制备药物凝集体的方法与流程
本发明是关于制备一球状药物凝集体的方法,其凝集体具有一特定大小。该球状药物凝集体包含高浓度药物,且具有良好流动性、安定性以及易后制处理的特性。该球状药物凝集体可用来当作药物组合物的成分,减少已知制程中冗长的药物加载、覆膜程序。该球状药物凝集体可经由再配制成控制释放剂型。
背景技术:
在制药领域,粉末的颗粒特性对于其产物具有决定性的因素,因此,在备制过程中需要严格控制,尤其是在口服药剂方面。颗粒大小与形状会影响到最终产物的特性以及后续制程,通常在配制过程中像是混合、压片以及锭包膜等,皆须从较小的颗粒开始。除此之外,药物颗粒大小分布也会影响药物药效〈例如:溶解度、生体可用率、成分均一度、安定性等等〉。
不良颗粒特性的问题可藉由粒化克服。粒化(granulation)为一种获得较大颗粒的方法,通过造粒得到的颗粒大小较为匀称,有助于接下来的配制。已知技术主要利用两种粒化程序,包含湿式造粒以及干式造粒。于湿式造粒制程中,添加造粒液体至一粉末床,通过搅拌翼〈高剪力造粒机或双轴挤压机〉或空气〈流体化床造粒机〉加以搅拌,形成粒状结构。由机器带动的机械搅拌以及制备程序的湿化过程使得粉末集结产生湿粒状结构。造粒液体包含一可挥发溶剂,该溶剂可通过干燥步骤移除。常见的造粒液体包含水、乙醇、异丙醇及其组合。当物质对于湿气或高温特别敏感,则可通过不使用液体的干式造粒制程使的形成粒状结构。干式造粒通过高压将粉末压聚至紧实。粉末可经重压打锭而形成一大型锭剂,或是粉末经滚轮压实机而形成一片状材质。在这两种情况下,中间产物被裁切或筛选至适当大小。然而,如果药物的物理或化学性质较不安定,上述技术则不适用于粒化,因为水气以及高温〈湿式造粒〉可能会加速药物降解,而高压〈干式造粒〉也可能导致药物降解。而且,上述粒化制程皆需昂贵器材才得以进行。
固定大小的药物载珠为另一种控制颗粒大小的方式。于一流体化床或离心式造粒机中,将一包含药物的溶液/悬浮液喷洒在固定大小的惰性珠粒上。然而,通常需要相当长的时间才能将药物载负在珠粒上,尤其药物剂量较大时,需要更长的时间。此外,在药物载珠上包覆一释放速率控制层,又需要更多的时间。
除了上述的方法之外,调整原料颗粒特性也是另一种方式用来改进或增加赋形剂的额外功效。不同种类及功效的赋形剂皆可向医药级供应商取得。然而,相较于已商品化的赋形剂,药物原料规格的选择仍然有限。
球状结晶〈Spherical Crystallization〉为一种用来获得较大球状颗粒的制程。在此一制程中,结晶以及凝集可同时进行,而且可增加结晶形药物的流动性与相容性〈Powder Technol.130,2003,298-306〉。根据不同产物颗粒大小,许多技术被用来形成球状结晶,举例来说,球状凝集〈Spherical Agglomeration〉、氨扩散〈Ammonia Diffusion〉、乳化液溶剂扩散〈Emulsion Solvent Diffusion〉以及中和反应〈Neutralization〉。
1974年,Kawashima以及Capes公开了在结晶步骤中利用凝集获得较大颗粒的概念。二氧化硅砂分散置入机械搅拌的四氯化碳中,并与氯化钙水溶液凝集,此一程序成为标准流程〈Power Technol.10,1974,85-92〉。之后,Kawashima又揭露一种在结晶过程中用来获得更大颗粒的方法,藉由控制结晶凝集掌控颗粒特性〈Science 4June 1982,1127-1128〉。Kawashima的方法利用三种溶剂,一种为药物溶解介质,例如一有效溶剂,另一种介质可让药物部分溶解并具有湿润的特性,例如一联结液体〈bridging liquid〉,以及一种与药物不互溶的液体,例如一非有效溶剂。药物颗粒先溶解于有效溶剂,接着倒入非有效溶剂造成再结晶,之后加入联结溶剂使得粒子凝集。然而,在药物溶解-再结晶的过程中控制结晶性质十分困难,因为药物可能无法形成理想的形状或大小,尤其形状与大小取决于药物本身特性以及制程参数设定。除此之外,上述球状凝集体是否能在制药程序中进行包膜也是未知。
