吸入用吡咯衍生物干粉制剂的制作方法与工艺
曲霉病是由曲霉菌引起的病症谱,该曲霉菌为丝状真菌,更具体地为按照半知菌亚门的细分形式分类的子囊菌。侵袭性曲霉病(IA)是一种分生孢子萌发后的曲霉菌定植的高级状态,并且其是在免疫功能低下(IC)的患者中引起发病率和死亡率相关的感染性疾病的常见原因。在过去的二十年里,IA感染的发病率大幅升高。例如,从80年代到1997,与侵袭性曲霉病相关的死亡率的趋势显示增幅为357%。作为机会性疾病(opportunisticdisease),当前临床实践中遇到的IC病人的数量的增加可以解释上述增幅。该病菌的主要途径(80%-90%的IA)并且经常作为侵袭的起点能够导致播散状态,在多于90%的情况下导致死亡。该真菌能够在侵袭肺组织之后播散通过血流到达肝脏、脾脏、肾脏、脑和其他器官。该侵袭状态主要针对分生孢子吸入后没有足够的免疫防御(主要是巨噬细胞)以阻止其萌发并因此通过组织和血液毛细血管在污染区菌丝增殖(主要是中性粒细胞)的IC人群。临床指南建议使用两性霉素B用作肺侵袭性曲霉病的初步治疗。但是两性霉素B耐受性差,表现出多种严重的副作用。此外,吸入型两性霉素B在化疗或自体骨髓移植后的长期中性粒细胞减少的病人的预防中表现为无效。由于这些原因,通常两性霉素B是禁用的并且一线治疗作为黄金标准级别的是唑类衍生物(伊曲康唑,伏立康唑,泊沙康唑,雷夫康)。尽管有这些现有的治疗(口服的和静脉内的),一旦进入侵袭性阶段,死亡率便从50%长至90%(在关于群体的类别和研究中)。对于多数IC患者的病发进程可以是惊人地快速的(例如从起始到死亡为7-14天)。多种因素的结合可以解释上述的高失败率。首先,侵袭性肺曲霉病在疾病在第一阶段时难以诊断出并且一旦首先表现出症状往往已经进入高级侵袭阶段。失败的另外一个重要原因是现有的治疗(口服,静脉的),由于其需要高全身暴露(highsystemicexposure)以达到所需的肺部浓度从而引发多种副作用和代谢相互作用。此外,由于唑类衍生物水溶性差(例如伊曲康唑水溶性<1μg/ml),口服治疗显示出生物利用度方面的高度个体内和个体间差异,能够导致其在肺组织中的浓度低于治疗浓度。另外一个重要因素也需要考虑以解释治疗的高失败率。实际上,为实现最佳抗真菌活性,需要在肺上皮细胞中和肺组织中保持最低抑菌浓度(MIC)。传统治疗(口服,静脉的),尽管有高全身浓度(highsystemicconcentrations),但浓度可能无法达到真菌病灶内。由于这些原因,肺部给药可以作为预防和/或治疗侵袭性肺曲霉病的感兴趣的可选手段。通过将抗真菌药物直接在肺部的感染位点给药,高于MIC90%的浓度能够有效地并且直接地保持在肺组织中,同时减少药物的全身暴露(systemicexposure),从而减少副作用和代谢相互作用。但是,为达到上述结果,水溶性差的活性成分必须有效的施用于肺部以及必须尽可能多地在原位溶解。历年来,已经广泛开发了肺部的药物给药。无数的其克服的问题和其在特殊情况下提供的优势能够证明对该特殊的给药途径的兴趣。实际上,肺部药物给药对全身给药和局部给药以治疗全身性或肺部疾病都是有效的。该非侵袭性给药途径避免了肝脏的首过效应,该首过效应例如能导致活性药物成分(API)失活或形成有毒的代谢产物。已经证明肺部药物给药需要比口服途径更小的剂量便达到相当的肺部治疗效果。这在通过抗感染药物(如唑类衍生物)吸入剂治疗的并呈现全身副作用和代谢相互作用的肺部感染疾病的情况下尤其有趣。实际上,肺部药物给药能够允许最大限度地减少全身浓度,从而减少副作用,同时直接在感染位点保持有效的肺浓度。将抗感染药物直接施与肺能够使得全身浓度最大限度的减少,因此最大限度地减少常规抗真菌药物中的非常明显的药物全身副作用和代谢相互作用。这些相互作用和副作用通常是不同患者群体中治疗失败的原因。有许多实现口服吸入(肺部给药)的方法。吸入装置可分为三种不同类型,包括液体喷雾剂,加压气溶胶计量吸入器(pMDIs),和干粉分散装置。前两种类型的装置由于其劣势人们对其已逐渐丧失兴趣,而该劣势能够通过使用干粉吸入器(DPIs)而克服。液体喷雾主要遇到的问题是储存期间的药物不稳定性,达到总雾化的相对较长的时间,有细菌污染的风险,高成本,低效率和重复性差。关于pMDIs的给药过程失败的其中一个主要原因是活化剂和呼吸之间的同步化的必要性。由于这些原因,DPIs在当前肺部给药领域中是最感兴趣的研究重点。关于上述问题,需要解决的问题是给患者提供一种抗真菌吸入组合物,其能够提供高肺部沉积以及能够使得水溶性差的活性成分在原位具有足够的溶解曲线,从而能够使得药物产品具有优化的功效。另外,该吸入组合物应该表现出可接受的安全性,应该是稳定的,应该是以可重复的和精准的方式容易地给药的。所述组合物的制备过程应该是短的,简单的,价格便宜的,生态的,可靠的,并且环境友好的(非USP一类二类溶剂)。首先,制剂必须具备一个重要的特点是具有改进的最佳的体外溶解曲线(与未经配制的药物相比)。该制备过程必须表现出控制活性成分的溶解速度的灵活性,能够获得最佳的药代动力学特征从而提供最佳的治疗反应。最佳的药代动力学特征对应表现为肺部停留时间的最大化并同时最大限度的减少全身吸收和清除。唑类化合物是水溶性差的物质(例如伊曲康唑的溶解度为pH7<1μg/ml)并且吸入不溶性粉末能够导致(ⅰ)低耐受性和/或(ⅱ)缺乏功效。水溶性差的活性成分的低湿润性能够引起吸入后肺粘膜的刺激和感染。吸入微粒的湿润性必须加强。而且,为了有效,抗真菌药物必须在给药后(在该情况下为吸入给药)达到高于相关真菌的MIC的肺部浓度。通常认为药物的活性形式是溶解状态。也即,吸入剂量的溶解比例必须在肺表皮细胞和肺组织中保持高于曲霉菌的MIC浓度。那么药物的溶解率会直接影响具有抗真菌活性作用的沉积的剂量比例。如上所述,唑类化合物是难溶性的并且是具有极差溶解速度的微粉化松散材料。该溶解速度和湿润性的改进在此处对于防止通过下呼吸道中肺泡巨噬细胞过量清除药物的不溶部分,以及上呼吸道中粘液纤毛的清除是必要的。但是,活性成分的溶解速度的加快最好限制在一定的范围内,因为过快的溶解速度会导致溶解的部分在全身区域过量吸收并且从而可能导致不良事件。本发明应该满足的一个需求是改变干粉组合物从而改进和/或调节其溶解速度,但同时保持良好的粉末流动性和高分散性的可能性。该活性成分的溶解速度必须保持在确定的范围内并且其应该能够变化溶解曲线(在溶解范围内同一时间点较多或较少量的溶出的活性物质),以改变原位的溶解速度因此改变治疗和副作用。其次,口服吸入后抗真菌唑类化合物必须到达感染位点。所述干粉应该表现出最佳的空气动力学行为。也就是干粉必须到达潜在的分生孢子的沉积位点,该位点真菌能够生长并侵袭外周组织区。关于这点,显然从干粉吸入器启动剂量后,生成颗粒的特定的部分必须表现出真菌孢子的类似的空气动力学直径范围(1.9-6μm之间),从而提供给肺部一个适当的抗真菌剂量。该在呼吸状态下从吸入装置生成的颗粒必须表现为具有高比例的小于6μm的空气动力学直径的颗粒。该比例能够直接影响实际到达肺部的剂量。该颗粒的空气动力学行为由其大小和组成决定。如上所述,制剂必须表现出最优的溶解曲线以获得最优的体内药代动力学性质。一旦开发了最优的组合物,其应该能够改变其空气动力学行为以调节粉末细颗粒级分以达到能够正确发挥其真菌活性(取决于其溶解速度曲线)的适当的沉积剂量。第三,另一个原始观点也需要考虑。实际上,在吸入所述干粉后必须表现出良好的安全性并且必须与肺膜兼容以防止高反应性、咳嗽、气道痉挛或感染。溶解速度的提高,在该颗粒情况下有必要的,通常使用特定的能够引起不良反应或不适合肺部给药用的赋形剂。由于关于吸入赋形剂的安全性的文献非常有限,为避免吸入后肺部毒性,生理成分的使用,公认安全(GRAS)的且许可的赋形剂必须在肺部配方中是特许的(例如美国食品和药物管理局(FDA数据库))。这是一个现实的限制,因为许可的赋形剂非常有限并且主要地肺部的内源性或内源性物质的衍生物被认为是GRAS赋形剂。此外再次考虑到制剂的安全性,优选地在制备过程中应该避免使用美国药典委员会(USP)和欧洲药典1类和2类溶剂,该类溶剂具有高毒性和在药物制剂中低耐受的残留量。从生态角度考虑,仅使用3类溶剂和节省赋形剂能够显著减少污染并且减少运营商有害污染的风险,这是不可忽视的受益。这也通过减少必须实施以防止运营商的可能污染或泄漏的资源从而减少了制造成本。第四,干粉吸入器用的粉末必须显示出良好的流动性,低聚集的趋势以用于工业规模的简单制备过程。最后,所述制备过程必须简单,可连续并且设计为可通过一或两个步骤实现以获得最终的干品。在此需要开发一种简单的,灵活的过程仅使用GRAS许可的赋形剂和低毒性潜在溶剂生产吸入用干粉以治疗肺侵袭性曲霉病(ⅰ)能够改进和/或控制活性成分的溶解速度(ⅱ)能够改变颗粒的空气动力学行为并同时保持其溶解速度的改进和/或改变(ⅲ)表现出良好的流动性(ⅳ)包括简单的、可靠的、可重复的并且相对价格便宜的制备过程。本发明能够生产具有高比例颗粒的干粉,其表现出相同吸入分生孢子同样的空气动力学直径。该颗粒级分表现出与未配制的药物相比,具有改进的和/或可控的溶解曲线。该释放曲线能够通过仅使用内源性或GRAS物质和低毒性潜在溶剂而改变。其整个过程是一或两步操作。
