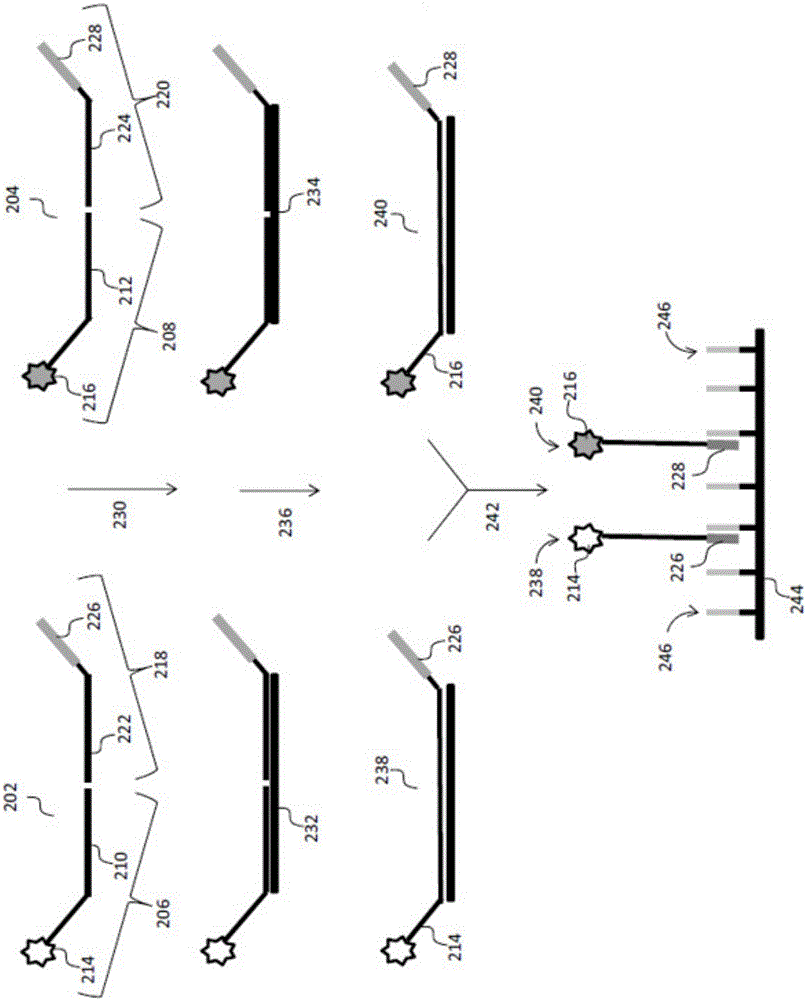
在某些方面,第一组固定序列寡核苷酸对第一染色体或第一靶基因组区域有选择性并且第二组固定序列寡核苷酸对不同染色体或第二靶基因组区域有选择性。图2示出一个实施方案,其中每一组标记的固定序列寡核苷酸与不同染色体上的基因座杂交并且在包含捕获探针的阵列上进行连接产物的竞争性评价。提供两组经标记的固定序列寡核苷酸202、204,每一组具有包含与选定基因座210、212互补的序列和标记214、216的第一固定序列寡核苷酸206、208以及包含与选定基因座222、224互补的序列和捕获区域226、228的第二固定序列寡核苷酸218、220。对于每一组固定序列寡核苷酸而言标记214、216是不同的,以允许检测期间第一靶基因组区域(在这种情况下,为第一染色体)和第二靶基因组区域(在这种情况下,为第二染色体)之间的区别。在步骤230中,将所述组的固定序列寡核苷酸202、204引入样品中并且允许与两条不同染色体上的基因座232、234杂交。杂交之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤236中,连接固定序列寡核苷酸以产生包含捕获区域226、228和标记214、216的连接产物238、240。虽然在图2中将固定序列寡核苷酸示出为在基因座中相邻杂交,但是也可存在空位,可以例如使用延伸反应或使用在固定序列寡核苷酸之间相邻杂交的桥接寡核苷酸来填补空位。在步骤242中,将连接产物238、240引入到包含多个捕获探针246的杂交阵列244中,其中连接产物238、240的捕获区域226、228与捕获探针246竞争性杂交。在一个优选实施方案中,226和228具有基本上相同的序列。连接产物与阵列杂交之后,优选从阵列中去除未杂交的连接产物(未示出)。然后可以根据所用标记的类型使用适当的检测机构来检测标记214、216并且可以量化对应于第一染色体和第二染色体中的每一条的基因座以确定遗传样品中每条染色体的存在和量。
根据本发明,“核苷酸”可以是未标记的或通过公知技术可检测地标记。荧光标记及其与寡核苷酸的附连在许多综述中有描述,包括Haugland,Handbook of Fluorescent Probes and Research Chemicals,第9版,Molecular Probes,Inc.,Eugene OR(2002);Keller和Manak,DNA Probes,第2版,Stockton Press,New York(1993);Eckstein编辑,Oligonucleotides and Analogues:A Practical Approach,IRL Press,Oxford(1991);Wetmur,Critical Reviews in Biochemistry and Molecular Biology,26:227-259(1991);等等。在以下参考文献样本中公开了适用于本发明的其他方法:Fung等人,美国专利第4,757,141号;Hobbs,Jr.等人,美国专利第5,151,507号;Cruickshank,美国专利第5,091,519号;Menchen等人,美国专利第5,188,934号;Begot等人,美国专利第5,366,860号;Lee等人,美国专利第5,847,162号;Khanna等人,美国专利第4,318,846号;Lee等人,美国专利第5,800,996号;Lee等人,美国专利第5,066,580号;Mathies等人,美国专利第5,688,648号;等等。标记也可用量子点进行,如以下专利和专利出版物中所公开那样:美国专利第6,322,901、6,576,291、6,423,551、6,251,303、6,319,426、6,426,513、6,444,143、5,990,479、6,207,392、2002/0045045和2003/0017264号。可检测标记包括,例如放射性同位素、荧光标记、化学发光标记、生物发光标记和酶标记。核苷酸的荧光标记可以包括但不限于荧光素、5-羧基荧光素(FAM)、2'7'-二甲氧基-4'5'-二氯-6-羧基荧光素(JOE)、罗丹明(rhodamine)、6-羧基罗丹明(R6G)、N,N,N',N'-四甲基-6-羧基罗丹明(TAMRA)、6-羧基-X-罗丹明(ROX)、4-(4'-二甲基氨基苯偶氮基)苯甲酸(DABCYL)、CASCADE(芘氧基三磺酸)、OREGON GREENTM(2',7'-二氟荧光素)、TEXAS REDTM(磺基罗丹明101酸氯)、花青和5-(2'-氨乙基)氨基萘-1-磺酸(EDANS)。经荧光标记的核苷酸的具体实例包括可从Perkin Elmer,Foster City,Calif.获得的[R6G]dUTP、[TAMRA]dUTP、[R110]dCTP、[R6G]dCTP、[TAMRA]dCTP、[JOE]ddATP、[R6G]ddATP、[FAM]ddCTP、[R110]ddCTP、[TAMRA]ddGTP、[ROX]ddTTP、[dR6G]ddATP、[dR110]ddCTP、[dTAMRA]ddGTP和[dROX]ddTTP,可从Amersham,Arlington Heights,IL获得的FluoroLink脱氧核苷酸、FluoroLink Cy3-dCTP、FluoroLink Cy5-dCTP、FluoroLink FluorX-dCTP、FluoroLink Cy3-dUTP和FluoroLink Cy5-dUTP;可从Boehringer Mannheim,Indianapolis,IN获得的荧光素-15-dATP、荧光素-12-dUTP、四甲基-罗丹明-6-dUTP、IR770-9-dATP、荧光素-12-ddUTP、荧光素-12-UTP和荧光素-15-2'-dATP;及可从Molecular Probes,Eugene,OR获得的染色体标记的核苷酸,BODIPY-FL-14-UTP、BODIPY-FL-4-UTP、BODIPY-TMR-14-UTP、BODIPY-TMR-14-dUTP、BODIPY-TR-14-UTP、BODIPY-TR-14-dUTP、CASCADE-7-UTP(芘氧基三磺酸-7-UTP)、CASCADE-7-dUTP(芘氧基三磺酸-7-dUTP)、荧光素-12-UTP、荧光素-12-dUTP、OREGON GREENTM 488-5-dUTP(2',7'-二氟荧光素-5-dUTP)、RHODAMINE GREENTM-5-UTP((5-{2-[4-(氨甲基)苯基]-5-(吡啶-4-基)-1H-i-5-UTP))、RHODAMINE GREENTM-5-dUTP((5-{2-[4-(氨甲基)苯基]-5-(吡啶-4-基)-1H-i-5-dUTP))、四甲基罗丹明-6-UTP、四甲基罗丹明-6-dUTP、TEXAS REDTM-5-UTP(磺基罗丹明101酸氯-5-UTP)、TEXAS REDTM-5-dUTP(磺基罗丹明101酸氯-5-dUTP)和TEXAS REDTM-12-dUTP(磺基罗丹明101酸氯-12-dUTP)。
在本发明的某些方面,使来自样品的核酸与底物缔合—例如,使用结合对,例如生物素和链霉亲和素,使遗传物质附连到底物表面或直接共价附连—将所述组的固定序列寡核苷酸添加到样品中之前。简言之,结合对的第一成员(例如,生物素)可与来自样品的核酸缔合,并且核酸经由底物表面上结合对的第二成员(例如,亲和素或链霉亲和素)附连到底物上。在固定序列寡核苷酸和/或桥接寡核苷酸与基因座杂交之后,来自样品的核酸的附连在去除未杂交的寡核苷酸中特别有用。简言之,来自样品的核酸可与固定序列寡核苷酸杂交,随后杂交复合物再与底物结合。可选地,来自样品的核酸可以在固定序列寡核苷酸杂交之前或同时附连到固体载体上。不管怎样,在核酸杂交并附连到固体载体之后,或可选地在杂交寡核苷酸连接之后,可处理载体的表面以去除任何未杂交或未连接的寡核苷酸,例如通过洗涤或其他去除方法,如降解寡核苷酸,如Willis等人,美国专利第7,700,323和6,858,412号中所讨论那样。当有两个固定序列寡核苷酸在相同探针上时降解寡核苷酸是优选方面,使得连接产生环化探针。然后可使用外切核酸酶降解非环化核酸,包括过量的探针和样品DNA。
存在许多可用于缔合核酸与结合对的方法。例如,许多方法可用于以生物素标记核酸,包括随机的光生物素化(photobiotinylation)、用生物素进行末端标记、用生物素化核苷酸复制和用生物素标记的引物复制。
在本发明的方法中对每条染色体分析的基因座数量可以在分析的每个靶基因组区域为两个至20,000个或更多个之间变化。在一个优选方面,每个靶基因组区域的基因座数量介于48和1000个之间。另一方面,每个靶基因组区域的基因座数量为至少100个。另一方面,每个靶基因组区域的基因座数量为至少400个。另一方面,每个靶基因组区域的基因座数量不超过1000个。另一方面,每个靶基因组区域的基因座数量为至少500个但不超过2000个。
