基于量子点探针的三唑磷检测试剂盒的制作方法
本发明涉及农药检测领域,具体而言,涉及一种基于量子点探针的三唑磷检测试剂盒。
背景技术:
有机磷杀虫剂是使用最广泛的杀虫剂,包括农业中使用的一些毒性最大的化学品。三唑磷(triazophos)已广泛用作高毒性有机磷的优良替代品,以控制谷物、水果、蔬菜上的害虫。由于化学和光学稳定性及半衰期长,三唑磷可能在自然环境中残留时间长。据文献报道,三唑磷诱导大鼠氧化应激和组织病理学改变,可能对人类健康也有潜在的危害。自2016年底,中国政府已禁止在蔬菜上使用三唑磷。因此,有必要开发一种有效的分析方法,用于定期监测农业和环境样品中的三唑磷残留。
目前三唑磷检测方法有气相色谱(gc),气相色谱与质谱联用(gc-ms),液相色谱与质谱联用(lc-ms),以及基于酶/抗体的免疫分析。但成本高,操作复杂,分析时间长、仪器尺寸大等是使用gc/lc-ms进行现场直接检测的主要缺陷。另一方面,抗体的生产,试剂的稳定性和实验动物的使用限制了免疫测定的广泛应用。因此有必要建立一种灵敏,稳定,廉价的三唑磷检测方法,特别适用于现场快速检测,以满足快速响应和控制的需要。
分子印迹是模仿抗原-抗体相互作用原理,是一种在合成分子印迹聚合物(mips)中产生特异性识别位点的技术,是天然抗体的替代品。mip具有很多优点,如高化学稳定性和良好的刚性,特异性识别,低成本和易于合成等。因此,mips已被广泛用于许多仿生免疫吸附测定中,特别是仿生酶联免疫吸附测定。然而,酶是大分子物质,酶标抗原可能引起空间位阻,影响竞争效应,进一步降低方法的灵敏度。而用量子点(qds)替代酶标记半抗原探针尺寸小且结构较简单。qds灵敏度高,亮度强和光稳定性好,发射波长可调,具有宽激发光谱及窄发射光谱。用qd标记半抗原可以增加方法的灵敏度。
有鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于克服传统的三唑磷检测方法存在检测时间长、仪器价格昂贵、特异性抗体难制备等缺陷,提供一种基于量子点标记三唑磷仿生免疫吸附检测方法。
为了实现本发明的上述目的,特采用以下技术方案:
本发明涉及一种基于量子点探针的三唑磷检测试剂盒,包括:
包被有分子印迹聚合物膜的固相载体以及量子点探针标记的三唑磷半抗原;
所述分子印迹聚合物膜以三唑酮为模板分子制备得到。
为了防止分子模板泄漏,本申请选择三唑酮(三唑磷的结构类似物)作为模板,在固相载体(例如96孔板)表面合成分子印迹膜作为识别材料,同时合成cdse/znsqds标记的探针。本发明开发了基于微阵列印迹膜的直接竞争cdse/zns量子点荧光测定法,并用于测定样品(例如蔬菜与水果,如甘蓝和苹果)中的三唑磷。
根据本发明的另一方面,本发明还涉及使用如上所述的试剂盒进行三唑磷检测的方法,包括:
使用待检测物与所述量子点探针标记的三唑磷半抗原竞争性地与固相载体上包被的分子印迹聚合物膜结合,读取所述量子点探针的荧光值以确定所述待检测物中三唑磷的含量。
与现有技术相比,本发明的有益效果为:
1).本发明提供的三唑磷吸附功能材料具有较高的选择性,采用虚拟模板可解决模板渗漏问题,可以代替生物抗体应用于荧光免疫技术;该吸附功能材料由化学方法制备,具有较高的稳定性、较长的使用寿命和较强的抗恶劣环境的能力,克服了传统生物抗体制备周期长、易失活、成本高等缺点。