背景技术:
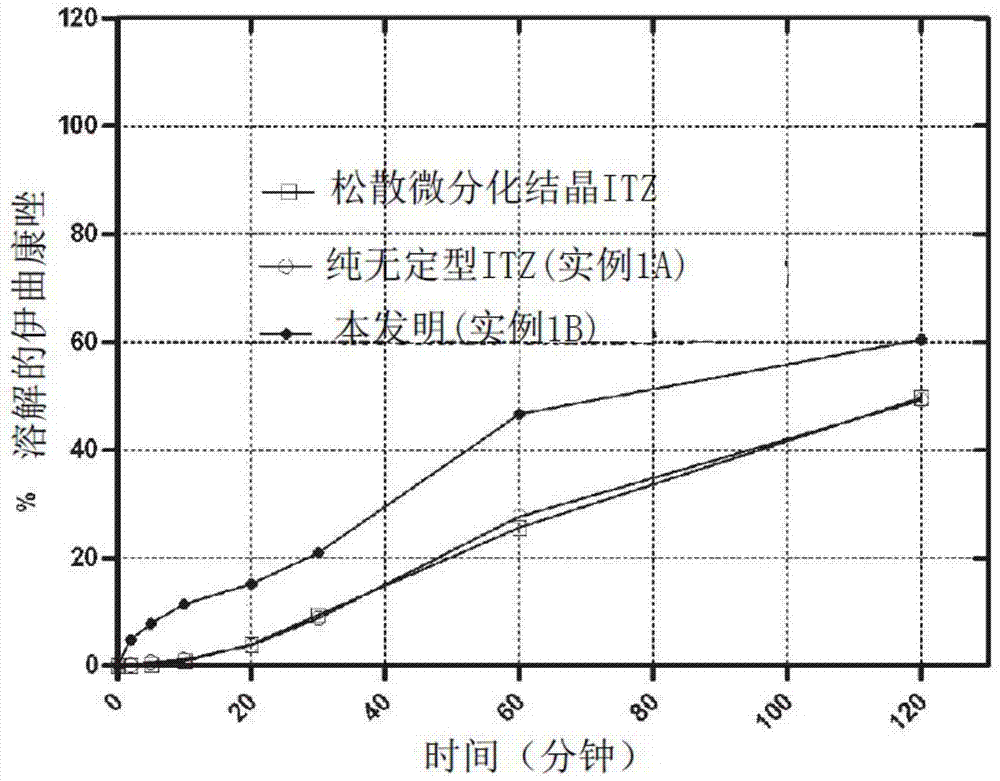
已经开发了多种方法用于开发适合难溶性组合物的肺部给药的制剂。多数此类发明披露了制备过程或制剂的一种策略,但没有任何发明能够满足前述的所有需求。关于上述问题,需要解决的问题是给患者提供一种抗真菌吸入组合物,其能够提供高肺部沉积并且同时能够使得难溶性活性成分在原位具有足够的溶解曲线,从而能够使得药物产品具有优化的效果。另外,该吸入组合物应该表现出可接受的安全性,应该是稳定的,应该是能够以可重复的并且精准的方式容易的给药。所述组合物的制备过程应该是短的、简单的、价格便宜的、生态的、可靠的、并且环境友好的(非USP一类二类溶剂)。很多发明人发明了悬液、纳米悬液和适合雾化用难溶的活性成分的溶液(US专利号6,264,922B1,德国专利公布号10145361A1,PCT国际公布号WO03035031,PCT国际公布号WO2009/137611A2)。但是如前所述的通过喷雾的肺部给药方式表现出多种问题和缺点,例如药物不稳定性,达到总雾化的时间长,细菌污染的风险,高成本,低效率和重复性差。此外,由于纳米颗粒药物具有固有溶解度,这些策略使得无法优化药物的溶解速度。PCT国际公布号WO2009/106333A1公开了一种新型抗真菌唑类衍生物纳米悬液具有改进的纯度性能。该高纯度性能通过高质量的制备过程从而保证,该制备过程最大化的减少了可能来自于设备的制剂污染。这就保证了可由无机不溶性杂质产生的最小毒性。加拿大公布号2014401A1涉及药学组合物用于治疗吸入的侵袭性真菌感染。其公开了吸入用干粉,其中微粉化活性成分与可接受载体混合。这种组合物能够使得活性成分深层渗透到肺但是不能促进溶解速度。许多其他的研究小组的研发兴趣在于制备吸入用干粉药物的开发,其表现出溶解度的改进。美国专利号6,645,528B1公开了一种多孔药物基质的制备方法其表现出比同样药物的松散材料(bulkmaterial)和无孔药物基质更快的溶解速度。该基质产品能够作为干粉通过吸入给药。在制备过程中,该活性物质溶解于挥发性溶剂以形成药物溶液。将成孔剂与该药物溶液结合以形成乳液,悬液或第二溶液。之后去除该挥发性溶剂和成孔剂(优选的通过喷雾干燥)从而得到药物多孔基质。该成孔剂可以是挥发性液体或挥发性固体,优选的为与挥发性溶剂不混溶的挥发性盐。作者描述了成孔剂的使用是增加活性成分的溶解速度的关键特性。但是并没有提及或阐述通过该方法,能够优化吸入颗粒的体外溶解速度和空气动力学行为。此外其也没有公开为吸入干粉特别设计的体外碰撞实验和溶解度测试的实例。另外一方面,其所有实例中使用的溶剂和赋形剂不满足肺部给药领域对毒性的要求。该制剂的构思优选的开发用来生产肠外制剂,其给药前需要一个在溶液中重新分散的步骤。美国专利公开号2004/0105821A1的申请使用了该吸入用干粉构思以生产吸入用缓释制剂并且在说明书中包括抗真菌剂如唑类衍生物的应用,但没有提供任何实例。美国专利号7,521,068B2公开了制剂和相关的制备方法生产纳米颗粒分散气雾剂,干粉纳米颗粒气雾剂,和基于推进剂的气溶胶制剂的制备。其中描述的水分散体或干粉包括难溶性药物微粒(包括唑类衍生物)其表面具有表面改性剂。其包括表面改性剂,有机和无机药用赋形剂。该赋形剂包括多种聚合物,低分子量低聚物,天然产物和表面活性剂。该干粉直接通过干燥液体纳米悬液而制得。干燥前,该药物的水性分散体和表面改性剂可包括溶解的稀释剂如糖。关于我们的特殊情况(最大程度保持肺部停留时间同时最大程度减少全身吸收和去除)其中溶解速度是原始的,该发明具有一些缺点。实际上,其正确的强调了优点,即尺寸减小能够改进溶解速度,由于固体API溶解速度和其表面区域之间具有的比例,可用于Nernst–Brunner/Noyes–Whitney方程式所描述的溶解。但是不可能通过该制备方法改变制剂中表现的固体纳米颗粒的溶解速度。吸入后的该固体API的溶解速度是固有的纳米颗粒溶解速度,其能够导致全身区域内过量吸收因此增加了不良反应、药物-药物相互作用和代谢相互作用的可能性,其能够引发治疗失败。纳米颗粒溶解速度一般极快并且该发明没有清楚的阐述活性成分溶解速度的延迟,减少或控制的可能性。另外,表面改性剂对纳米悬液的稳定是必需的,并且它能够增加颗粒表面湿润并且因此增加其溶解速度。此外,能在水性纳米悬液的干燥步骤前加入的稀释剂和赋形剂被限制为亲水性组分,且所描述的分散剂的水溶性特征不能为疏水性的。一旦该稀释剂将被吸入后与水溶性肺表面活性物质接触,其将快速溶解且纳米微粒的溶解速度不可能改变因此其全身吸收导致多度消除。PCT国际公布号WO2004/060903A2公开了特别是吸入后两性霉素B的肺部的有效浓度和停留时间,用以治疗真菌感染或提供预防。但是近期数据表明该制剂类型相关的毒性不能被接受,这成为肺部给药的严重阻碍(Spickard和Hirschmann,内科学文献1994,154(6),686)。此外,两性霉素B用于对化疗后或自体骨髓移植后的长期中性粒减少的患者的预防是无效的。其中所述的制剂为两性霉素B脂质复合物制剂,由于其难溶性使得成为唑类衍生物的缺点。该申请的说明书中包括了唑类衍生物但是没有提供该药用类型的实例。也没有给出具体的能够优化该浓度和停留时间的制造方法。其虽然描述了基于脂质/磷脂的制剂制备方法但是该制备仅特定针对两性霉素B(复合物形式)并且其没有应用于如唑类衍生物的不同组合物。美国专利申请公开号2007/0287675A1公开了可吸入组合物及制造该组合物的方法。组合物由一或多个包含一或多个水溶性差的活性剂的可吸入聚合体组成。吸入后,该组合物能够达到至少0.25μg/g的最高肺部浓度并且能够维持一定时间。该发明人描述了一系列能够用于制备该可吸入聚合体的方法。这些方法包括超快速冷冻(美国专利申请公开号2004/0137070),喷雾冷冻成液体(美国专利号6,862,890),蒸发沉淀成水溶液(美国专利号6,862,890),控制(美国专利申请公开号2003/0049323),高内相溶液(美国专利号5,539,021和5,688,842)。他们用比较试验证明了提供在体外具有不同溶解速度但是具有相同的制备过程的聚合体的可能性。其制备过程涉及使用特定比例的表面活性剂。该比例是固定的以生成受控的颗粒大小并且不转变药物的溶解性能。其没有公开任何撞击试验的实例和专为吸入用干粉设计的体外实验结果。在其提供的所有的实例中使用了吸入后有毒的1类和2类溶剂,这需要全部溶解伊曲康唑。药物在共溶剂或胶束溶液中的增溶是改进和/或改变难溶的活性成分溶解速度的另一种可能。但是这一类制剂也设计以喷雾而非吸入用干粉的方式给药。环化糊精络合是在制备吸入用干粉时改进难溶物质的溶解速度的另一策略。但是环化糊精在吸入后表现出诱发炎症反应的前兆并且其安全性在现今仍不够清楚。