虽然图2中说明的实施方案使用与可检测标记直接偶合的固定序列寡核苷酸,但是可由与固定序列寡核苷酸的连接产物或其扩增子或裂解的扩增子(如下所述)杂交的单独、经标记的寡核苷酸提供标记以允许检测。任选地,经标记的寡核苷酸在与固定序列寡核苷酸或扩增子或裂解的扩增子杂交之后与捕获探针连接。图3是对本发明的一个实施方案的说明,其中在经标记的寡核苷酸杂交和标记检测之前使固定序列寡核苷酸与感兴趣的基因座杂交、连接、扩增并引入到阵列中。在图3描绘的方法中,提供了两组固定序列寡核苷酸302、304,其中所述组包含第一固定序列寡核苷酸306、308,其各自包含与选定基因座310、312互补的序列、标记结合区域314、316和通用引物区域318、320;和第二固定序列寡核苷酸322、324,其包含与选定基因座326、328互补的序列、捕获区域330、332和通用引物区域334、336。在许多实施方案中,捕获区域330、332包含基本上相同的序列并且都将与阵列上的同一捕获探针杂交。标记结合区域314、316包含对于每组固定序列寡核苷酸302、304而言不同的序列,允许对于每个靶基因组区域而言与基因座缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸302、304而言捕获区域330、332相同以允许连接产物与阵列上的捕获部件的竞争性杂交。在步骤338中,将所述组的固定序列寡核苷酸302、304引入样品中并允许其与不同靶基因组区域的基因座340、342杂交。杂交之后,优选将未杂交的固定序列寡核苷酸与遗传样品的其余部分分开(未示出)。
在步骤344中,连接所述组的固定序列寡核苷酸302、304以产生连接产物346、348。在步骤358中,将通用引物350、352、354和356引入连接产物346、348中,连接产物分别与通用引物区域318、334、320和336结合,并且产生各自包含捕获区域364、366和标记结合区域368、370的扩增子360、362。在某些优选实施方案中,350和354具有基本上相同的序列,该序列与318和320两者互补,并且352和356具有基本上相同的序列,该序列与334和336两者互补。将扩增子引入包含多个捕获探针374的杂交阵列372中,其中扩增子360、362的捕获区域364、366与捕获探针374杂交。在步骤378中将经标记的寡核苷酸380、382引入阵列372中,其中扩增子360、362的标记结合区域368、370与经标记的寡核苷酸380、382的靶识别区域384、386杂交。任选地,使经标记的寡核苷酸与阵列上的捕获探针连接(未示出)。经标记的寡核苷酸杂交之后,优选从阵列中分离未杂交的经标记的寡核苷酸(未示出)。然后可检测标记并且量化对应于每个靶基因组区域的基因座以提供关于样品中每个靶基因组区域的存在和量的信息。
在某些实施方案,如图3所示的实施方案中,在连接产物或扩增子或裂解的扩增子与阵列杂交之后,使经标记的寡核苷酸与固定序列寡核苷酸或其扩增子或裂解的扩增子杂交。在某些其他实施方案中,在与阵列杂交之前,使经标记的寡核苷酸与固定序列寡核苷酸或其扩增子或裂解的扩增子杂交。
为促进连接产物或其扩增子的杂交,可以在与阵列杂交之前减小连接产物或扩增子的尺寸。在某些实施方案中,裂解连接产物(例如,使用限制性内切核酸酶或其他酶裂解机制)以减小待检测的连接产物的尺寸,留下例如可用于检测的捕获区域和标记结合区域。对裂解的经标记的连接产物或裂解的扩增子的检测用作代替检测整个连接产物的替代法。
减小连接产物、扩增子和/或经标记的连接产物的尺寸可例如通过改善杂交动力学和通过降低位阻来促进在阵列上的结合。在某些实施方案中,连接产物尺寸的减小通过使用限制酶裂解连接产物或扩增子来实现。例如,在某些实施方案中,每组固定序列寡核苷酸中的一个固定序列寡核苷酸包含邻近捕获区域或标记结合区域(或根据该实施方案为相应互补序列)的限制酶识别位点。限制酶可用于在限制酶识别位点裂解连接产物,留下可用于杂交和检测的标记或标记结合区域和捕获区域。限制酶识别位点可位于在裂解之后留下可用于检测的捕获区域和标记结合区域的任何位置。例如,限制酶识别位点可直接紧靠捕获区域或标记结合区域或位于来自捕获区域或标记结合区域中任一个的几个碱基内。优选地,在经标记的寡核苷酸与连接产物或扩增子杂交之前进行裂解。在某些其他方面,在与阵列杂交之后进行裂解以便检测。
图4是对本发明的一个具体实施方案的说明,其中在与阵列杂交之前裂解连接产物。在图4描绘的方法中,提供两组固定序列寡核苷酸402、404。每一组包含第一固定序列寡核苷酸406、408,其包含与基因座410、412互补的序列、标记结合区域414、416、捕获区域418、420、通用引物区域422、424和限制酶识别位点区426、428。标记结合区域414、416包含对于所述组的固定序列寡核苷酸402、404而言不同的序列,以允许与每个不同靶基因组区域缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸402、404而言此实施方案中的捕获区域418、420相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点426、428对于两组固定序列寡核苷酸402、404而言可以是相同的或对于每一组而言是不同的。
在步骤442中,将所述组的固定序列寡核苷酸402、404引入样品中并且允许与选定基因座444、446杂交。杂交和/或连接之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤448中,连接所述组的固定序列寡核苷酸402、404以产生连接产物450、452。在步骤454中,将通用引物456、458、460和462引入连接产物450、452中,连接产物分别与通用引物区域422、438、424和440结合,以产生包含标记结合区域472、474、捕获区域468、470和限制酶识别位点476、478的扩增子464、466。在步骤480中,将限制酶引入扩增子464、466中,扩增子与限制酶识别位点476、478结合并且裂解扩增子,留下包含标记结合区域472、474和捕获区域468、470的裂解的扩增子482、484。同样在步骤480,将裂解的扩增子482、484引入包含多个捕获探针488的杂交阵列486中,其中裂解的扩增子482、484的捕获区域468、470与捕获探针488竞争性杂交。在步骤492中,将经标记的寡核苷酸492、494引入阵列486中,其中每个裂解的扩增子482、484的标记结合区域472、474与经标记的寡核苷酸492、494的靶识别区域496、497杂交。经标记的寡核苷酸与裂解的扩增子杂交之后,优选从阵列中分离未杂交的经标记的寡核苷酸(未示出)。然后可检测经标记的寡核苷酸492、494的标记498、499并且可量化对应于每个靶基因组区域的裂解的扩增子以提供关于样品中靶基因组区域的存在和量的信息。注意:图4中经标记的寡核苷酸492、494毗邻捕获探针488,并且因此经标记的寡核苷酸492、494可与捕获探针488连接,以例如增加结合稳定性。然后可通过洗涤从阵列中消除裂解的扩增子482、484。
图5是对本发明的另一个具体实施方案的说明,其中连接产物在与阵列杂交之前被裂解。在图5描绘的方法中,提供两组固定序列寡核苷酸502、504。每一组包含第一固定序列寡核苷酸506、508,其包含与基因座510、512互补的序列、标记结合区域514、516、捕获区域518、520、通用引物区域522、524和限制酶识别位点区526、528。标记结合区域514、516包含对于所述组的固定序列寡核苷酸502、504而言不同的序列,以允许与每个不同靶基因组区域缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸502、504而言此实施方案中的捕获区域518、520相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点526、528对于两组固定序列寡核苷酸502、504而言可以是相同的或对于每一组而言是不同的。
在步骤542中,将所述组的固定序列寡核苷酸502、504引入样品中并且允许与基因座544、546杂交。杂交之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤548中,连接所述组的固定序列寡核苷酸502、504以产生连接产物550、552。在步骤554中,将通用引物556、558、560和562引入连接产物550、552中,连接产物分别与通用引物区域522、538、524和540结合,以产生包含标记结合区域572、574、捕获区域568、570和限制酶识别位点576、578的扩增子564、566。在步骤580中,将限制酶引入扩增子564、566中,扩增子与限制酶识别位点576、578结合并且裂解扩增子,留下包含标记结合区域572、574和捕获区域568、570的裂解的扩增子582、584。裂解产物与其各自的标记582、584结合(580)并且引入包含多个捕获探针588的杂交阵列586中,其中裂解的扩增子的捕获区域568、570与捕获探针590竞争性杂交588。