2).本发明将分子印迹技术与荧光免疫技术联用,建立对三唑磷具有高灵敏度的仿生荧光免疫吸附检测方法。该方法的最低检出限为0.01μgl-1,大大低于最高残留限量值,能够满足检测需要,适用于快速检测三唑磷。
3).本发明制备的仿生抗体可以重复使用,成本大大降低;并且前处理简单,灵敏度高,实验操作简单,适用于各种食品中三唑磷的快速检测。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
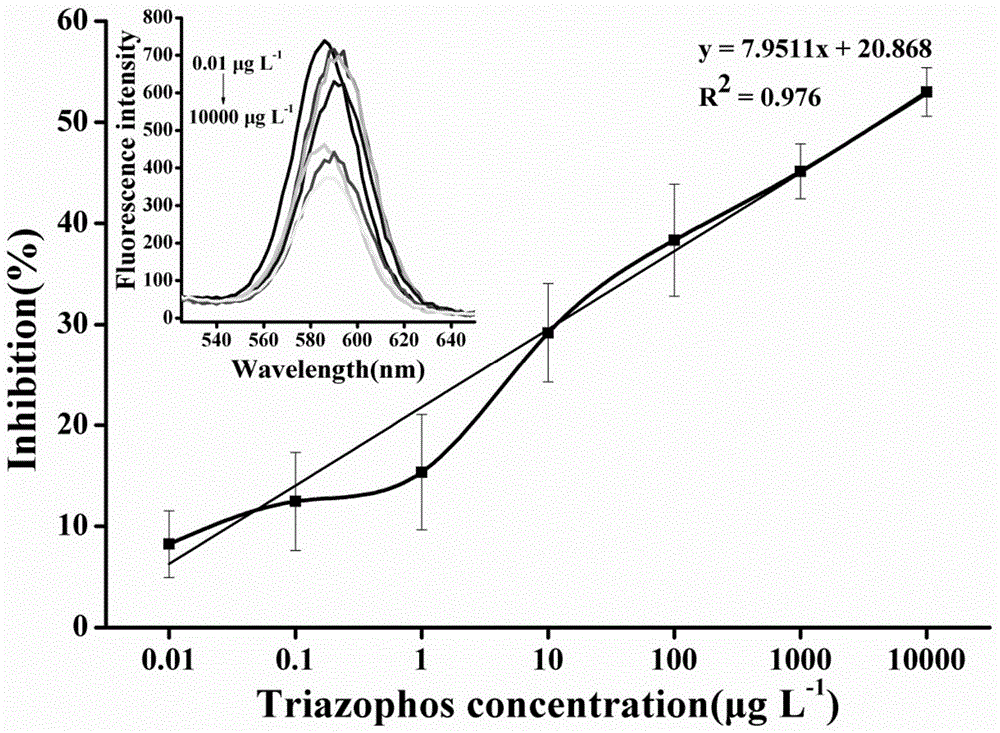
图1为本发明一个实施例中三唑磷的量子点标记仿生免疫分析方法标准曲线。
具体实施方式
本发明涉及一种基于量子点探针的三唑磷检测试剂盒,包括:
包被有分子印迹聚合物膜的固相载体以及量子点探针标记的三唑磷半抗原;
所述分子印迹聚合物膜以三唑酮为模板分子制备得到。
在一些实施方式中,所述量子点探针标记的三唑磷半抗原的制备方法包括:
将三唑磷半抗原依次与nhs、edc活化反应后得到活化酯,将所述活化酯与所述量子点探针进行偶联。
在一些实施方式中,所述三唑磷半抗原与nhs、edc的摩尔比为1:(1.7~2.3):(2.7~3.3);
在一些实施方式中,所述三唑磷半抗原与nhs、edc的摩尔比为1:2:3。
在一些实施方式中,所述活化酯和所述量子点探针的摩尔比为(460~500):1;也可以选择(470~490):1,或480:1。
在一些实施方式中,所述量子点探针为cdse/znsqds。
在一些实施方式中,所述分子印迹聚合物膜的制备在所述固相载体中进行,其方法包括:
将模板分子于有机溶剂中,与甲基丙烯酸(maa)进行预反应;
随后预反应产物在有机溶剂中与三羟甲基丙烷三甲基丙烯酸酯(trim),偶氮二异丁腈(aibn)接触反应;洗脱模板分子后即得所述分子印迹聚合物膜。
在一些实施方式中,所述预反应的反应条件为室温磁力搅拌25min~35min。
在一些实施方式中,所述接触反应的条件为:
继续磁力搅拌使混合液预聚合反应40~80min得到预聚合液,向所述固相载体中加入预聚合液,真空35℃~39℃下反应20h~28h以完成包被。
在一些实施方式中,洗脱模板分子所用的洗脱液为甲醇:乙酸(7:1)溶液。
在一些实施方式中,所述模板分子与所述甲基丙烯酸的摩尔比为1:(5~7);也可以选择1:6。
在一些实施方式中,所述模板分子的浓度为1.5~2.5mmol;也可以选择2mmol。
在一些实施方式中,所述模板分子与三羟甲基丙烷三甲基丙烯酸酯的摩尔比为1:(2.5~3.5);也可以选择1:3。
在一些实施方式中,所述有机溶剂为乙腈;
在一些实施方式中,所述模板分子:乙腈=2mol:15~25ml;也可以选择为2mol:20ml。
在一些实施方式中,所述模板分子:偶氮二异丁腈=1mol:20~30mg;也可以选择为1mol:25mg。
在一些实施方式中,所述固相载体为酶标板。
根据本发明的另一方面,本发明还涉及使用如上所述的试剂盒进行三唑磷检测的方法,包括:
使用待检测物与所述量子点探针标记的三唑磷半抗原竞争性地与固相载体上包被的分子印迹聚合物膜结合,读取所述量子点探针的荧光值以确定所述待检测物中三唑磷的含量。
在一些实施方式中,所述方法具体包括:
在包被有分子印迹聚合物膜的固相载体(例如酶标板)上设置空白对照孔、阴性对照孔以及检测孔;
所述检测孔中加入量子点探针标记的三唑磷半抗原稀释液,以及三唑磷标准液或样品提取液,且不同孔中所加入的三唑磷标准液或样品提取液按梯度稀释;