聚合的表面活性剂如聚氧化乙烯和聚氧化丙烯已经用于很多DPI制剂并且表现出改进的体外溶解速度(McConville等,2006)在吸入毒性研究中已经指出这些聚合物在暴露两周后引起轻微的肺泡炎。具有增强的溶解性的盐形式制剂和固体分散体制剂也是制剂领域用来改进难溶性物质的溶解速度的常见方法。改进药物溶解速度的另外的一种可能是改变干活性成分的物理形式。纳米化干结晶颗粒和药物无定形干燥形式的制剂能够引起物质溶解速度的改进。但是干燥微粒通常会引起聚集,进而由于可用于溶解介质的总表面积的减小使得溶解速度的改进有损失。此外,在此需要具有确定的空气动力学直径的颗粒形式以在吸入后到达曲霉菌定植位点的感染位点(与其空气动力学直径有关)。将这些纳米尺寸结晶和/或无定形颗粒分散在可接受的赋形剂内用以吸入是形成颗粒的一种有趣的方法,该颗粒具有适当的空气动力学直径并且一旦沉积在肺粘膜上便能够保持生成干颗粒的改进的溶解速度。基质试剂的性质应该具有增强或延缓活性成分的溶解速度的性质(与其他的制剂相比)。使用的所有的赋形剂和溶剂必须是生理上可耐受的或确认为吸入后或制备过程中最大程度减小潜在毒性的,并且能够减小有害的环境污染。本发明提供了一或两步制备过程以生产该类型的干粉,其仅使用安全和经许可的赋形剂/溶剂。该干粉表现出良好的流动性。该生产的干粉一旦从干粉吸入装置中发射之后,表现出适当的空气动力学特征(关于吸入分生孢子)。该制剂的构思使得能够改进和/或改变/控制难溶性活性成分的溶解速度,从而获得能够最大程度减少全身吸收且同时最大程度增加其在肺部停留时间从而最大化其功效的制剂。
技术实现要素:
本发明的主题已在所附的独立权利要求中加以限定。优选的实施方式在从属权利要求中加以限定。在第一个实施方式中,本发明的主题是一种吸入用喷雾干燥颗粒组合物(X)包括(a)重量百分比为5%-50%之间的至少一种非无定型状态的唑类衍生物和(b)至少一种基质试剂来用于组合物中,所述基质试剂选自由多元醇、单糖、二糖、胆固醇、及其任意混合物组成的组,所述多元醇为例如山梨糖、甘露糖醇和木糖醇,所述单糖为例如葡萄糖和树胶醛醣,所述二糖为例如乳糖、麦芽糖、蔗糖和右旋糖。优选的,所述基质试剂为甘露糖醇或胆固醇。优选的,唑类衍生物/基质试剂的重量比为0.5/99.5-40/60之间,优选地为1/99-35/65之间,更优选地为10/90-35/65之间。所述唑类衍生物不包含由奥美拉唑,埃索美拉唑,兰索拉唑(lansaprazole),泮托拉唑和雷贝拉唑组成的组中的化合物。特别地,所述颗粒进一步包括表面活性剂并且优选的包括重量比为0.1%-5%的表面活性剂。优选地,所述表面活性剂选自卵磷脂,磷脂衍生物如磷酸盐、磷脂酰胆碱(饱和或不饱和)、磷脂酰乙醇胺、磷脂酰甘油、磷脂酰丝氨酸、磷脂酰肌醇、二油酰磷脂酰胆碱、二豆蔻酰磷脂酰胆碱、二棕榈酰磷脂酰胆碱、二硬脂酰基磷脂酰胆碱、二花生酰基磷脂酰胆碱、二苯甲酰基磷脂酰胆碱、双二十三烧酸基磷脂酰胆碱、双二十四酰磷脂酰胆碱、双豆蔻酰磷脂酰乙醇胺、二棕榈酰磷脂酰乙醇胺、二棕榈油酰磷脂酰乙醇胺、二硬脂酰磷脂酰乙醇胺、双豆蔻酰磷脂酰甘油、二棕榈酰磷脂酰甘油、二棕榈油酰磷脂酰甘油以及更多优选的氢化衍生物或修饰的维生素包括α-生育酚衍生物。本发明的另一个主题是吸入用喷雾干粉组合物包括颗粒(X),其中所述组合物包括至少50%的基质试剂,并且使得在美国药典2型溶解装置检测中,以每分钟50转,在37℃,900ml的调pH1.2且含0.3%月桂醇硫酸钠盐的溶解介质中,检测所述唑类衍生物具有以下溶解速度,溶解速度为至少10分钟内5%,20分钟内10%和60分钟内40%。按照“吸入用制剂:细颗粒的评估”中所述的方法,使用多级液体空气采样器,装置C-欧洲药典第2.9.18章,所述组合物优选的提供了干粉中占总标称剂量唑类至少35%的唑类衍生物细颗粒级分。优选地,所述组合物进一步包括颗粒(Y),所述的颗粒(Y)包括(a)重量百分比5%-50%之间的至少一种无定形状态的唑类衍生物(b)至少一种基质试剂,和(c)表面活性剂。所述颗粒(Y)优选的包括重量比为0.5%-5%的表面活性剂。优选地,所述组合物进一步包括颗粒(Z),所述颗粒(Z)包括重量比达20%的晶体结构的唑类衍生物纳米颗粒,其具有0.1-1μm的平均大小。优选地,所述组合物中使得唑类衍生物具有以下溶解速度,溶解速度为5分钟内5%-50%,10分钟内10%-60%,20分钟内15%-90%和60分钟后40%-100%。优选地,所述的唑类衍生物选自咪康唑、氟康唑、伊曲康唑、泊沙康唑、伏立康唑、异康唑、酮康唑、奥昔康唑、联苯苄唑、芬替康唑、噻康唑、特康唑、硫康唑、雷夫康唑、益康唑、特康唑,优选的伊曲康唑。本发明另一主题是所述的喷雾干粉颗粒和组合物的制备方法,其包括以下步骤:a)制备液体组合物包括:ⅰ.选自根据欧洲药典第三类溶剂的液体载体,例如乙酸、庚烷、丙酮、乙酸异丁酯、苯甲醚、乙酸异丙酯、1-丁醇、乙酸甲酯、2-丁醇、3-甲基-1-丁醇、乙酸丁酯、甲乙酮、叔丁基甲基醚、甲基异丁基酮、异丙基苯、2-甲基-1-丙醇、二甲亚砜、戊烷、乙醇、1-戊醇、乙酸乙酯、1-丙醇、乙醚、2-丙醇、甲酸乙酯、甲酸丙酯、甲酸,或其混合物,或该溶剂与水的混合物;ⅱ.在所述液体载体溶液中至少一种唑类衍生物;并且ⅲ.在所述液体载体溶液中至少一种基质试剂,其中,唑类衍生物/基质试剂的重量比为0.5/99.5和40/60之间,优选的1/99和35/65之间,更优选的10/90和35/65之间,b)喷雾干燥所述液体组合物用以制备干粉组合物颗粒。优选地,所述方法进一步包括步骤:c)制备另一液体组合物,其包括液体载体和至少一基质试剂于所述液体载体溶液中,其中所述液体载体选自第三类溶剂,或两或更多含水或不含水的所述溶剂的任意混合物,其中所述液体组合物进一步包括:ⅰ.至少一唑类衍生物于所述液体载体溶液中和至少一表面活性剂;和/或ⅱ.平均大小在0.1-1μm之间的至少一唑类衍生物纳米颗粒,d)喷雾干燥步骤(c)得到的液体组合物用以制备干粉组合物颗粒。e)物理混合步骤(b)得到的颗粒和步骤(d)得到的颗粒。本发明的另一主题是一种液体组合物包括:ⅰ.选自根据欧洲药典第三类溶剂的液体载体,例如乙酸、庚烷、丙酮、乙酸异丁酯、苯甲醚、乙酸异丙酯、1-丁醇、乙酸甲酯、2-丁醇、3-甲基-1-丁醇、乙酸丁酯、甲乙酮、叔丁基甲基醚、甲基异丁基酮、异丙基苯、2-甲基-1-丙醇、二甲亚砜、戊烷、乙醇、1-戊醇、乙酸乙酯、1-丙醇、乙醚、2-丙醇、甲酸乙酯、甲酸丙酯、甲酸,或其混合物,或该溶剂与水的混合物;ⅱ.在所述液体载体中溶液中至少一种唑类衍生物;并且ⅲ.在所述液体载体中溶液中至少一种基质试剂,其中,唑类衍生物/基质试剂的重量比为0.5/99.5和40/60之间,优选的1/99和35/65之间,更优选的10/90和35/65之间。优选地,所述液体组合物其进一步包括至少一表面活性剂和/或平均大小在0.1-1μm之间的至少一唑类衍生物纳米颗粒。附图说明图1是喷雾干燥伊曲康唑的MDSC加热曲线。图2是微粉化晶体松散伊曲康唑、纯无定形伊曲康唑和根据本发明(实例1B)的包含亲水基质和伊曲康唑的喷雾干粉制剂的体外溶解曲线。图3是根据本发明(实例2A-2D)的喷雾干粉制剂的体外沉积模式(平均±S.D,n=3),其通过的MsLI而测定。结果(以标称剂量函数显示)以从装置中回收的和冲击器的每部分的伊曲康唑的百分比显示(喉部、1、2、3、4级和滤器)。使用以下条件:100ml/分钟,2.4秒,每个实验使用填充了相当于2.5mg伊曲康唑的制剂量的三个3号HPMC胶囊。图4是松散结晶伊曲康唑和根据本发明(实例2A-2D)的喷雾干燥制剂的体外溶解曲线。图5是根据本发明(实例3A-3E)的喷雾干粉制剂和喷雾干燥伊曲康唑(实例3F)放大x1000倍的SEM图像。图6是根据本发明(实例3A-3E)的喷雾干粉制剂和喷雾干燥伊曲康唑(实例3F)和喷雾干燥甘露醇的MDSC加热曲线。图7是根据本发明(实例3A-3E)的喷雾干粉制剂的体外沉积模式(平均±S.D,n=3),使用的MsLI测定。结果(以标称剂量函数显示)以从装置中回收的和冲击器的每部分的伊曲康唑的百分比显示(喉部、1、2、3、4级和滤器)。