在本发明此实施方案的某些方面(并且如关于图4所说明和讨论的),可使经标记的寡核苷酸与杂交阵列的捕获探针连接以增加结合稳定性。此实施方案需要将经标记的寡核苷酸与捕获探针并列。捕获探针、裂解的连接产物和经标记的寡核苷酸可配置成使得经标记的寡核苷酸和捕获探针彼此相邻杂交,或可延伸经标记的寡核苷酸和捕获探针或如本文别处所述可采用空位填补寡核苷酸。
拷贝数变异的检测
如上所述,本发明提供了鉴定样品中靶基因组区域(相对较短的基因组区域、较大的基因组区域、亚染色体区域和染色体)的拷贝数变异、突变和多态性的方法。本发明提供了用于鉴定包含母体和胎儿DNA的母体样品中的胎儿染色体非整倍性特别有效的方法。
本发明的方法也可用于分析多条染色体上的多个基因座并且对来自特定染色体的基因座的频率一起进行平均。可对一个或多个靶序列进行频率的归一化或标准化。
在一些方面,本发明的方法可用于对混合样品如母体血清样品中两个来源的每条染色体上的基因座的频率求和,例如通过检测标记的总信号,然后将来自一条染色体上的基因座的标记的总和与来自另一条染色体上的基因座的标记的总和做比较以确定是否存在染色体异常。可选地,可以分析每条染色体上的基因座亚组以确定是否存在染色体异常。可以在来自相同染色体的区域之间或在来自不同染色体的区域之间进行比较。
用于确定基因座频率的数据可将离群数据排除在外,所述离群数据似乎是由于实验误差而产生的,或基于特定样品中的特发性遗传偏差而具有升高或降低的水平。在一个实例中,用于求和的数据可排除在一个或多个样品中具有特别高的频率的DNA区域排除。在另一个实例中,用于求和的数据可将一个或多个样品中发现丰度特别低的基因座排除。
可随机选择基因座亚组,但是数量足以在确定是否存在染色体异常中产生统计显著性结果。可在混合样品中和在不同阵列上进行不同基因座亚组的多种分析以产生更高统计功效。例如,如果对于染色体21而言存在100个基因座并且对于染色体18而言存在100个基因座,可进行一系列分析,在不同阵列上对每条染色体评价少于100个基因座。在此实例中,未选择性排除靶序列。
某些染色体上可检测的不同基因座的量可根据许多因素变化,包括母体样品中胎儿基因座的总体表现、代表母体样品中的胎儿基因座的不同基因座的降解速率、样品制备方法等。
串联连接测定
在某些方面,本发明的方法采用串联连接方法,其包括使用与靶基因组区域,例如感兴趣的染色体或参考染色体内的基因座互补的第一固定序列寡核苷酸和第二固定序列寡核苷酸,和与第一固定序列寡核苷酸和第二固定序列寡核苷酸之间且与其直接相邻的基因座区域互补的一个或多个短的桥接寡核苷酸(也称为“夹板(splint)”寡核苷酸或“空位”或“空位填补”寡核苷酸)。杂交的固定序列寡核苷酸之间的这些寡核苷酸的杂交,之后是这些寡核苷酸的连接提供了连接产物,若需要,连接产物转而提供用于扩增的模板。串联连接测定往往比使用单个寡核苷酸探针或使用仅仅两个固定寡核苷酸探针更有辨别力,因为在两个连接位点处必须存在固定序列和桥接寡核苷酸与选定基因座的完美互补性才能进行连接。
在优选方面,在串联连接方法中使用单个桥接寡核苷酸,其在固定序列寡核苷酸之间相邻杂交。可选地在一些方面,串联连接方法使用多组的两个固定序列寡核苷酸与一组的两个或更多个桥接寡核苷酸,桥接寡核苷酸在与第一固定序列寡核苷酸和第二固定序列寡核苷酸互补的区域之间的核酸区域中相邻杂交。这些桥接寡核苷酸彼此相邻杂交并且与固定序列寡核苷酸杂交。所述两个固定序列寡核苷酸和两个桥接寡核苷酸在连接反应期间连接,产生用作扩增模板且最终用于检测的单一连接产物。
在本发明的其他方面,所述方法采用多组与选定基因座内的非相邻区域结合的固定序列寡核苷酸,并且利用引物延伸在连接步骤之前产生一组连续的杂交寡核苷酸。可选地,所述方法可采用多组固定序列寡核苷酸、一个或多个桥接寡核苷酸及一个或多个杂交寡核苷酸的延伸。延伸和连接的组合提供了连接产物,连接产物转而用作扩增模板,接着用于检测和量化。
在一个优选方面,本发明的方法采用与一组的三个或更多个寡核苷酸的多路复用反应用于每个选定基因座。此一般方面在图6中有说明,其中提供了两组固定序列寡核苷酸602、604。每一组固定序列寡核苷酸602、604包含第一固定序列寡核苷酸606、608,其各自包含与感兴趣的核酸区域610、612互补的序列、标记结合区域614、616和通用引物区域618、620;和第二固定序列寡核苷酸622、624,其包含与感兴趣的核酸区域626、628互补的序列、捕获区域630、632和通用引物区域634、636。在步骤638中,将所述组的固定序列寡核苷酸602、604引入样品中并允许其与靶基因组区域内的基因座640、642的互补部分特异性杂交。杂交之后,优选将未杂交的固定序列寡核苷酸与遗传样品的其余部分分开(未示出)。去除未杂交的固定序列寡核苷酸可选地可以在下面的连接步骤之后与遗传样品的其余部分分离。在步骤644中,引入多组桥接寡核苷酸646、648并允许与第一固定序列寡核苷酸606、608和第二固定序列寡核苷酸622、624之间的基因座区域特异性杂交。可选地,桥接寡核苷酸646、648可与固定序列寡核苷酸同时引入。
在步骤650中,连接所述组的固定序列寡核苷酸602、604和桥接寡核苷酸630、632以产生与基因座640、642互补的连接产物652、654。同样,在步骤650中,将通用引物658、660、662和664引入连接产物652、654中,其中通用引物分别与通用引物区域618、634、620和636结合,并且扩增连接产物以产生包含标记结合区域666、668和捕获区域662、664的扩增子678、680。在某些优选实施方案中,通用引物658和662具有基本上相同的序列,该序列与618和620两者互补,并且通用引物660和664具有基本上相同的序列,该序列与634和636两者互补。将扩增子678、680引入包含多个捕获探针672的杂交阵列670中,其中扩增子658、660的捕获区域662、664与捕获探针672上的靶捕获区域674竞争性杂交。在步骤676中,经标记的寡核苷酸688、690的靶识别区域682、684与扩增子678、680的标记结合区域666、668特异性杂交。经标记的寡核苷酸杂交之后,优选从阵列中去除未杂交的经标记的寡核苷酸(未示出)。然后可检测经标记的寡核苷酸688、690的标记686、688并且任选地量化基因座以提供关于遗传样品中每个基因组区域的存在和量的信息。
在某些方面,桥接寡核苷酸可由在每个位置具有简并性的寡核苷酸的混合物组成,使得桥接寡核苷酸的混合物将与多路复用测定中需要指定长度的桥接的所有反应相容。在一个实例中桥接寡核苷酸是随机物,其中合成了桥接寡核苷酸的所有组合。例如,在使用5-碱基寡核苷酸的情况下,独特的桥接寡核苷酸的数量将是4^5=1024。这将不依赖于靶向区域的数量,因为所有可能的桥接寡核苷酸将存在于反应中。另一方面,桥接寡核苷酸可以具有不同长度,使得寡核苷酸的混合物与需要不同长度的桥接寡核苷酸的连接反应相容。
在另一个方面,桥接寡核苷酸可以在特定位置—即,已知的多态性位点具有简并性—且串联连接反应限于需要在多态性位点提供的特异性序列的那些。
在另一个实例中,桥接寡核苷酸具特异性,被合成为匹配空位中的序列。例如,在使用5-碱基寡核苷酸的情况下,所述测定中提供的独特寡核苷酸的数量将等于或小于基因座的数量。如果在两个或更多个基因座之间共有空位序列,则可以获得数量小于基因座数量的桥接寡核苷酸。在此实例的一个方面,可能有目的地选择基因座且特别是空位序列,使得在空位序列中存在尽可能多的相同重叠,最小化多路复用反应所必需的桥接寡核苷酸的数量。
另一方面,设计桥接寡核苷酸的序列并选择基因座,使得所有基因座在桥接寡核苷酸的每个末端上共有相同的碱基。例如,可以选择基因座及其空位位置,使得所有空位在第一位置共有“A”碱基且在空位的最后一个位置共有“G”碱基。可以基于诸如研究的基因组、该区域中的序列变异的可能性等的因素利用第一个碱基和最后一个碱基的任何组合。在此实例的具体方面,可以通过桥接寡核苷酸内部位置上的碱基的随机简并性合成桥接寡核苷酸,其中桥接寡核苷酸第一个位置和最后一个位置上具有特定核苷酸。在5-核苷酸的情况下,第二、第三和第四个位置将为简并性的,并且桥接寡核苷酸末端处的两个特定寡核苷酸将被固定。在这种情况下,独特的桥接寡核苷酸的数量将为4^3=64。