所述阴性对照孔中加入量子点探针标记的三唑磷半抗原稀释液以及缓冲液;
其中,所述量子点探针标记的三唑磷半抗原稀释液由量子点探针标记的三唑磷半抗原标准溶液用缓冲液稀释得到;
所述空白对照孔中加入所述缓冲液;
各孔按上述方式加入完毕后室温孵育50min~70min,洗板后在各孔中分别加入底物液,室温孵育30min~60min后终止反应;酶标仪读取各孔吸光度并按照下列公式计算抑制率:
ic%=[1-(f样品-f空白)÷(f对照-f空白)]×100;
式中:
ic%——三唑磷对抗原抗体结合反应的抑制率;
f对照——所述阴性对照孔平均荧光值;
f样品——所述检测孔中三唑磷标准液或样品提取液的平均荧光值;
f空白——所述空白对照孔的平均荧光值;
以三唑磷标样梯度稀释液浓度的对数值为横坐标,相应的抑制率百分数为纵坐标,分别绘制标准曲线;
将所述样品提取液对应的吸光度值代入所述标准曲线,即得所述样品提取液浓度。
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
本实施例提供了一种三唑磷量子点标记仿生免疫分析方法,包括以下步骤:
1.半抗原的合成:
(1)o-乙基硫代磷酰二氯(tzm-1)的合成
称取三氯硫磷(pscl3)68g(约0.4mol)置于带有低温温度计的三口烧瓶中,以冰盐水浴将其冷却至-10~-5℃,在剧烈搅拌下滴加入无水乙醇55g(约1.2mol),严格控制滴加速度,使反应液温度始终不大于0℃。滴加完毕后在10℃下继续反应2h。反应毕,以(0±5)℃蒸馏水洗涤反应液(100ml×2),分出油层并以无水na2so4干燥,再经水泵减压蒸馏,收集65~75℃馏分,得到51.8g无色透明油状液体(收率72.3%,以三氯硫磷计)。
(2)o-乙基-o-[3-(1-苯基-1,2,4-三唑基)硫代磷酰一氯(tzm-2)的合成称取tzm-136g(约0.2mol)置于250ml三口烧瓶中,搅拌下加入约16g(约0.1mol)。1-苯基-1,2,4-三唑醇,再加入15mltea和80mldcm。待固体全部溶解后,用冰水浴将该溶液降温至20℃以下,加入微量催化剂并逐滴加入55ml2mol/l的naoh水溶液,继续反应1h。反应完毕后,加入50ml5%naoh冰水溶液,振荡后分出水层,油层以冰水洗涤至中性,再经无水na2so4干燥,减压浓缩,得少量棕色油状物,该油状物用石油醚萃取(50ml×2),提取液再减压浓缩,得10.6g黄色液体(收率35%,以三唑醇计)。
(3)三唑磷半抗原的合成称取1.03g(约10mmol)4-氨基丁酸,溶解于10mlnaoh溶液(1mol/l)中,冰水浴冷却至0~10℃,搅拌下缓慢加入1.51g(约5mmol)溶解于10ml二氧六环的tzm-2,加入微量催化剂,再滴加入10mlnaoh水溶液(1mol/l)。升温至15~25℃,反应4h。反应完毕后,加入50ml水,再用石油醚洗涤反应混合物(40ml×2),弃石油醚层,以2mol/lhcl调节水相ph约为3,再以乙酸乙酯萃取(40ml×2),提取液用少量水洗涤后经无水na2so4干燥,减压浓缩,残余物于4℃密封存放过夜,有无色品体析出。以乙酸乙酷-石油醚重结晶,过滤,干燥,得0.52g白色固体(thbu,收率27%,,以中间体tzm-2计)。
2.量子点标记半抗原探针的制备:
(1)活化酯的制备三唑磷半抗原(15mmoll-1,溶解在dmf中)和nhs(12.3mg/ml,溶解在dmf中)加入离心管中涡旋混匀1min。然后,加入edc(5mg/ml,溶解在dmf中),用dmf定容到1ml。混合液在室温下震荡反应12h以上。半抗原、nhs、edc摩尔比为1:1.7:3.3。
(2)标记反应反应前加入1.5μl吐温-20减少量子点团聚。50μlcdse/znsqds(用340μlbbs溶液溶解)和80μl活化酯加入到离心管中室温震荡反应12小时以上。活化酯和量子点摩尔比为460:1。
(3)标记物的纯化标记后的反应液在pbs(10mmoll-1,ph=7.4)溶液中室温透析1天(每4h更换一次透析液)。
3.分子印迹聚合物膜的制备:三唑酮(2mmol)溶解在15~25ml乙腈中,然后加入maa(10mmol)。室温磁力搅拌25min~35min后,依次加入trim(5mmol)和40mgaibn。继续磁力搅拌使混合液预聚合反应40min。向黑色96孔板中每孔加入100μl预聚合液,真空39℃下反应20h。反应结束后分别用超纯水和甲醇洗板3次以除去未反应的溶液。用甲醇:乙酸(7:1)溶液超声分子印迹膜进行洗脱,直至用hplc检测不出模板,随后换用甲醇超声4h除去残留的乙酸。37℃干燥后得分子印迹聚合物膜。
4.直接竞争cdse/znsqds荧光仿生免疫分析方法的建立:步骤3)制备的印迹聚合物膜作为仿生抗体;步骤2)制备的量子点标记半抗原标准溶液用bbs(ph=5)溶液稀释100倍,作为荧光标记稀释液。
首先,用pbst溶液洗板3次。然后,分别向空白孔和对照孔加入200μl和100μl的10%甲醇-bbs(ph=5)溶液,实验孔中加入100μl标准溶液或样品提取液。接着除了空白孔外,向其他每个孔中加入100μlcdse/znsqds标记探针溶液,将96孔板在室温下避光震摇60min。pbst溶液洗板10次,用酶标仪读取每孔荧光值(激发波长368nm,发射波长586nm),分别计算抑制率;
按照下列公式计算不同浓度三唑磷对抗原抗体结合反应的抑制率值:
ic%=[1-(f样品-f空白)÷(f对照-f空白)]×100;
式中:
ic%——三唑磷对抗原抗体结合反应的抑制率;
f对照——对照孔平均荧光值;
f样品——三唑磷标准液或样品提取液的平均荧光值;
f空白——空白孔平均荧光值。
以三唑磷标样梯度稀释液浓度的对数值为横坐标,相应的抑制率百分数为纵坐标,分别绘制标准曲线。
所述pbs(0.01mol/l,ph=7.4)的配制为:kh2po40.27g,无水na2hpo41.14g,nacl8.0g,kcl0.2g用超纯水定容至1l。
所述pbst溶液的配制为:在pbs(0.01mol/l,ph=7.4)缓冲液中加入0.05%吐温-20摇匀。
所述bbs(ph=5)溶液的配制为:分别准确称取硼酸1.237g,加超纯水至100ml,称取硼砂1.907g,加超纯水至100ml,再分别取体积比为4.5:5.5混合,调ph至5。
实施例2
本实施例提供了一种三唑磷量子点标记仿生免疫分析方法,包括以下步骤:
1.同实施例1
2.量子点标记半抗原探针的制备:
(1)活化酯的制备三唑磷半抗原(15mmoll-1,溶解在dmf中)和nhs(12.3mg/ml,溶解在dmf中)加入离心管中涡旋混匀1min。然后,加入edc(5mg/ml,溶解在dmf中),用dmf定容到1ml。混合液在室温下震荡反应12h以上。半抗原、nhs、edc摩尔比为1:2.3:2.7。