使用以下条件:100ml/分钟,2.4秒,每个实验使用填充了相当于2.5mg伊曲康唑的制剂量的三个3号HPMC胶囊。图8是微粉化结晶松散伊曲康唑、喷雾干燥无定形伊曲康唑(实例3F)和根据本发明(实例3A-3E)的喷雾干粉制剂的体外溶解曲线。图9是根据本发明(实例3A-3E)的喷雾干粉制剂的体外溶解曲线,其中曲线A显示了10分钟内5%、20分钟内10%和60分钟内40%的溶解速度。图10是根据本发明(实例3A-3E)的喷雾干粉制剂的体外溶解曲线,其中曲线B和B’分别显示了5分钟内5%、10分钟内10%、20分钟内15%和60分钟内40%的溶解速度,以及5分钟内50%、10分钟内60%、20分钟内90%和60分钟内100%的溶解速度。图11是微粉化结晶松散伊曲康唑和根据本发明(实例3A-3E)的包含伊曲康唑、胆固醇和磷脂(实例4)的喷雾干粉制剂的体外溶解曲线。图12是微粉化结晶松散伊曲康唑和根据本发明的包含伊曲康唑和甘露醇的喷雾干粉制剂的体外溶解曲线,也即不含结晶纳米颗粒伊曲康唑的颗粒(实例5A)和包含结晶纳米颗粒伊曲康唑的颗粒(实例5B)。具体实施方式本发明涉及唑类衍生物吸入用干粉制剂及其制备方法,其条件是所述唑类衍生物不是由奥美拉唑,埃索美拉唑,兰索拉唑,泮托拉唑和雷贝拉唑家族组成的组中的化合物。所述的唑类衍生物选自由咪康唑、氟康唑、伊曲康唑、泊沙康唑、伏立康唑、异康唑、酮康唑、奥昔康唑、联苯苄唑、芬替康唑、噻康唑、特康唑、硫康唑、雷夫康唑、益康唑、特康唑组成的组。本申请所述的干粉在从吸入装置吸入后能够最大程度的表现高分散性能,颗粒的比例具有合适的空气动力学直径范围。合适的空气动力学范围指具有吸入分生孢子的空气动力学直径。通常呼吸状态下来自吸入装置的颗粒必须具有与曲霉菌分生孢子相同的空气动力学范围(1.9-6μm)以到达潜在感染位点从而实现最佳治疗靶向性和有效性。有利地,所述干粉组合物基于使用特定的生理组分赋形剂,安全的,通常认为安全的赋形剂(GRAS),FDA许可的吸入治疗用赋形剂以保证吸入后良好的安全性并且与肺膜兼容以防止高反应性、咳嗽、气道痉挛或感染。该制备过程需要一或两个步骤以获得最终干燥产物,并且其使用的所有技术均能够用于简单规模的高达工业批量大小的生产。所述干粉其本身设计为具有增强的流动特性以用于工业规模的简单制备过程。所述干粉尤其设计为口服吸入以治疗或给肺侵袭曲霉病提供预防。所述唑类衍生物以通过不同程度改进和/或通过改变干粉组合物从而改变其溶解速度的形式。所述改进能够通过改变干粉组合物和/或活性药学成分(API)物理状态或通过结合本申请不同实施方式的现有给药方式而加以控制。这是非常有利的,因为所述溶解速度的改变能够克服体内清除和导致感染位点的药物比例降低的吸收机制。所述干粉由基质颗粒组成,所述基质颗粒由安全的生理组分或吸入的FDA许可的赋形剂组成,其中所述活性成分分散为改进的物质状态。这些颗粒吸入后,基质溶解或腐蚀后,所述活性成分将以比相同剂量纯的喷雾干活性成分颗粒更高的表面积暴露于肺粘膜,从而产生改进的溶解速度。基质固有的性质直接影响活性成分的溶解曲线。所述基质试剂可是(i)亲水的使得与肺粘膜接触时能够直接释放所述活性成分(ii)疏水的使得延缓活性成分的释放(iii)亲水和疏水(以不同比例)混合的试剂使得其具备中间释放曲线。基质试剂是生理组分赋形剂,GRAS赋形剂;吸入治疗用FDA许可的赋形剂以尽可能的防止肺部或全身毒性。所述基质试剂能够结合在一起从而使所述干粉具有所需的流动,空气动力学和溶解的性能。所述基质试剂在组合物中是必需的。基质试剂可以选自由糖醇,如山梨糖、甘露糖醇和木糖醇的多元醇,和结晶糖包括单糖(葡萄糖和树胶醛)和二糖(乳糖,麦芽糖,蔗糖和右旋糖)和胆固醇组成的组。在本发明的一个实施方式中,所述API多为无定形状态。该无定形活性成分的比例(以占本发明活性成分总量的百分比)为51%-100%,优选地为70%-100%之间,更优选地为100%。获得无定形组合物的一个方式是将其从溶液中喷雾干燥,由于在干燥过程中快速的溶剂挥发使得没有足够的时间再结晶固体颗粒。但是唑类组合物以及特别是伊曲康唑仅难溶于如二氯甲烷,氯仿的氯化溶剂,由于其高毒性不推荐用于制备药用制剂,本发明提供了通过从溶液中喷雾干燥API以获得无定形产品的方法,该溶液仅使用第3类溶剂。这种溶剂认为是低毒的潜在溶剂并且之后其能够在吸入残存的情况下提供更安全的性能。这一类的溶剂包括乙酸、庚烷、丙酮、乙酸异丁酯、苯甲醚、乙酸异丙酯、1-丁醇、乙酸甲酯、2-丁醇、3-甲基-1-丁醇、乙酸丁酯、甲乙酮、叔丁基甲基醚、甲基异丁基酮、异丙基苯、2-甲基-1-丙醇、二甲亚砜、戊烷、乙醇、1-戊醇、乙酸乙酯、1-丙醇、乙醚、2-丙醇、甲酸乙酯、甲酸丙酯、甲酸,或其混合物,或该溶剂与水的混合物。通过喷雾干燥活性成分有机溶液,能够有可能在干燥过程后获得具有适于吸入治疗用几何尺寸(<5μm)的无定形状态的该活性成分。从药物饱和有机溶液中可完成该过程。但是如伊曲康唑的唑类衍生物在第3类溶剂中的溶解度极低。该低浓度使其不能作为在喷雾干燥后能够使干粉良好地恢复的优选。为了获得喷雾干燥后干粉良好的恢复,选择具有高溶解性的唑类衍生物替代伊曲康唑。基质试剂可在喷雾干燥该类溶液前加入,以增加总溶质浓度。可在磁力搅拌下在预热的第3类有机溶剂中加入酸,以增加如伊曲康唑的难溶唑类化合物的溶解度。可在磁力搅拌下加热含唑类化合物的有机溶液至高温,以获得溶解度增强的唑类类化合物。这些选择仅能够将疏水性赋形剂在溶液中溶解。可将一定量的水加入到这些溶液类型的其中一种,以溶解难溶的活性物质,亲水的和疏水的赋形剂。这将对改进活性成分的溶解速度,颗粒尺寸,空气动力学行为和流动性是尤其有趣的,优选的水和有机溶剂(体积-体积百分比)的比例为0-50%,优选的为0-30%,更优选的为介于10和30%之间,更优选的为介于20和30%之间。从热力学角度,由于其无组织的结构,无定形化合物表现出具有比相同结晶化合物更高的溶解度的优势。在实践中,溶解过程中,无定形化合物经常重结晶为具有比起始产品溶解性低的低能结晶状态。本发明提供的制剂,其活性化合物呈无定形状态,并且制备使得其在药物重结晶完成前发生溶解,从而使其成为具有改进的溶解速度的产品。实际上,到达患者局部位点的干粉制剂的改进和其表面积的增大可以通过喷雾干燥活性成分溶液连同亲水基质试剂而获得,该基质试剂提供了分散于基质试剂内的无定形形式的含活性成分的颗粒。这种表面积的改进可加快活性成分的溶解速度以防止溶解前过度重结晶。无定形药物的重结晶也可在储存期间发生,其会导致产品溶解性能降低。本发明的一个方面提供了稳定的无定形产品,其制备为基质试剂中的活性成分固体分散体。本发明的一组合物,可以加入到基质试剂中的唑类衍生物的量以重量比剂为0.5-40%,优选地为1-35%,更优选地为10-35%。令人惊讶的,能够通过改变喷雾干燥溶液的浓度或基质试剂/API比例而改变生成的颗粒的空气动力学行为。改变溶液中的浓度或基质试剂/API比例能够直接改变干燥颗粒的几何直径和密度,从而改变其空气动力学直径,也会直接改变其空气动力学行为。改变这些参数之一会导致颗粒制剂具有不同的空气动力学行为并同时具有相似的溶液速度。这将帮助提供一种具有优化的溶解速度的干粉,其能够以足够的量渗透入肺部从而以预定的标称剂量提供合适的抗真菌剂量。这些参数的改变使得喷雾干粉的细颗粒剂量(FPD)最优化,同时保持了其改进的溶解速度。优选地,唑类衍生物在液体组合物中的量以唑类衍生物的重量在液体组合物总体积的百分比(g/100mL)计为0.1%-5%之间,优选地为0.5%-2%之间。可在根据本发明的含颗粒的干粉制剂的基质中加入表面活性剂,以改进活性成分溶解速度的增加。表面活性剂为两亲化合物同时具有亲水性和疏水性的特性。通过喷雾干燥含活性成分基质溶剂和表面活性剂的溶液能够制备分散有活性成分和表面活性剂的基质颗粒。所述表面活性剂能够对活性成分起增加湿润度的作用,从而减少颗粒聚集并且与无表面活性剂的基质颗粒相比能够加快/改进其溶解速度。所述表面活性剂可选自由生理化合物,GRAS(公认为安全的)赋形剂,FDA许可的吸入治疗用赋形剂以减少肺部或全身毒性。