在人类基因组中,二核苷酸CG的频率远低于由各自的单核苷酸频率所预期的频率。这提供了利用桥接寡核苷酸的特定混合物来增强测定的特异性的机会。在此方面中,可以选择具有5'G和3'C的桥接寡核苷酸。这种碱基选择允许每个寡核苷酸在人类基因组中具有高频率,但是使得两个桥接寡核苷酸彼此相邻杂交成为稀有事件。然后,减少在测定中没有靶向的基因组位置中连接多个寡核苷酸的可能性。
在某些方面,在所述组的固定序列寡核苷酸已经杂交之后,且在任选去除所有未杂交的固定序列寡核苷酸之后,将桥接寡核苷酸添加到反应中。优选地,优化杂交反应的条件接近桥接寡核苷酸的Tm,以防止与核酸区域不完全互补的寡核苷酸的错误杂交。如果桥接寡核苷酸具有显著低于固定序列寡核苷酸的Tm,则优选地添加桥接寡核苷酸作为连接酶反应的一部分。
使用短的桥接寡核苷酸的优点是,只有当桥接寡核苷酸的所有碱基匹配空位序列时,才可能出现任一末端上的连接。使用短的桥接寡核苷酸的另一优点是,必需的不同桥接寡核苷酸的数量可少于基因座的数量,提高了寡核苷酸有效浓度,以便允许更快发生完美匹配。较少数量的桥接寡核苷酸提供了节约成本和质量控制的优点。使用固定的第一个碱基和最后一个碱基以及之间的随机碱基的优点包括能够利用较长的桥接寡核苷酸得到更高的特异性,,同时减少反应中总的桥接寡核苷酸的数量。
多态性的检测
在某些方面,本发明的方法检测包括多态性的一个或多个靶基因组区域。在一些实施方案中,这种方法不一定被设计为鉴定特定等位基因,例如,母体相对于胎儿的等位基因,而是确保对应于靶基因组区域内的基因座的不同等位基因都被包括在本发明的量化方法内。然而,在某些方面,可能期望使用等位基因信息来为靶基因组区域内的所有基因座计数以及使用等位基因信息,例如计算母体样品内所含的胎儿DNA的量,或鉴定来自癌症患者的遗传样品中具有特定突变的等位基因百分比。因此,本发明的方法旨在涵盖用于通过量化直接确定拷贝数变异而检测含SNP的基因座以及用于确保测定的整体效率的SNP检测的两种机制。
因此,在本发明的特定方面,通过固定序列寡核苷酸或通过用于检测基因座的桥接寡核苷酸提供等位基因辨别(allele discrimination)。在此类实施方案中,标记结合区域可用作嵌入一组的第一固定序列寡核苷酸或第二固定序列寡核苷酸中的等位基因指示物。在某些具体方面,在第一固定序列寡核苷酸和第二固定序列寡核苷酸中存在等位基因指示物(例如,标记结合区域)以检测基因座内的两种或更多种多态性。此类方面中所用的固定序列寡核苷酸的数量可对应于选定基因座正评估的可能等位基因的数量,并且与等位基因指示物缔合的标记的检测可检测遗传样品中特定等位基因的存在、量或不存在。
图7说明本发明的一个方面,其中使用与阵列的竞争性杂交检测多态性。在图7中,提供了两组固定序列寡核苷酸702、704,其中每一组的第一固定序列寡核苷酸706、708包含与分别包含例如A/T或G/C SNP的感兴趣的基因座710、712互补的序列及标记714、716。第二固定序列寡核苷酸722、724包含与选定基因座726、728互补的序列和捕获区域730、732。在一些实施方案中,分别除SNP 718和720及标记714和716外,固定序列寡核苷酸706与固定序列寡核苷酸708相同;并且固定序列寡核苷酸722与固定序列寡核苷酸724相同。即,所述组的固定序列寡核苷酸之间的差异是探查的SNP和对应于SNP的标记。在步骤734中,将所述组的固定序列寡核苷酸702、704引入样品中并允许其与包含SNP 740、742的感兴趣的基因座736、738特异性杂交。杂交或连接之后,优选将未杂交的固定序列寡核苷酸与遗传样品的其余部分分开(未示出)。在步骤744中,连接所述组的固定序列寡核苷酸702、704以产生连接产物746、748。重要的是注意,在此实施方案中连接是等位基因特异性的,只要等位基因特异性核苷酸靠近连接接点即可。通常,等位基因特异性核苷酸必须在连接的末端的5个核苷酸内;然而,在优选方面,等位基因特异性核苷酸是末端碱基。
在步骤750中,将连接产物746、748引入到包含多个捕获探针754的杂交阵列752中,其中连接产物746、748的捕获区域730、732与捕获探针754杂交。连接产物杂交之后,优选将未杂交的连接产物与遗传样品的其余部分分开(未示出)。然后可检测标记714、716并且量化对应于每个靶基因组区域的基因座的特异性等位基因以提供关于遗传样品中每个等位基因和靶基因组区域的存在和量的信息。
在另一个实施方案中,等位基因特异性核苷酸置于第一固定序列寡核苷酸和/或第二固定序列寡核苷酸中并且桥接寡核苷酸用于产生连接产物。图8说明了本发明的此方面。在图8中,提供两组固定序列寡核苷酸802、804,其中两组固定序列寡核苷酸均配置用于探查相同选定基因座处的不同SNP。每一组的第一固定序列寡核苷酸806、808包含与分别包含例如A/T或G/C SNP的选定基因座810、812互补的序列、标记结合区域814、816和通用引物区域818、820;并且第二固定序列寡核苷酸826、828各自包含与选定基因座830、832互补的序列、捕获区域834、836和通用引物区域838、840。在多态性测定中,除SNP位点和标记结合区域814、816外,第二固定序列寡核苷酸826和828具有基本上相同的序列并且第一固定序列寡核苷酸806、808具有基本上相同的序列。在步骤842中,将所述组的固定序列寡核苷酸802、804引入样品中并允许其与包含SNP 848、850的基因座844、846杂交。杂交之后,优选将未杂交的固定序列寡核苷酸与遗传样品的其余部分分开(未示出)。
在步骤852中,将一组固定序列寡核苷酸854、856引入样品中并允许其与介于每一组的第一和第二固定寡核苷酸之间的基因座844、846杂交。在一个优选实施方案中,854和856具有基本上相同的序列。在步骤858中,连接杂交的固定序列寡核苷酸和杂交的桥接寡核苷酸以产生连接产物860、862。在步骤864中,将通用引物866、868、870和872引入连接产物860、862中,连接产物分别与通用引物区域818、838、820和840结合,并且扩增连接产物860、862以产生包含标记结合区域882、884和捕获区域878、880的扩增子874、876。将扩增子引入包含捕获探针888的杂交阵列886中,其中扩增子874、876的捕获区域878、880与捕获探针888杂交。在步骤892中,将经标记的寡核苷酸894、895在允许经标记的寡核苷酸894、895的靶识别区域896、897与扩增子874、876的标记结合区域882、884选择性杂交的条件下引入杂交阵列886中。经标记的寡核苷酸杂交之后,优选从阵列中去除未杂交的经标记的寡核苷酸(未示出)。然后可对标记898、899进行检测并且量化对应于靶基因组区域的基因座相对应的等位基因以提供关于遗传样品中等位基因座处的等位基因和靶基因组区域的存在和量的信息。
在本发明的某些方面,通过桥接寡核苷酸提供等位基因辨别。在此方面,桥接寡核苷酸位于SNP上,优选足够靠近连接接点的一端以便提供等位基因特异性。
在一个实例中,在相同反应混合物中存在两种等位基因桥接寡核苷酸变异体并且等位基因检测由缔合的标记与杂交阵列的随后杂交所引起。图9说明了此方面。
在图9中,提供两组固定序列寡核苷酸902、904,其中两组固定序列寡核苷酸均配置用于探查相同选定基因座并且除标记结合区域以外是相同的。在此实施方案中,桥接寡核苷酸探查SNP,并且必须至少在两个单独容器中进行测定,直至连接步骤已经发走之后。每一组中的第一固定序列寡核苷酸906、908包含与基因座910、912互补的序列、捕获区域914、916和通用引物区域918、920。每一组的第二固定序列寡核苷酸922、924包含与基因座926、928互补的序列、标记结合区域930、932和通用引物区域934、936。在步骤938中,将所述组的固定序列寡核苷酸902、904在允许每一组902、904与基因座940、942特异性杂交的条件下引入样品中,其中选定基因座包含SNP 944、946。在第一固定序列寡核苷酸和第二固定序列寡核苷酸杂交之后(或可选地在连接之后),优选将未杂交的固定序列寡核苷酸与遗传样品的其余部分分开(未示出)。在步骤948中,引入对应于A/T SNP或G/C SNP的桥接寡核苷酸950、952并允许其与介于第一固定序列寡核苷酸和第二固定序列寡核苷酸之间的基因座940、942区域结合。可选地,桥接寡核苷酸可与固定序列寡核苷酸同时引入样品中。