(2)标记反应反应前加入1.5μl吐温-20减少量子点团聚。50μlcdse/znsqds(用340μlbbs溶液溶解)和80μl活化酯加入到离心管中室温震荡反应12小时以上。活化酯和量子点摩尔比为500:1。
(3)标记物的纯化标记后的反应液在pbs(10mmoll-1,ph=7.4)溶液中室温透析1天(每4h更换一次透析液)。
3.分子印迹聚合物膜的制备:三唑酮(2mmol)溶解在15~25ml乙腈中,然后加入maa(14mmol)。室温磁力搅拌25min~35min后,依次加入trim(7mmol)和60mgaibn。继续磁力搅拌使混合液预聚合反应80min。向黑色96孔板中每孔加入100μl预聚合液,真空35℃下反应28h。反应结束后分别用超纯水和甲醇洗板3次以除去未反应的溶液。用甲醇:乙酸(7:1)溶液超声分子印迹膜进行洗脱,直至用hplc检测不出模板,随后换用甲醇超声4h除去残留的乙酸。37℃干燥后得分子印迹聚合物膜。
4.同实施例1
实施例3
本实施例提供了一种三唑磷量子点标记仿生免疫分析方法,包括以下步骤:
1.同实施例1
2.量子点标记半抗原探针的制备:
(1)活化酯的制备160μl三唑磷半抗原(15mmoll-1,溶解在dmf中)和45μlnhs(12.3mg/ml,溶解在dmf中)加入离心管中涡旋混匀1min。然后,加入255μledc(5mg/ml,溶解在dmf中),用dmf定容到1ml。混合液在室温下震荡反应12h以上。半抗原、nhs、edc摩尔比为1:2:3。
(2)标记反应反应前加入1.5μl吐温-20减少量子点团聚。50μlcdse/znsqds(用340μlbbs溶液溶解)和80μl活化酯加入到离心管中室温震荡反应12小时以上。活化酯和量子点摩尔比为480:1。
(3)标记物的纯化标记后的反应液在pbs(10mmoll-1,ph=7.4)溶液中室温透析1天(每4h更换一次透析液)。
3.分子印迹聚合物膜的制备:三唑酮(2mmol)溶解在20ml乙腈中,然后加入maa(12mmol)。室温磁力搅拌30min后,依次加入trim(6mmol)和50mgaibn。继续磁力搅拌使混合液预聚合反应1h。向黑色96孔板中每孔加入100μl预聚合液,真空37℃下反应24h。反应结束后分别用超纯水和甲醇洗板3次以除去未反应的溶液。用甲醇:乙酸(7:1)溶液超声分子印迹膜进行洗脱,直至用hplc检测不出模板,随后换用甲醇超声4h除去残留的乙酸。37℃干燥后得分子印迹聚合物膜。
4.同实施例1
实验例
称取10g打碎的甘蓝或苹果样品加入50ml离心管中,加入10ml乙腈涡旋2min。再加入4gmgso4和1gnacl涡旋2min,5000rpm下离心5min。取2ml上清液加入到10ml称有100mgpsa,100mgc18和300mgmgso4的离心管中,涡旋2min,10000rpm下离心5min。将上清液氮吹干,用10%甲醇-bbs(ph=5)溶液稀释20倍。
将样品提取液代替实施例3步骤4中所述三唑磷标样梯度稀释液,重复步骤4)操作,根据抑制率由标准曲线计算出待测物中三唑磷的含量。图1为三唑磷的量子点标记仿生免疫分析方法标准曲线。由图1可知,该分析方法对三唑磷的最低检出限为0.01μgl-1,满足检测需要。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!