加入表面活性剂的量可影响唑类化合物的溶解速度的改进,优选的表面活性剂的量包括占干粉组合物的重量比为0.12-5%。优选地,表面活性剂可为磷脂,卵磷脂,脂质或GRAS改进维生素,或以上表面活性剂的组合。可使用磷脂包括磷酸盐、磷脂酰胆碱(饱和或不饱和)、磷脂酰乙醇胺、磷脂酰甘油、磷脂酰丝氨酸、磷脂酰肌醇。此类磷脂的例子包括二油酰磷脂酰胆碱、二豆蔻酰磷脂酰胆碱(DMPC)、二棕榈酰磷脂酰胆碱(DPPC)、二硬脂酰基磷脂酰胆碱(DSPC)、二花生酰基磷脂酰胆碱(DAPC)、二苯甲酰基磷脂酰胆碱(DBPC)、双二十三烧酸基磷脂酰胆碱(DTPC)、双二十四酰磷脂酰胆碱(DLPC)、双豆蔻酰磷脂酰乙醇胺(DMPE)、二棕榈酰磷脂酰乙醇胺(DPPE)、二棕榈油酰磷脂酰乙醇胺、二硬脂酰磷脂酰乙醇胺(DSPE)、双豆蔻酰磷脂酰甘油(DMPG)、二棕榈酰磷脂酰甘油(DPPG)、二棕榈油酰磷脂酰甘油以及更多优选的氢化衍生物。GRAS修饰的维生素包括α生育酚衍生物。制剂中过高量的表面活性剂可引起喷雾干燥过程中重要的颗粒尺寸的增加。由于其低熔点,表面活性剂可在喷雾干燥过程中软化或融化以增加颗粒尺寸。将表面活性剂在基质溶剂中稀释可掩盖该效果从而使得能够产生具有合适性能的更小的颗粒。本发明一个具体实施方式包括使用本领域描述的方法获得结晶纳米颗粒形式的活性成分。本发明使用的术语“纳米颗粒”是指尺寸为1nm-1000nm范围的固体离散颗粒。喷雾干燥颗粒中的结晶纳米唑类衍生物和含结晶纳米颗粒的颗粒的重量比可通过使用粉末X-光衍射,以及差示扫描量热法结合高效液相色谱(HPLC)药物定量法加以测定。这些纳米颗粒进而分散于基质试剂中以使该制剂具有合适的颗粒尺寸、流动性、溶解速度和空气动力学行为。这些纳米颗粒的溶解速度是瞬间发生的(5分钟内)具有非常显著的突释效应且由于该纳米颗粒的固有的溶解速度而不能够延迟。制备该制剂类型(如含活性成分结晶化纳米颗粒的颗粒和基质试剂)的制备过程包括两个步骤。第一步是制备药物纳米颗粒以及第二步是干燥步骤。该纳米颗粒可通过现有技术描述的方法制备。优选地,纳米颗粒通过高压匀浆制备。所述基质试剂可在尺寸减小步骤前或喷雾干燥过程前加入。在本发明的一个具体实施方式中,所述活性成分以结晶纳米颗粒和无定形化合物的形式分散于基质试剂中。该实施方式可以作为喷雾干燥溶液中的基质试剂和活性成分连同其中活性纳米颗粒的结果。该实施方式的另一个方面是根据本发明的干粉制剂是通过简单混合所述活性成分纳米颗粒和基质试剂制备,该活性成分纳米颗粒由喷雾干燥含结晶纳米颗粒的悬液获得,或者通过机械球磨结晶活性成分和通过喷雾干燥溶液中的活性成分获得的无定形基质制剂制备而成。该混合粉将填入胶囊,泡或多剂量装置。期望的结果是通过优化制剂中纳米颗粒/无定形化合物的比例从而使得所述制剂具有可控的溶解曲线。该溶解曲线仅通过制剂中的纳米颗粒无法实现。纳米颗粒/无定形化合物的比例的调整使得能够改变溶解曲线。优选地,无定形基质颗粒/纳米结晶基质组合物的比例(w/w)包括100/0-80/20。在另一个具体实施方式中,所述活性成分以纳米颗粒或微米颗粒的形式分散于相同的活性成分的基质中。所述活性成分基质为无定型状态。纳米悬液可与含前基质(matrixformer)的活性成分溶液进行共同喷雾干燥。无定形和纳米颗粒溶解速度之间存在的差异使得能够改变所述制剂的溶解速度。溶液中API可用作前基质以包裹纳米颗粒。这能够提供具有有趣的溶解速度和最优化的空气动力学特性的制剂。实例实例1:起始材料由体积平均直径为3.5μm且90%的颗粒小于6.2μm的结晶微粉化伊曲康唑(ITZ)组成。使用Büchi微型喷雾干燥机B-191a(Büchi实验室-技术,瑞士),通过喷雾-干燥法制备实验室规模的纯的无定形伊曲康唑(实例1A)和亲水性伊曲康唑干粉基质制剂(实例1B;本发明)。制备两种原料母液后在以下条件下分别进行喷雾干燥:喷雾空气流,800l/h;干燥空气流,35m3/小时;溶液进料速度,2.7g/分钟;喷嘴尺寸,0.5mm;进口温度,90℃;使得出口温度为53℃。所述原料母液组合物如表1所示。每种组分在磁力搅拌下(600rpm)溶解于水-醇溶液(20水-80异丙醇)并在70℃下加热。喷雾干燥溶液过程中保持温度在60-70℃之间。表1:实例1中的喷雾干燥溶液组合物。通过MDSC(调制温度差示扫描量热法)和PXRD(粉末X-射线衍射)评估干燥样品的结晶图谱。该两种技术互为补充且能够最大化的提供样品多态性信息。MDSC实验使用配备冷却系统的Q2000DSC(TA仪器)进行。MDSC与标准DSC在对样品同时使用两种加热速度的可能性方面是不同的,MDSC在线性升温坡线中加入正弦调制。总测量的热流量与传统DSC中的标准热流量相对应。MDSC升温条件为可逆和不可逆热流量的重叠提供了可能性,其中能够显著地检测出特定的温度事件。进而在不可逆热流量中观察到结晶现象,在可逆热流量中观察到玻璃化转变,在总热流量中观察到熔融。所有的样品在相同的条件下进行分析。在低质量范围的铝封闭锅中精准的称量2-3mg样品。以5C°/分钟的温速,每60秒调制+/-0.8℃,将样品从25℃升温至185℃。以铟为标准校准仪器温度。使用标准蓝宝石样品(sapphire)校准热流量和热容量信号。使用UniversalAnalysis2000软件分析每一个热事件。PXRD技术是广泛用于评估各种化合物的晶体形式的强有力的工具。它有助于测定产品的结构物理状态。特定的晶格能够对应特定的PXRD谱,并且相反地,特定的无序系统(作为无定形状态)则不会出现衍射峰。因此其有助于评估喷雾干燥后获得的多晶型(polymorphicform)并且在第二时间内判断样品内的无定型相的比例。通过德拜-谢乐法(Debye-Scherrer法)分析该干粉。该样品经铜Kα线,单色辐射放置上述Bragg-Brentano反射的衍射仪(西门子D5000,德国)与单色仪和通道项目Diffracplus软件相连。40KV,40mA下,2°-60°角的范围的2θ,以0.02°步,通过每步计算速度为1.2秒,样品旋转速度为15rpm。每个样品在封闭塑料容器中置于8、25、40℃储存。喷雾干燥后,在不同温度下储存2个月之后,直接分析该样品。可将特定的化合物的晶相比例量化。已经开发了多种计算技术。在本情况下,通过测量曲线下的面积来测定样品中的无定形相的百分比。确实,样品中基线(Ac)的偏差以上的衍射峰曲线下面的面积的比率和表示晶相总量的衍射图谱的总面积(Atot)之间成比例关系。为计算样品中的结晶度,不综合离基线的偏差而只测量衍射峰的曲线下的面积已经足够,因为其来自样品中的噪音和无定形区域。然后综合衍射图谱曲线(AT)下的总区域。晶相的比例表示为方式1。该以%表示的无定形量以100%减去测量的结晶度得出。方程式1:MDSC分析(图1)显示无定形伊曲康唑(实例1A)在49℃呈现玻璃化转变。100℃和125℃之间观察到重结晶放热峰,随后在约164℃有吸热峰,其与早期形成的晶体材料的熔融相对应。当在相同条件下分析时(约168℃),该结晶伊曲康唑的熔融温度低于松散材料。这些温度事件是玻璃化伊曲康唑的特性。PDRX验证了实例1A和1B的无定型状态的伊曲康唑。在T0月实例1A的衍射图中没有出现衍射峰。大约计算样品中的非晶相等于100%。这证实了样品中缺少任何晶体结构。表2:基于DRX评估非晶样品含量。制剂T0月T2月8℃实例1A(Cex)100%100%实例1B(INV)52%52%25℃实例1A(Cex)100%100%实例1B(INV)52%55%40℃实例1A(Cex)100%63%实例1B(INV)52%55%8℃和25℃下储存两个月后没有发生重结晶。无定形相的比例保持在100%并且在衍射图谱中没有观察到结晶伊曲康唑衍射峰的表征。40℃下储存无定形伊曲康唑发生重结晶,并且无定形相转为约63%。松散结晶伊曲康唑的原始衍射角的重结晶峰说明了无定形伊曲康唑重结晶为其原始的更稳定形式。在T0,实例1B的衍射图谱呈现不同衍射峰。但是没有任何峰与结晶伊曲康唑对应。