在步骤954中,连接杂交的固定序列寡核苷酸和桥接寡核苷酸以产生连接产物956、958。此时可任选地将两个单独测定合并至单个容器中但并非必需。在步骤960中,将通用扩增引物962、964、966和968引入连接产物956、958中,其中通用引物与通用引物区域918、934、920和936结合,并且扩增连接产物956、958以产生各自包含标记结合区域980、982和捕获区域974、976的扩增子970、972。将扩增子970、972引入包含多个捕获探针986的杂交阵列984中,其中扩增子970、972的捕获区域974、976与捕获探针986杂交。在步骤990中,将经标记的寡核苷酸992、994引入阵列984中,其中经标记的寡核苷酸992、994的靶识别区域996、997与扩增子970、972的标记结合区域980、982杂交。经标记的寡核苷酸杂交之后,优选从阵列中去除未杂交的经标记的寡核苷酸(未示出)。然后可检测标记998、999并且量化又对应于靶基因组区域的基因座的相对应的等位基因以提供关于样品中每个等位基因和靶基因组区域的存在和量的信息。
另外的实施方案
图10是对本发明的另一个具体实施方案的说明,其中使用桥接寡核苷酸并且连接产物在与阵列杂交之前被双重裂解。在图10描绘的方法中,提供两组固定序列寡核苷酸1002、1004。每一组包含第一固定序列寡核苷酸1006、1008,其包含与基因座1010、1012互补的序列、标记结合区域1018、1020、捕获区域1014、1016、通用引物区域1022、1024和限制酶识别位点区1026、1028。标记结合区域1018、1020包含对于所述组的固定序列寡核苷酸1002、1004而言不同的序列,以允许与每个不同靶基因组区域缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸1002、1004而言此实施方案中的捕获区域1014、1016相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点1026、1028对于每个固定序列寡核苷酸上的两个裂解位点而言可以是相同的,和/或对于不同组的固定序列寡核苷酸1002、1004而言可以是相同的。每一组中还存在第二固定序列寡核苷酸1030、1032,其中每个第二固定序列寡核苷酸包含与基因座1034、1036互补的序列和通用引物区域1038、1040。
在步骤1042中,将所述组的固定序列寡核苷酸1002、1004引入样品中并且允许与基因座1044、1046杂交。杂交(或可选地连接)之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤1048中,向每一组添加桥接寡核苷酸1026、1028并且在杂交的固定序列寡核苷酸之间相邻杂交。在步骤1054中,连接所述组的寡核苷酸以产生连接产物1050、1052。在步骤1054中,将通用引物1056、1058、1060和1062引入连接产物1050、1052中,连接产物分别与通用引物区域1022、1038、1024和1040结合,以产生包含标记结合区域1018、1020、捕获区域1014、1016和限制酶识别位点1072、1074、1076和1078的扩增子1064、1066。在步骤1070中,将一种或多种限制酶引入扩增子1064、1066中并且扩增子经双重裂解以产生包含标记结合区域1018、1020和捕获区域1014、1016的裂解的扩增子。将裂解产物引入包含多个捕获探针1088的杂交阵列1086中,其中裂解的扩增子的捕获区域1014、1016与捕获探针1090竞争性杂交。然后将杂交的捕获区域引入1088与裂解产物上的互补序列结合的经标记的寡核苷酸1082、1084中并进行检测。
图11是对本发明的另一个具体实施方案的说明,其中桥接寡核苷酸用于鉴定相同基因座内的不同等位基因并且连接产物在与阵列杂交之前被双重裂解。在图11中,对于具有可能的多态性的单个基因座提供两组固定序列寡核苷酸1102、1104。每一组包含第一固定序列寡核苷酸1106、1108,其包含与选定基因座1110、1112的一个等位基因互补的序列、标记结合区域1118、1120、捕获区域1114、1116、通用引物区域1122、1124和限制酶识别位点区1126、1128。标记结合区域1118、1120包含对于所述组的固定序列寡核苷酸1102、1104而言不同的序列,以允许与每个不同等位基因缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸1102、1104而言此实施方案中的捕获区域1114、1116相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点1126、1128对于每个固定序列寡核苷酸上的两个裂解位点而言可以是相同的,和/或对于不同组的固定序列寡核苷酸1102、1104而言可以是相同的。每一组中还存在第二固定序列寡核苷酸1130、1132,其中每个第二固定序列寡核苷酸包含与选定基因座1134、1136互补的序列和通用引物区域1138、1140。
在步骤1142中,将所述组的固定序列寡核苷酸1102、1104引入样品中并且允许与选定基因座1144、1146杂交。杂交(或可选地连接)之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤1148中,向每一组添加桥接寡核苷酸1126、1128并且在杂交的固定序列寡核苷酸之间相邻杂交。在步骤1154中,连接所述组的寡核苷酸以产生连接产物1150、1152。在步骤1154中,将通用引物1156、1158、1160和1162引入连接产物1150、1152中,连接产物分别与通用引物区域1122、1138、1124和1140结合,以产生包含标记结合区域1118、1120、捕获区域1114、1116和限制酶识别位点1172、1174、1176和1178的扩增子1164、1166。在步骤1170中,将一种或多种限制酶引入扩增子1164、1166中并且扩增子经双重裂解以产生包含标记结合区域1118、1120和捕获区域1114、1116的裂解的扩增子。将裂解产物引入包含多个捕获探针1188的杂交阵列1186中,其中裂解的扩增子的捕获区域1118、1120与捕获探针1190竞争性杂交。然后将杂交的捕获区域引入1188与裂解产物上的互补序列结合的经标记的寡核苷酸1182、1184中并进行检测以辨别基因座的不同等位基因。
图12是对本发明的另一个具体实施方案的说明,其中使用桥接寡核苷酸并且连接产物在与阵列杂交之前被裂解。在此实施方案中,用于扩增连接产物的引物经差异性标记。在图12中描绘的方法中,提供两组固定序列寡核苷酸1202、1204。每一组包含第一固定序列寡核苷酸1206、1208,其包含与基因座1210、1212互补的序列、引物结合区域1218、1224(不同)、捕获区域1214、1220和限制酶识别位点区1226、1228。引物结合区域1218、1224包含对于所述组的固定序列寡核苷酸1202、1204而言不同的序列,以允许与每个不同靶基因组区域缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸1202、1204而言此实施方案中的捕获区域1214、1220相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点1226、1228对于每个固定序列寡核苷酸上的两个裂解位点而言可以是相同的,和/或对于不同组的固定序列寡核苷酸1302、1304而言可以是相同的。每一组中还存在第二固定序列寡核苷酸1230、1232,其中每个第二固定序列寡核苷酸包含与基因座1234、1236互补的序列和引物区域1238、1240。
在步骤1242中,将所述组的固定序列寡核苷酸1202、1204引入样品中并且允许与基因座1244、1246杂交。杂交(或可选地连接)之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤1248中,向每一组添加桥接寡核苷酸1226、1228并且在杂交的固定序列寡核苷酸之间相邻杂交。在步骤1254中,连接所述组的寡核苷酸以产生连接产物1250、1252。在步骤1254中,将引物1256、1258、1260和1262引入连接产物1250、1252中,连接产物分别与引物区域1218、1238、1224和1240结合,以产生包含引物结合区域1278、1284、捕获区域1276、1280和限制酶识别位点1272和1274的扩增子1264、1266。引物1256和1260经差异性标记,从而扩增子一旦与阵列杂交,就允许来自不同基因座的扩增子之间的区别。在步骤1270中,将一种或多种限制酶引入扩增子1264、1266中并且扩增子经裂解以产生包含标记结合区域1278、1284和捕获区域1276、1280的裂解的扩增子。在步骤1290将裂解产物引入包含多个捕获探针1288的杂交阵列1286中,其中裂解的扩增子的捕获区域1276、1280与捕获探针1288竞争性杂交。