出现了α,β和δ甘露醇的衍射曲线。样品中无定形相总量约为52%。该值比实际样品中的伊曲康唑的量高。其可能来自喷雾干燥后无定形甘露醇的比例。当在8℃,25℃和40℃储存时,只观察到样品中大约无定形相有很小的变化(见表2)。与实例1A相反,在其特征衍射角没有显现出伊曲康唑重结晶的证据。甘露醇中的分散无定形伊曲康唑(通过喷雾干燥含两种组分的溶液)趋向于无定形API的稳定。从干粉吸入器引发剂量后产生的颗粒的空气动力学行为通过多级液体碰撞器(MsLI)进行测试。使用的干粉吸入器为(SMB实验室)。以2.4秒内100L/分种的流速(调至4kPa的压力降)通过装置以做每一次引发。该装置填充装有大约与2.5mg伊曲康唑相当的干粉的量的HPMCn°3胶囊。每个测试释放三次。三个剂量引发后,使用合适有效的HPLC方法定量冲击器的每一部分中的总沉淀的干粉。每个实验重复三次。按照欧洲药典7.2关于细颗粒空气动力学评估,仪器C(MxLI)所描述的方法评估每个实验的所述细颗粒剂量(FPD)。显示的结果与2.5mg恒定的伊曲康唑标称剂量加权。细颗粒级分(FPF)以FPD的标称计量百分比表示。使用Malvern激光衍射装置在空气动力细颗粒测试实验中测定颗粒尺寸分布(PSD)。激光束直接置于喉部和冲击器之间以测量产生的干粉云的PSD,然后其在模拟吸入条件下,在MsLI中沿空气动力学直径分裂。每个样品重复三次测得平均PSD。结果以D[4,3],d(0.5)和d(0,9)显示,其体积平均直径(volunmmeandiameter)和以微米尺寸中50%和90%的颗粒比剩余的分布小。结果如表3所示。表3:不同制剂的尺寸和空气动力学性质:通过测得颗粒尺寸性质(平均值±SD,n=3)和细颗粒级分(具有dae<5μm的颗粒%)以标称计量函数的表示(FPF;平均值±SD,n=3)通过冲击试验(MsLI)测得。颗粒大小分析显示本发明的体积平均直径小于5μm,这是深度肺沉积的第一原则。空气动力学细颗粒评价测试也验证了该结论。本发明显示出为46.9±1.9%的高FPF。使用USP33的2型桨装置(Distek溶解系统2100C,DistekInc.,美国)进行溶解实验。该溶解介质由含3%的月桂基硫酸钠的pH1.2(HCl0.063N)的去离子水组成。该溶解使得实验始终能够保持SINK的条件。该介质加热至37℃并在实验中保持该温度。所述桨的速度设定为50rpm并且溶解容器中装入900ml的溶解介质。精确称量相当于10mg伊曲康唑的干粉分散于溶解介质中(=T0)。使用合适有效的HPLC法以预设间隔定量(0,2,5,10,20,30,60,和120分钟)伊曲康唑。从溶解容器中取5ml溶解介质并直接更换为新鲜的溶解介质。将该5ml直接通过0.2μm直径的滤器过滤以避免设定时间间隔中的不溶颗粒的量。药物释放累积量以起始上药量和与时间绘制的比例进行计算和表示。每个实验重复三次。图2显示溶解曲线。结晶微粉化(松散ITZ)和纯无定形ITZ(实例1A)的溶解曲线的对比显示药物释放曲线没有差别。该结论非常有趣,无定形ITZ原本与结晶ITZ相比应该具有更快的溶解曲线。这可能因为药物的高疏水性能够通过阻碍改善药物溶解的水性溶解介质从而导致低湿润度。无定形ITZ的渐进重结晶还可发生在溶解阶段,无定形形式的延迟溶解。但是,很惊讶的发现根据本发明实例1B的制剂中,ITZ分散于甘露醇颗粒中给ITZ的溶解速度提供了显著的改进,即,与松散微粉化结晶ITZ和纯无定形ITZ相比,其溶解速度为10分钟为11.4%,20分钟为15.2%,以及60分钟为46.7%。在分散于甘露醇颗粒时给无定形ITZ溶解介质提供增加的表面积能够解释该溶解速度的显著增加(图2)。甘露醇几乎瞬间溶解,这认为是由于剩余的ITZ与纯喷雾干燥无定形颗粒相比可以在溶解介质中暴露更大的表面积。甘露醇形成分散有无定形ITZ的球状基质。一旦该甘露醇溶解,由于其通过甘露醇溶解形成的无数的孔隙,多孔无定形ITZ颗粒便在溶解容器中释放。给溶解介质提供增加的表面积则能够加快溶解速度和防止过度重结晶,从而提高溶解度和加快溶解速度。实例2:本实例的目的在于说明本发明改善干粉空气动力学行为但同时不改变其溶解速度方面的能力,其通过改变赋形剂/API的比例和用于喷雾干燥的液体组合物的总溶质实现。使用Büchi微型喷雾干燥机B-191a(Büchi实验室-技术,瑞士),通过喷雾-干燥法制备实验室规模的四种制剂。分别制备四种原料母液并进行喷雾干燥。将设定质量的伊曲康唑和甘露醇(见表4)在磁力搅拌下(600rpm)溶解于100ml的水-醇溶液(20水-80异丙醇)并在70℃下加热。实例2A和2B的溶液中的总干燥产品的量相似(1.56g)。该两种制剂之间唯一的区别是伊曲康唑/甘露醇的比例不同。对于制剂2A、2C和2D,伊曲康唑/甘露醇的比例是恒量,但是溶液中溶质的总量在液体组合物是不同的。喷雾干燥的条件与实例1的条件相同。表4:实例2中喷雾干燥用液体组合物中的伊曲康唑和甘露醇的量。通过PXRD(粉末X-射线衍射),在与实例1所述相同的条件下评估干燥样品的结晶图谱。所述四种制剂的衍射图谱显示出一些衍射峰。但是没有任何衍射峰与结晶伊曲康唑相对应。这说明伊曲康唑在这些制剂中以无定形形式存在。甘露醇多数以结晶状态存在。该三种不同的多晶形(α,β和δ)以不同的比例存在于所有样品中,以δ型为多数。通过实例1描述的Carr’s可压性指数(CI)评估粉末的流动性。Carr’s指数值在40%以上即为粉末流动性差,而在20%以下则是指粉末流动性非常好。其中四种的CI值为20.9%-28.8%。该值显示两种制剂均具有良好的粉末流动性。粉末颗粒尺寸分布通过MalvernMastersizer(Malvern设备)激光衍射,采用Sirocco(Malvern设备)干燥采样分散设备,测量粉末的颗粒尺寸分布。4Bar压强,采样速度振动为40%,进行+/-50mg样品的颗粒尺寸测量。由于这些非常显著的分散条件,以上条件能够测得部分去聚集粉末和全部去聚集粉末(desagglomeratedpowder)的颗粒尺寸分布。选择颗粒折射率的实部为1.48虚部为0.1。这些值保证了低加权残余(<2%)违背了结果的完整性。使用实例1所述的Malvern对于两个实验,每个样品重复三次测得平均PSDs。结果以D[4,3],d(0.5)和d(0,9)显示,其体积平均直径和以微米尺寸中50%和90%的颗粒比剩余的分布小。结果如表5所示。产生的颗粒的空气动力学行为通过如实例1所述的撞击测试进行评估。所述细颗粒级分为以具有小于5μm空气动力学直径的标称剂量(FPF)的%表示的FPD。计算发射剂量并且对应于源自吸入端口的恢复剂量和测试中的MsLI的五级。发射剂量以标称剂量表示并且对应于有效的离开装置和胶囊的标称剂量比例。结果如表6和图3所示。Malvern测定显示四种制剂呈现相似的质量中值直径d(0,5)并且制剂2B和2C的体积平均直径值(D[4,3])高于另外两种制剂,如表5所示。在该两种制剂中似乎有略微较大的颗粒的形成。此外,在模拟吸入条件下分析制剂2B和2C获得的更高d(0,5)和D[4,3]值显示,去聚集化似乎更困难。表5:实例2中不同制剂的尺寸特性:通过Malvern和测量颗粒尺寸特性(平均值±SD,n=3)。尽管制剂具有较高的颗粒尺寸和低去聚集效率,制剂2B和2C具有比制剂2A和2D更高的FPF。这直接涉及两种制剂(2B和2C)的更高的喷射量。由于该极精细的颗粒测定法,尽管具有更低的去凝聚趋势和相对略微大的颗粒尺寸,这两种制剂(2B和2C)比制剂2A和2D在撞击器中渗透的更深从而导致较高的FPF。表6:冲击试验过程(MSLI,100l/分钟,2.4秒,每次试验流出3,2.5mg下称重的标称计量,n=3)获得的颗粒沉积,FPD和FPF(平均值±SD,n=3)和喷射剂量(%标称计量)。实例2A实例2B实例2C实例2D平均FPD(mg)1.17±0.051.40±0.011.36±0.091.19±0.04平均FPF(%)49.6±1.956±0.454.4±1.847.6±1.6喷射剂量nom(%)53.3±1.971±0.573.5±6.353.3±1.5如实例1所述进行溶解度测试。得出的溶解曲线如图4所示。该四种制剂呈现不同的溶解速度,并且比松散微粒化结晶ITZ的溶解速度快(图4)。该实例2A、2B、2C、2D的溶解曲线相似。基于该结果,能够通过改进活性成分/基质前比例,溶质总量,或喷雾干燥溶液的溶液中活性成分的浓度,从而改进产生的颗粒的空气动力学行为同时保持相似的溶解曲线。不改变赋形剂类型或喷雾干燥参数改进空气动力学行为。这说明了改变颗粒的空气动力学行为同时不改变API溶解速度的一个灵活步骤的可能性。使用的所有赋形剂为GRAS。该四种制剂表现出良好的粉末流动性。实例3:本实例的目的在于显示本发明在改善制剂溶解速度的加速同时保持良好流动性能和空气动力学特征方面的能力。使用Büchi微型喷雾干燥机B-191a(Büchi实验室-技术,瑞士),通过喷雾-干燥采样母液制备实验室规模的三种制剂。对于5个实例,将设定量的伊曲康唑、甘露醇和含有多于90%氢化磷脂酰胆碱的氢化大豆卵磷脂(Phospholipon90H)(见表7)在磁力搅拌下(600rpm)溶解于100ml的水-醇溶液(20水:80异丙醇)并在70℃下加热。喷雾干燥的条件与实例1的条件相同。表7:实例3中使用的喷雾干燥过程的喷雾干燥溶液,干燥制剂的理论组成。进行药物量测定以比较预期的和实际的药物量。设定量的干粉溶于稀释相超声处理20分钟。该溶液进行HPLC–UV分析以测定所述药物量(wt%)。五次测定计算出平均量(wt%)和标准偏差。不同制剂的伊曲康唑量测定结果如表8所示。测量值与预期的非常接近,具有-3.9%和3.0%之间的范围的相对误差。较低的伊曲康唑量和在制剂中引入的磷脂导致该相对误差降低。所述活性成分似乎在颗粒内均匀分布,因为其样品随机选择且所有五种样品的变异系数不高于3.25%。在空气动力学颗粒尺寸分析中使用该精确的ITZ量值以确定精确的标称剂量。在喷雾干燥过程中似乎没有发生ITZ降解。纯喷雾干燥伊曲康唑的测得的和预期的ITZ量之间的相对误差(实例3F)为0.7%。表8:实例3中喷雾干粉通过高效液相色谱测定法测定ITZ的量(平均值±SD,n=5)通过扫描电镜PhilipsXL30ESEM-FEG(FEI,荷兰),进行定性形态分析。将样品分散于碳粘合带上,然后用金层包覆,氩条件6.10-2mbar,40mA,90秒。根据样品情况使用5-25KV的加速电压进行观察。关于喷雾干燥制剂的定量组成,甘露醇为主要的组分并处理以形成其中分散有ITZ的基质颗粒,如合适情况下分散有PL。该形态分析显示含有甘露醇和伊曲康唑不含PL的喷雾干燥溶液中形成非常小的并且具有光滑表面的球形颗粒(11-2μm出现亚微米颗粒)(实例3A和2B,图5)。尽管有不同比例的无定形含量和甘露醇多晶型,这些制剂中没有观察到形态差异。但是实例3B似乎由较大的球形颗粒。PL的加入导致形成更大的颗粒外观的颗粒。对于具有最高PL含量的制剂(实例3D),观察到更明显的颗粒外观和颗粒间连接(图5)。这些连接可能在喷雾干燥过程中由于PL软化和熔融引起部分聚集而形成。PL量的减少能够在颗粒感方面极大的降低。使用Q500设备(TAinstruments,NewCastle,美国)进行热重分析(TGA)和Universal分析,2000version4.4A软件(TAInstruments,Zellik,比利时),测定残留水分和不同的干粉的溶质的量。该残留水分和溶质的量在25℃和125℃之间的重量损失计算并且以起始样品百分比表示。按照操作25℃-300℃,10℃/分的加热速度,对10mg的样品质量进行设定,操作进行三次。测量的在25℃和125℃样品加热过程中的重量损失在每种制剂中都非常低(<0.5%)。如实例1所述进行MDSC,结果如图6所示。如前所述(实例1A),在所述条件下的喷雾干燥MTDSC分析显示在喷雾干燥过程之后,伊曲康唑以特定的无定形玻璃状态重现(在此为实例3F)。该特殊性质在含最高比例伊曲康唑的制剂(实例3A,3C,3D,图6)的MDSC温谱图中也可观察到。在可逆热流量中显示在约49℃玻璃化转变,在不可逆热流量中约100℃冷结晶放热。该热时间在含有最小比例的伊曲康唑的制剂(~10%,实例3B和3E)中没有观察到,可能是由于稀释的组合物缺乏热检测灵敏度导致。喷雾-干燥甘露醇和伊曲康唑在大约相同的温度下熔融(总热流量)。四种制剂都在约164℃下观察到单个熔融点。实例3E在约150℃观察到补充吸热峰随后一放热峰。该转变对应于δ-甘露醇的熔融随后结晶化进入β多形体。另外一种制剂没有呈现该热事件可能是由于甘露醇在该制剂中仅以几乎全为δ-形式存在(见PXRD结果)。如实例1所述对所有喷雾干燥粉末进行PXRD分析。采用衍射图下的面积计算无定形含量,如表9所述。具有更高ITZ含量的制剂表现出更高的无定形含量。在无定形量和通过HPLC(R2>0.9)测得的伊曲康唑量之间观察到良好的相关性。通过参比强度比法计算每种甘露醇多晶的参与量对总结晶网络制剂的比例。DiffracplusEVA软件进行计算。该半定量评估方法由通过对比参照模式(ICDD数据库)测定样本中不同相和通过对比识别相位的峰密度相对测定多相样品中不同相的比例组成。表9:基于PXRD评估非晶含量和α、β和Δ甘露醇。为每个多晶选定一个特定的衍射峰,在干燥粉末衍射中不存在其他结晶结构。43.92,16.81和22.09°2θ的特定的衍射峰分别用于α,β和δ甘露醇,并且根据该衍射射线调整其分别的ICDD图谱用以计算。结果用以评估制剂中每种多晶如表9所示。如实例2所示,通过测定Carr’s可压性指数评估流动性。良好的流动性是最终工业规模简易制备过程的必要的性质。此外,更尤其针对吸入用干粉,良好的流动性关乎源自吸入仪的干粉的足够熔融的产生,分散和流动。所有制剂都显示出15.6%和26.4%之间的CI值(见表10),这说明该制剂类型具有良好的潜在流动性。通过两种不同的方法进行颗粒尺寸分析。第一种方法(使用Malvern)提供了与完全个体化颗粒相对应的尺寸结果。第二种方法(使用Malvern)能够以去聚集率评估来自吸入装置的分散后的产生的颗粒尺寸。Malvern的结果显示所有制剂呈现具有体积平均直径1.00μm-2.04μm范围,并且质量体积中值直径包括0.74μm和1.81μm之间的非常精细粒度(表10)。实例3A和3B的不含PL的制剂的PSD分别非常接近于0.74μm和0.88μm的d(0,5)值。但是如SEM中所观察的,实例3B中形成小量的较大颗粒,这如D[4,3]和d(0,5)所显示的增加。表10:通过不同溶液获得的制剂的尺寸,空气动力学和流动性能:使用和测定颗粒尺寸性能(Mean±SD,n=3),发射剂量(以标称剂量%表示),以及通过碰撞试验(Mean±SD,n=3,Carr’s指数值(CI)(Mean±SD,n=3)测定细颗粒分级(颗粒d<5μm的百分比)。如实例2所述进行空气动力学细颗粒测定。结果如表10所示。对于所有的制剂,计算得实例3B和3E中其FPF达40%甚至高达60%。也即,多于40%的仪器的加样制剂能够在装置发射后沉积于吸入真菌孢子的潜在沉积位点。沉积模式如图7所示。如实例1所述的条件进行溶解实验。每种制剂表现出不同的溶解速度,并且都比无定形喷雾干燥伊曲康唑(实例3F)和结晶松散ITZ(图8)的溶解速度快。如图9所示,所有根据本发明3A-3E的ITZ的溶解速度为至少10分钟内5%,20分钟内10%和60分钟内40%,实验根据美国药典2型溶解装置在每分钟50转,37℃在pH1.2滴定的含有0.3%的月桂基硫酸钠的9000ml水性溶解介质中,也即在曲线A的上部区域观察到该溶解速度,其表示了10分钟内5%,20分钟内10%和60分钟内40%的溶解速度。如图10所示,据3A-3E的ITZ的溶解速度还包括曲线B和B’之间的区域,其分别表示了5分钟内5%,10分钟内10%,20分钟内15%和60分钟内40%的溶解速度,并且该5分钟内50%,10分钟内60%,20分钟内90%和60分钟内100%的溶解速度,实验根据美国药典2型溶解装置在每分钟50转,37℃在pH1.2的含有0.3%的月桂基硫酸钠的9000ml水性溶解介质中。