图13是对本发明的另一个具体实施方案的说明,其中使用桥接寡核苷酸并且连接产物在与阵列杂交之前被裂解。图13说明了与图12中的实施方案非常相似的实施方案。在此实施方案中,用于扩增连接产物的引物经差异性标记。在图13中描绘的方法中,提供两组固定序列寡核苷酸1302、1304。每一组包含第一固定序列寡核苷酸1306、1308,其包含与基因座1310、1312互补的序列、引物结合区域1318、1324(不同)、捕获区域1314、1320(相同)和限制酶识别位点区1326、1328。引物结合区域1318、1324包含对于所述组的固定序列寡核苷酸1302、1304而言不同的序列,以允许与每个不同靶基因组区域缔合的固定序列寡核苷酸的差异性标记,而对于两组固定序列寡核苷酸1302、1304而言此实施方案中的捕获区域1314、1320相同以允许与杂交阵列的捕获部件的竞争性杂交。根据该实施方案,限制酶识别位点1326、1328对于每个固定序列寡核苷酸上的两个裂解位点而言可以是相同的,和/或对于不同组的固定序列寡核苷酸1302、1304而言可以是相同的。每一组中还存在第二固定序列寡核苷酸1330、1332,其中每个第二固定序列寡核苷酸包含与基因座1334、1326互补的序列和引物区域1338、1340。
在步骤1342中,将所述组的固定序列寡核苷酸1302、1304引入样品中并且允许与基因座1344、1346杂交。杂交(或可选地连接)之后,优选将未杂交的固定序列寡核苷酸与样品的其余部分分开(未示出)。在步骤1348中,向每一组添加桥接寡核苷酸1326、1328并且在杂交的固定序列寡核苷酸之间相邻杂交。在步骤1354中,连接所述组的寡核苷酸以产生连接产物1350、1352。在步骤1354中,将引物1356、1358、1360和1362引入连接产物1350、1352中,连接产物分别与引物区域1318、1338、1324和1340结合,以产生包含引物结合区域1378、1384、捕获区域1376、1380和限制酶识别位点1372和1374的扩增子1364、1366。引物1356和1360经差异性标记,从而扩增子一旦与阵列杂交,就允许来自不同基因座的扩增子之间的区别。在步骤1370中,将一种或多种限制酶引入扩增子1364、1366中并且扩增子经裂解以产生包含标记结合区域1378、1384和捕获区域1376、1380的裂解的扩增子。在步骤1390将裂解产物引入包含多个捕获探针1388的杂交阵列1386中,其中裂解的扩增子的捕获区域1376、1380与捕获探针1388竞争性杂交。
在图10-13的实施方案中,引入阵列中的裂解的扩增子不包括靶基因组区域的任何部分或与之互补的序列。捕获区域用于与不同靶基因组区域缔合并且经标记的寡核苷酸用于在区域之间进行辨别。以这种方式,裂解的扩增子与常见捕获探针竞争性杂交,并且对靶基因组区域的量化通过对应于与阵列结合的裂解产物的被检测标记来确定。
如先前所述,图2-13所示的本发明的实施方案描绘了用于探查每个基因座或等位基因的两个单独的固定序列寡核苷酸。然而,在一些方面,可使用单个探针,其包含与基因座(包括环前探针)互补的两个或更多个不同的非相邻固定序列寡核苷酸。此类环前探针,例如在美国专利第5,854,033和6,316,229中由Lizardi描述,并且可在与阵列杂交之前通过例如在探针中包括限制性内切核酸酶的位点而线性化。
通用扩增
在本发明的某些方面,在固定序列寡核苷酸杂交并连接之后,直接或在桥接寡核苷酸延伸或引入之后使用通用扩增来扩增连接产物。在多路复用测定系统中,优选使用与每一组的第一固定序列寡核苷酸和第二固定序列寡核苷酸上的通用引物区域杂交的通用引物,通过通用扩增来完成扩增。来自固定序列寡核苷酸的通用引物区域变成连接产物的一部分,然后可以在单个通用扩增反应中扩增连接产物。虽然优选经由固定序列寡核苷酸引入通用引物序列,但是也可经由连接将其添加到连接产物的近端。通用引物区域引入固定序列寡核苷酸中允许在阵列杂交之前对连接产物的全部或一部分进行后续受控的通用扩增。由此扩增过程产生的扩增子可直接使用或在引入阵列中进行检测之前进一步加工。在具体实施方案中,裂解扩增子,并且将包含捕获区域的部分引入到阵列中进行杂交和检测。可以在DNA扩增期间引入偏差和变异性,如在聚合酶链式反应(PCR)期间观察到的。在多路复用扩增反应的情况下,存在基因座以不同速率或效率扩增的可能。基于引物和模板DNA的序列情况、缓冲条件和其他条件,指定基因座的多组引物表现可能有所不同。在某些方面,所述方法中使用的通用引物区域经设计成与利用一般引发机制同时分析大量核酸的常规多路复用测定方法相容。此类“通用”引发方法允许存在于遗传样品的核酸区域的量的有效、高容量分析,且允许此类样品内的核酸区域的存在的全面量化。
整个连接反应或连接反应等分试样可用于通用扩增。使用等分试样允许使用相同或不同条件(例如,聚合酶、缓冲液等)进行平行扩增反应,例如以确保不会由于实验条件而无意中引入偏差。另外,引物浓度的变化可用于有效地限制序列特异性扩增循环的次数。多路复用测定方法的实例包括但不限于Oliphant等人,美国专利第7,582,420.号中描述的那些。
如本文所详细描述,本发明的许多方法利用用于多路复用检测的偶联反应,其中来自多步骤工艺的早期阶段的寡核苷酸含有可用于该工艺的后期阶段的序列。可以单独或联合使用本领域已知的用于扩增和/或检测样品中的核酸的工艺,包括但不限于下面描述的方法。在某些方面,本发明的方法利用下列组合的选择性和通用扩增技术中的一种:(1)LDR偶联PCR;(2)初级PCR偶联次级PCR,偶联次级PCR偶联LDR;和(3)初级PCR偶联次级PCR。这些技术组合中的每一种均可利用来自所述工艺的早期阶段的探针区域,探针区域可在所述工艺的晚期阶段用作引物序列。
Barany等人,美国专利第6,852,487、6,797,470、6,576,453、6,534,293、6,506,594、6,312,892、6,268,148、6,054,564、6,027,889、5,830,711、5,494,810号,描述了连接酶链式反应(LCR)测定用于检测各种核酸样品中的核苷酸的特定序列的用途。
Barany等人,美国专利第7,807,431、7,455,965、7,429,453、7,364,858、7,358,048、7,332,285、7,320,865、7,312,039、7,244,831、7,198,894、7,166,434、7,097,980、7,083,917、7,014,994、6,949,370、6,852,487、6,797,470、6,576,453、6,534,293、6,506,594、6,312,892和6,268,148号,描述了连接酶检测反应(“LDR”)和与聚合酶链式反应(“PCR”)偶联的检测反应用于核酸检测的用途。
Barany等人,美国专利第7,556,924和6,858,412号,描述了扣锁探针(也称为“环前探针”或“多倒置探针”)与偶联的连接酶检测反应(“LDR”)和聚合酶链式反应(“PCR”)用于核酸检测的用途。
Barany等人,美国专利第7,807,431、7,709,201和7,198,814号描述了组合的内切核酸酶裂解和连接反应用于检测核酸序列的用途。
Willis等人,美国专利第7,700,323和6,858,412号描述了环前探针在多路复用核酸扩增、检测和基因分型中的用途。
Ronaghi等人,美国专利第7,622,281号描述了用于使用包含独特引物和条码的衔接子,标记和扩增核酸的扩增技术。
使用杂交和阵列来鉴定感兴趣的核酸区域。用于进行用于检测的多核苷酸杂交测定的方法得到良好开发并且在本领域中是公知的。杂交测定程序和条件将根据应用而改变并且根据已知的一般结合方法来选择,包括在以下提到的那些方法:Maniatis等人,Molecular Cloning:A Laboratory Manual(第2版Cold Spring Harbor,N.Y.,1989);Berger和Kimmel Methods in Enzymology,第152卷,Guide to Molecular Cloning Techniques(Academic Press,Inc.,San Diego,Calif.,1987);Young和Davis,P.N.A.S,80:1194(1983)。用于实施重复且受控制的杂交反应的方法和装置在美国专利第5,871,928、5,874,219、6,045,996和6,386,749、6,391,623中已有描述,其各自以引用的方式并入本文。
在一些实施方案中,阵列包含在溶液中的多种底物,如例如在美国申请第20140057269号和美国申请第20140042366号及美国申请第20140024550号中教导的那些。
本发明还在某些优选方面中考虑到了配体间杂交的信号检测。参见美国专利第5,143,854、5,578,832、5,631,734、5,834,758、5,936,324、5,981,956、6,025,601、6,141,096、6,185,030、6,201,639、6,218,803和6,225,625号,美国专利申请60/364,731和PCT申请PCT/US99/06097(公布为WO99/47964)。
用于信号检测和强度数据处理的方法和装置公开于,例如,美国专利第5,143,854、5,547,839、5,578,832、5,631,734、5,800,992、5,834,758、5,856,092、5,902,723、5,936,324、5,981,956、6,025,601、6,090,555、6,141,096、6,185,030、6,201,639、6,218,803和6,225,625号中,美国专利申请60/364,731和PCT申请PCT/US99/06097(公布为WO99/47964)中。
在某些方面,来自单一样品的连接产物或其扩增子的捕获区域含有索引序列,其将该连接产物鉴定为来自特定样品。阵列的部件将包含探针,其包括与不同样品的索引序列互补的序列以允许鉴定来自特定样品的基因座,基因座来源于该特定样品。
母体样品中胎儿DNA比例的估计
母体样品中胎儿DNA的比例可用作本发明风险计算的一部分,因为胎儿比率提供了关于染色体剂量的预期统计存在的重要信息。与预期统计存在的偏差可指示胎儿非整倍性,尤其是特定染色体的胎儿三体性或单体性。
可以使用本领域中已知用于估计母体样品中胎儿DNA的百分比的任何方法,包括如果胎儿为雄性则量化Y序列或检查表观遗传学标志物(参见,例如Chim等人,PNAS USA,102:14753-58(2005))。使用胎儿比率作为风险计算的一个分量在母体样品中的胎儿DNA水平较低的情况下特别有用。另外,当处于胎儿DNA百分比的某一下限时可能的情况是系统无法可靠地进行分析,因此对胎儿DNA百分比的了解可用于确定可对样品进行哪些另外的分析(如果有的话)。在其他方面,确定母体样品中的胎儿DNA比例可另外地影响检测胎儿非整倍性的确定性或功效的水平。
虽然描述以下方法是为了测定母体样品中胎儿含量的总比例,但是该比例也可以在染色体的基础上测定。例如,可以相比于胎儿染色体18确定胎儿染色体21的频率信息。在另一个实例中,两条或更多条染色体可用于检测胎儿比例,例如可以使用染色体1和2上的基因座的频率。在某些方面,用于确定胎儿比例的染色体是被探查可能的非整倍性的染色体。另一方面,用于确定胎儿比例的染色体明确不是被探查可能的非整倍性的染色体。
来自胎儿的DNA其基因座大约50%遗传自母体而其基因座大约50%遗传自父体。确定哪些遗传基因座有助于来自非母体来源的胎儿(信息基因座)允许估计母体样品中的胎儿DNA比例,并且因此提供了用于计算对于感兴趣的染色体而言在染色体剂量上的统计显著性差异的信息。
在某些方面,非母体多态性的测定通过靶向SNP和/或突变分析而实现以估计母体样品中胎儿DNA的百分比—这是特别适用于本发明的阵列分析的过程。可以使用多路复用SNP检测量化母体样品中的胎儿细胞游离DNA百分比,而无需预先了解母本或父本基因型。在此方面,使用在每个区域中具有已知SNP的两个或更多个选定的多态性核酸区域。在一个优选方面,选定的多态性核酸区域位于不太可能为非整倍性的常染色体上,例如不是染色体21、18或13。扩增来自母体样品(例如,血浆)的选定的多态性核酸区域。在一个优选方面,扩增是通用的;并且在一个优选实施方案中,于一个容器中在一个反应中扩增选定的多态性核酸区域,并且在一个优选实施方案中,在一个容器中在一个反应中扩增选定的多态性核酸区域,用固定序列寡核苷酸的连接产物测定染色体非整倍性。测定并量化母体样品中选定的多态性核酸区域的每个等位基因。在一个优选方面,高通量测序用于此类测定和量化。
这样鉴定基因座,其中母体和非母体基因型不同;例如,母体基因型为纯合的并且非母体基因型为杂合的。信息基因座的这种鉴定通过对特定的选定核酸区域观察一个等位基因的高频率(>80%)和另一个等位基因的低频率(<20%且>0.15%)来实现。使用多个基因组特别有利,因为减小了基因座间等位基因丰度测量中的变化量。使用满足这种要求的所有基因座或亚组以通过统计分析测定胎儿贡献。一方面,通过对来自两个或更多个基因座的低频等位基因一起求和,再除以低频和高频等位基因的总和,然后乘以2来确定胎儿贡献。
对于许多等位基因而言,母体和非母体序列可为纯合的并且相同,并且因为这种信息因而不能区别母体和非母体DNA,所以在母体样品中胎儿DNA百分比的测定中无用。本发明在胎儿DNA百分比的计算中利用等位基因信息,其中在非母体和母体DNA之间存在可区别的差异(例如,胎儿等位基因含有至少一个不同于母体等位基因的等位基因)。因此不选择关于对于母体和非母体DNA而言相同的等位基因区域的数据用于分析,或在估计胎儿DNA比例之前从有关数据中去除以免掩盖有用数据。用于量化母体血浆中的胎儿DNA的附加示例性方法可以,例如在Chu等人,Prenat.Diagn.,30:1226-29(2010)中找到,其以引用的方式并入本文。
数据分析
一旦计算了胎儿细胞游离DNA百分比,就可将该数据与用于非整倍性检测的方法组合以确定胎儿可含非整倍性的可能性。一方面,使用利用随机DNA片段分析的非整倍性检测方法,例如在例如Quake,USSN 11/701,686;和Shoemaker等人USSN号12/230,628中描述的方法。在一个优选方面,使用利用选定核酸区域分析的非整倍性检测方法。在此方面,计算样品的胎儿细胞游离DNA百分比。如本文所述,测定该样品的染色体比率、正常群体的染色体比率和正常群体的染色体比率的变化。可选地,计算的染色体比率使用染色体正常样品的预期值和非整倍体样品的预期值。然后将样品的计算染色体比率与那些预期值做比较。
在一个优选方面,由具有相似胎儿DNA百分比的正常样品确定正常群体的染色体比率及其变化。通过加和来自非整倍染色体的百分比贡献而计算具有该胎儿细胞游离DNA百分比的DNA样品的预期的非整倍染色体比率。然后可将样品的染色体比率与正常群体的染色体比率和预期的非整倍染色体比率进行比较,以使用染色体比率的变化在统计上确定样品是更有可能是正常的还是为非整倍性的,以及正常或非整倍性的统计可能性。
在一个优选方面,母体样品的选定区域包括用于估计胎儿DNA含量的区域以及来自两条或更多条染色体的非多态性区域以在单一反应中检测胎儿非整倍性。当利用胎儿DNA含量帮助确定染色体异常的存在或不存在时,单一反应有助于将测定系统中可能在不同步骤期间引入的可能另外歪曲结果的污染或偏差风险降到最低。
在其他方面,可利用选定的一个或多个核酸区域用于估计胎儿DNA含量以及检测胎儿染色体异常。选定的核酸区域的等位基因可用于估计胎儿DNA含量并且这些相同的选定核酸区域然后可用于检测胎儿染色体异常,忽略等位基因信息。利用相同的选定核酸区域用于胎儿DNA含量和染色体异常的检测还可有助于将由于实验误差或污染引起的任何偏差减到最小。
在一个实施方案中,使用常染色体SNP测量母体样品中的胎儿源贡献,不管胎儿性别如何(参见,Sparks等人,Am.J.Obstet&Gyn.,206:319.e1-9(2012))。所用方法不需要预先了解父本基因型、,因为在所述方法期间在不考虑对父本遗传的了解的情况下鉴定非母体基因座。使用二项分布的最大似然估计可用于计算每个母体样品中若干信息基因座的胎儿核酸估计贡献。使用的计算胎儿核酸贡献的方法例如在美国公布第2013/0024127号中有描述。用于确定胎儿贡献的多态性区域可来自染色体1-12,并且优选不靶向血型抗原。当测试染色体为三体性时,这通知了统计检验,使用来自多态性测定的胎儿贡献估计值限定预期反应幅度。检验统计量可由两个分量组成:与样品为二体性时的预期比率的偏差量度;和与样品为三体性时的预期比率的偏差量度。每个分量呈Wald统计量的形式(例如,Harrell,Regression modeling strategies,(2001,Springer-Verlag),章节9.2.2和10.5),其将观测比例与预期比例做比较并除以观测值变化。
统计Wj可用于测量与样品j为二体性时的预期值的偏差,并且定义为
其中pj和p0如以上用Z统计量所定义的,并且是感兴趣的指定染色体的代表的观测比例的标准偏差。可以使用参数bootstrap抽样来估计标准偏差以基于我们感兴趣的染色体的平均计数和标准误差而产生pj比例分布。第二统计量是其用经调节的胎儿比例代替p0,参考比例定义为
其中fj是样品j的胎儿比例并且p0是如前所述的参考比例。这种调节说明了胎儿为三体性时测试染色体的代表增加。因为作为对于测试染色体使用多种非多态性测定的自然结果而测量许多基因座间计数的方差,所以取初期数据集合内的所有估计值,并且不需要正如对于近似预期比例的方差而言通常所需的外部参考样品或历史信息及对过程偏差控制的归一化调节。
使用的最终统计量为从概念上讲,同时评价与二体性预期和三体性预期的偏差并总结成此单一统计量。合并这两个指标的具体优点是,虽然与二体性的偏差可能较高,但是在特定胎儿贡献水平下可能不会达到对三体性所预期的偏差。分量在这种情况下将为负,从而实际上对与二体性的偏差进行罚分。Sj=0指示二体性与三体性的机会相等。