该额外的磷脂导致伊曲康唑溶解速度的加快,也即,5分钟内溶解速度>20%、10分钟>35%、20分钟>60%、60分钟>90%。结果如表11所示。表11:ITZ的溶解速度在制剂中增加加入磷脂的量能够引起API溶解速度的加快。确实,作为一个例子,实例3C含有1%(w/w)的磷脂(以伊曲康唑的重量表示)而实例3D含有10%(w/w)的磷脂。制剂3E也含有以伊曲康唑的重量表示的10%(w/w)的磷脂,显示了与同样含有10%(w/w)的磷脂的实例3D相似的溶解曲线。虽然在最终干燥形式中的磷脂的总量在实例3E(实例3E中为0.99%)中要低得多,该制剂并没有表现出与在最终干燥形式中含有更高磷脂的总量的实例3D(3.47%)所不同的溶解曲线。这表明,当在这些条件下进行测定时,所述伊曲康唑/磷脂比率似乎是提高API溶解速度的关键因素。因此能够通过改变该比率,在该范围改变或调制溶解速度。这是在体内提供药物肺内药代动力学不同可能性的一个优势。因此,能够使得生产具有高细颗粒级分,和比松散材料更快的溶解速度的制剂。但是还可能通过改变加入的表面活性剂的量从而控制/调节该促进。实例4:本实例的目的在于显示本发明使用高潜能健康安全疏水基质形成剂,制备具有高细颗粒级分,改进的湿润性,不同的溶解曲线和良好的流动性的基质干粉的能力。使用Büchi微型喷雾干燥机B-191a(Büchi实验室-技术,瑞士),通过喷雾-干燥法制备实验室规模的制剂。将设定量的伊曲康唑、胆固醇和含多于90%氢化磷脂酰胆碱的氢化大豆卵磷脂(Phospholipon90H)(见表12)在磁力搅拌下(600rpm)溶解于100ml的异丙醇并在70℃下加热。在以下条件下将溶液进行喷雾干燥:喷雾空气流,800l/小时,50℃下加热;干燥空气流,35m3/小时;溶液进料速度,2.7g/分钟;喷嘴尺寸,0.5mm;进口温度,70℃;使得出口温度为45℃。表12:实例4中的喷雾干燥溶液组合物如实例1所述测定CI值为18.9%,显示了良好的粉末流动性。颗粒尺寸测量(表13)分析显示制剂4Mastersizer测量具有大约1.1μm的体积中值粒径测量具有2.9μm粒径。制剂中表现的某些聚集现象似乎具有更高的d(0.9)值。其可能是在喷雾干燥过程中由于玻璃化转变的出口温度接近使得磷脂一定软化而形成。表13:通过激光衍射方法测得实例4的制剂的尺寸分布参数聚集现象的出现影响了如实例1所述的空气动力学评估细颗粒测试中评价的颗粒的沉积。但是实例4的44%的上样剂量达到冲击器三个低级阶段。表14:冲击试验过程(MSLI,100l/分,2.4秒,每次试验流出3,2.5mg下称重的标称计量,n=3)获得的颗粒沉积mg(平均值±SD)和以标称计量FPD%计的细颗粒片段。实例4装置(mg)0.73±0.05咽喉(mg)0.15±0.031期(mg)0.26±0.142期(mg)0.17±.083期(mg)0.31±0.034期(mg)0.50±0.055期(mg)0.28±0.03平均FPD(mg)1.1±0.1平均FPF(%)44±4如实例1所述进行溶解度测试,但溶解介质由含1%月桂基硫酸钠的pH1.2滴定的(HCl0.063N)去离子水组成(见图11)。制剂4表现出比结晶微粉化松散伊曲康唑更快的溶解速度。使用疏水GRAS前基质直接改变了分散的API释放曲线,也提供了良好的空气学性能和流动性。实例5:本实例的目的在于揭示制剂中API的物理状态(非晶和结晶纳米颗粒)的影响。制备并表征具有相同的定量组成的两种制剂。但是每种制剂的AIP出于不同的物理状态。通过喷雾干燥溶液或纳米悬液分别获得制剂5A和5B,使用Büchi微型喷雾干燥机B-191a(Büchi实验室-技术,瑞士)。实例5A的干粉通过喷雾干燥赋形剂和API原料母液制备,0.10g伊曲康唑,0.9g甘露醇和0.01g的TPGS1000,磁力搅拌(600rpm)下并70℃加热溶解于100ml水-醇溶液(20水:80异丙醇)。该溶液在以下条件中喷雾干燥:喷雾空气流,800l/小时;干燥空气流,35m3/小时;溶液进料速度,2.7g/分钟;喷嘴尺寸,0.5mm;进口温度,90℃;使得出口温度为53℃。实例5B的干粉通过喷雾干燥赋形剂原料母液制备,其在喷雾干燥前已加入重悬了一定体积的API纳米悬液。该过程由两步组成,第一步是将微粉化的API悬液的尺寸减小至纳米尺寸范围内的悬液。第二步是在含基质试剂的原料母液中重悬一定量的制备的纳米颗粒,以使之喷雾干燥。根据以下步骤制备纳米悬液。磁力搅拌下(600rp)将75mgTPGS1000溶解于75ml的水-醇溶液(异丙醇25:水50)。使用CAT高速匀浆法X620(HSH)(CATM.Zipperer,Staufen,德国)24,000rpm进行5分钟,将750mg微粉化的伊曲康唑重悬于该溶液中。进而将该悬液在高压匀浆器EmulsiFlexC5(AvestinInc.,渥太华,加拿大)中以24000PSI循环直至颗粒呈300nm下d(0,5)和2.5μm下d(0,9)。使用具有湿采样系统(Mastersizer,Hydro2000,Malvern仪器,英国)的激光衍射进行匀浆悬液的颗粒尺寸分布分析。将测量样品分散于含2%泊洛沙姆407的伊曲康唑饱和去离子水中,以避免颗粒溶解和聚集。使用1.61折射率和0.01吸收指数进行测量。使用热交换器进行高压匀浆,将其放置与匀浆阀的前部以保持样品温度低于10℃。制备由200ml异丙醇和70ml水组成的270ml的水-醇溶液,其中在磁力搅拌下溶解2.7g甘露醇。使该溶液保持冰浴并在磁力搅拌下(200rpm)加入30ml制备的纳米悬液。喷雾干燥该最终悬液。在喷雾干燥过程中使用以下条件:喷雾空气流,800l/小时;干燥空气流,35m3/小时;溶液进料速度,2.7g/分钟;喷嘴尺寸,0.5mm;进口温度,80℃;使得出口温度为45℃。最终干燥产物组合物如表15所示。表15:实例5中的最终干燥产物的定量组成。进行制备的纳米悬液的颗粒大小分布测定。所述悬液分别表现0.257+/-0.005μm和1.784+/-0.010μm的d(0,5)和d(0,9)。所述两种干燥样品表现出良好的粉末流动性。实例5A和5B的的卡尔指数值(Carr’sindex)分别为19.9%和24.7%。PDRX分析显示制剂5A结晶伊曲康唑未显示特征衍射峰但实例5B的衍射图明显显示有该峰。进而伊曲康唑在制剂5A中呈非晶形式但在制剂5B中呈纳米晶体形式。Malvern颗粒尺寸分析揭示了两种制剂具有相似的尺寸分布值。结果显示如表16所示。与该结果相反,Spraytec测定揭示了在从吸入装置释放后,制剂5B呈完全不同的尺寸分布图(见表16)。实际上,图像观察到严重的聚集现象,以及确认了d(0,9)值明显增加至64.50±19.9μm。表16:通过Malvern和激光衍射测定的实例5中制剂的尺寸分布参数。在模拟呼吸条件下,制剂5B似乎表现出比制剂5A更低的解聚效率。但是,尽管制剂5B表现出严重的聚集现象,其表现出如实例1描述的测定的更高的细颗粒级分(见表17)。表17:冲击试验过程(MSLI,100l/分,2.4秒,每次试验流出3,2.5mg下称重的标称计量,n=3)获得的颗粒沉积mg(平均值±SD)和以标称计量FPD%计的细颗粒级分。使用实例1描述的方法进行溶解度测试。所述两种制剂表现出不同的溶解速度。制剂5B表现出比制剂5A更快的溶解速度,但两种制剂均表现比松散伊曲康唑更快的溶解速度。实例6:本发明也可包括混合结晶纳米颗粒基质制剂和非晶基质制剂以在期望范围内改变所述活性成分的溶解曲线。该混合可在胶囊填充之前或过程中实现。纳米颗粒具有的突释效应能够产生确定的ITZ浓度,其可通过非晶基质制剂的溶解以期望的速度加以增强从而最优化其溶解速度。在最终的混合物中的基质纳米颗粒制剂的比例将决定突释效应(药物的快速初始溶解)表现为何种程度。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1