本发明方法的计算机执行
根据一个示例性实施方案,计算机执行计算胎儿比例的软件组件并且将此信息应用于基因组区域和/或染色体的剂量值中。在一个实施方案中,计算机可包括个人计算机,但是计算机可包括任何类型的包括至少一个处理器和存储器的机器。
软件组件的输出包括带有基因组区域和/或染色体具有剂量异常的可能性值的报告。在一个优选方面,此报告是区域或染色体具有两个拷贝(例如,为二体性)的可能性值与区域或染色体具有更多个拷贝(例如,为三体性)或更少拷贝(例如,为单体性)的可能性值的优势比。该报告可以是打印出来的纸张,或为电子版,其可显示在监测器上和/或经由e-mail、FTP、文本消息发送、发布在服务器上等与使用者电子通信。
虽然本发明的归一化过程显示为作为软件执行,但其也可以作为硬件和软件的组合执行。另外,用于归一化的软件可作为在相同或不同计算机上操作的多个组件执行。
服务器和计算机两者均可包括典型计算设备的硬件部件,包括处理器、输入设备(例如,键盘、打印设备、用于语音命令的麦克风、触摸屏等)和输出设备(例如,显示设备、扬声器等)。服务器和计算机可包括计算机可读介质,例如存储器和存储设备(例如,闪存、硬盘驱动器、光盘驱动器、磁盘驱动器等),其含有执行在被处理器执行时公开的功能性的计算机指令。服务器和计算机还可包括用于通信的有线或无线网络通信接口。
实施例
提出下列实施例,以便为本领域的普通技术人员提供如何制备和使用本发明的完整公开内容和描述,且并不旨在限制发明人认为是其发明的范围,也未旨在代表或暗示下面的实验是所有的实验或仅是进行的实验。本领域的技术人员将认识到,可以在不脱离本发明所广泛描述的精神或范围的情况下,对具体方面中所示的发明作出许多变化和/或改变。因此,现存的方面在所有方面中被认为是说明性的而非限制性的。
已经作出了确保关于所用数字(例如,量、温度等)的准确性的努力,但是应考虑某些实验误差和偏差。除非另外指出,否则份数是重量份,分子量是重均分子量,温度按摄氏度计,且压力是或接近大气压。
实施例1:受试物
分析总共878份母体静脉血样品,三体性状态分类如下:691份为二体性,18份为三体性13,37份为三体性18,且132份为三体性21。血样从至少18岁的单胎妊娠且妊娠期为10-34周的孕妇获得。先前已经为所有受试样品确定了三体性分类;最初使用来自Ariosa Diagnostics,Inc.的Harmony Prenatal Test(San Jose,CA)对486份样品进行试验,而396份样品来源于接受了羊膜诊断核型分析或产后新生儿检查,然后怀疑为三体性时接着进行核型分析的患者。
实施例2:样品制备
如Sparks等人,Am J.Obstet Gynecol.,207(5):374.e1-6(2012)所述进行样品制备和分析。从每位患者的血浆纯化细胞游离DNA并且由染色体13、18和21上的基因座制备DANSRTM(选定区域数字化分析)测定产物(例如,串联连接产物)。对于这种分析而言,采用连接测定,该测定使用对应于每条染色体13、18和21上的864个基因组区域的多组固定序列寡核苷酸和桥接寡核苷酸。使DNA样品附连到固体载体上并且在连接之前去除未杂交的寡核苷酸。另外,开发了使用对应于染色体1-12上的576个多态性位点的多组固定序列寡核苷酸和桥接寡核苷酸以评价每份样品中胎儿cfDNA的分数。使用通用引物扩增由每份样品产生的连接测定产物的一部分并测序,并且使连接测定产物的其余部分与定制生产的DNA阵列杂交。杂交之前,裂解扩增子。这些部分含有相同产物。
实施例3:使用阵列的连接产物量化
由Affymetrix,Inc.(Santa Clara,CA)生产定制DNA阵列以专门量化DANSR测定的产物。在AffymetrixMulti-Channel(MC)仪器上对DNA阵列成像。在单个定制DNA阵列上测定每份患者样品。以384个互连集合生产和加工DNA阵列。在Illumina2500(San Diego,CA)上产生新一代测序数据。在Illumina Cluster Station上使用TruSeqTM簇生成试剂(San Diego,CA)生成簇。每次测定获得平均1104个测序计数。
实施例4:数据分析
使用先前公布的名为胎儿分数优化的三体性风险评价(FORTETM)的算法指定风险评分(see,e.g.,Sparks,et al.,Am J.Obstet Gynecol.,207(5):374.e1-6(2012))。对染色体13、18和21的非多态性连接测定用于确定这些染色体中的每一条的染色体比例。多态性连接测定用于确定胎儿分数。测定可变性被定义为序列计数(测序)或样品上阵列的强度(DNA阵列)的变异系数(CV):优选较低的测定可变性。胎儿分数可变性被定义为测量的胎儿分数的相对标准误差。
实施例5:结果
对于测定三体性风险的878份血浆样品而言,使用连接测定和通过杂交来检测,在基于阵列的风险评分和三体性状态之间存在完全一致性。相比之下,在基于序列的风险评分和三体性状态之间的一致性为99.6%。对于在先前的NIPT筛选中已经收到低风险评分的两份样品而言,测序报告了高风险评分。这些数据证明由基于阵列的分析精确地获得三体性风险评分。
另外,阵列数据使测定可变性降低大约两倍。精确度(如通过降低测定可变性所测量的)对于基于阵列的杂交分析而言相比于基于测序的分析增加。阵列序列检测的中位数测定可变性显示比基于新一代测序的检测提高近两倍(0.051与0.099;p值<0.0001)(参见图14)。柱状图的柱示出了共有特定范围的测定可变性(x轴)的连接测定的数量(y轴)。阵列数据用深灰色标绘,测序数据用白色标绘。两个群体的数量重叠时,柱为浅灰色。基于阵列量化的连接产物具有显著更低的测定可变性,其中测定可变性越低越好。
阵列可用于降低胎儿分数可变性:血浆中胎儿DNA分数影响试验的精确度。本发明的方法使用FORTE算法测量、报告并利用胎儿分数以提供具有低假阳性率的高灵敏度结果(Sparks,et al.,Am J Obstet Gynecol.,206(4):319.e1-9(2012))。FORTE计算的胎儿分数在基于阵列和基于测序的分析数据之间具有极高的重现性(R^2>0.99)。因为阵列能够同时测量比多路复用测序更大数量的连接测定,所以使用基于阵列的分析的胎儿分数估计值更精确,相比于0.021,中位数相对标准误差为0.013。
实施例中呈现的数据显示,与新一代测序相比,对于阵列而言数据可变性的两个主要来源更低:1)使用多态性测定测量的胎儿分数的变化(胎儿分数可变性)和2)样品间非多态性测定的变化(测定可变性)。通过包括更多的多态性测定,已经设计了基于阵列的连接产物产前检查,其提供更低的胎儿分数可变性,产生更佳的精确性。通过降低测定可变性,可以测量更小的非整倍性变化,如在低胎儿分数样品中所观察到的更小的变化。数据证明阵列平台能够进行可靠的非整倍性评估。而且,阵列成像是一个快速过程并且用于样品量化的周转时间减少到每份样品少于1分钟。这些实施例中采用的特定检测系统(GENETITANTM多通道仪)每台时可以为>90个阵列成像。相比之下,即使样品在每条泳道96份样品的组中多路复用时,HISEQTM2500测序系统也具有15份样品/台时的通量。当使用阵列序列检测时,台时和复杂性的这种降低直接转换为资本成本的减少。
基于序列的分析利用样品多路复用以便实现可用序列容量的经济有效的使用。然而,无需归一化,单个样品就可消耗流动池内的大部分序列读数,减少了可用于确定其余样品中的三体性风险的读数。为了更精确地使样品多路复用,需要用于归一化输入DNA的量的费力且昂贵的方法。即使已经做出努力使样品输入相等时,正如最近研究中所报告的,对于12重反应而言观察到每份样品中位数读数变化四倍(Jensen,et al.,PLoS ONe,8(3):e57381(Jul 2014)。基于阵列的NIPT方法不需要样品多路复用。相反,每份样品与单个阵列单独杂交。处理通量通过将例如384个阵列物理连接到单个多阵列板上来提高,以进行方便的高通量处理。因为单独处理每份样品,所以节约了时间并降低了成本。
虽然通过很多不同形式的方面来满足本发明,如与本发明的优选方面有关所详细描述的,但是应该理解,本公开内容被认为是本发明原理的例证且并不旨在将本发明限于本文说明和描述的特定方面。在不脱离本发明精神的情况下,本领域的技术人员可以作出许多改变。本发明的范围将通过所附权利要求及其等效内容来度量。摘要和标题并不能被解释为限制本发明的范围,因为其目的是使有关当局,以及一般公众能够迅速地确定本发明的一般性质。在下面的权利要求中,除非使用术语“手段(means)”,否则本文叙述的特征或元件均不应依照35U.S.C.§112,被解释为手段加功能限定。
- 还没有人留言评论。精彩留言会获得点赞!