技术实现要素:
本发明的目的在于提供一种制备药物凝集体的方法,以获得具有一特定大小的球状药物凝集体。该球状药物凝集体包含高浓度药物,具有良好流动性、 安定性以及易后制处理的特性。该球状药物凝集体可用来当作药物组合物的成分,减少已知制程中冗长的药物加载、覆膜程序。该球状药物凝集体可经由再配制成控制释放剂型。
根据本发明部分实施例,提供一制备药物凝集体的方法。由该方法产生的球状药物凝集体包含高纯度药物。球状药物凝集体中的药物为结晶型或非晶型。凝集体颗粒大小介于约0.1至2.0毫米之间,较佳为0.1至1.5毫米之间,或0.1至1.0毫米之间。球状药物凝集体可选择性地被其他膜衣包覆。
根据本发明部分实施例,制备药物凝集体的方法包含添加一粉末状药物至一第一溶剂以形成一第一溶液。接着,向该第一溶液喷洒一第二溶剂以形成一第二溶液,同时该粉末状药物进行成核作用以形成一药物凝集体。再来,自该第二溶液分离该药物凝集体。
根据本发明部分实施例,该第一溶剂是指C4-C8直链烷。
根据本发明部分实施例,该第二溶剂是选自于氰甲烷、乙醇、纯水及其组合所组成的群组。
根据本发明部分实施例,制备方法还包含混合该第一溶剂与该第二溶剂,并且添加该粉末状药物。
根据本发明部分实施例,该第二溶剂的液滴直径大小介于0.1毫米至1.0毫米之间。
根据本发明部分实施例,该药物凝集体的直径大小介于0.1毫米至2.0毫米之间,且该药物凝集体的药物重量比大于95%。
根据本发明部分实施例,该粉末状药物颗粒直径大小〈d90〉小于0.05毫米。
根据本发明部分实施例,该粉末状药物是指一酸感性药物。
根据本发明部分实施例,该粉末状药物是指杜洛希酊〈duloxitine〉。
根据本发明部分实施例,提供一种口服药物组合物,包含直径大小介于0.1毫米至2.0毫米之间的药物凝集体,其中所述药物凝集体的药物纯度等于或高于95%(w/w)。
相较于已知方法获得的原料或粒子,本发明的球状药物凝集体包含高纯度药物,具有更佳的稳定度以及相容性。
附图说明
本发明的上述和其他态样、特征及其他优点参照说明书内容并配合附加附图得到更清楚的了解,其中:
图1是根据本发明部分实施例的一药物凝集体制备方法流程图;
图2A-图2D是根据本发明部分实施例产生的杜洛希酊凝集体数码显微镜画面;
图3A-图3B是不同态样的杜洛希酊数码显微镜画面;
图4A-图4D是不同态样的杜洛希酊数码显微镜画面;
其中,符号说明:
S110-130步骤。
具体实施方式
为了使本揭示内容的叙述更加详尽与完备,下文针对了本发明的实施态样与具体实施例提出了说明性的描述;但这并非实施或运用本发明具体实施例的唯一形式。以下所揭露的各实施例,在有益的情形下可相互组合或取代,也可在一实施例中附加其他的实施例,而无须进一步的记载或说明。在以下描述中,将详细叙述许多特定细节以使读者能够充分理解以下的实施例。然而,可在无此等特定细节的情况下实践本发明的实施例。
本说明书中「药物」一词是指一种物质用来治疗或预防疾病,或用来辅助生理或心理状态。药物可以以单镜像异构物、混合立体异构物、自由碱基或其他医药级可接受的盐类方式存在。药物包含但不局限于以下几种特征:安定性不佳〈例如:易受肠胃系统环境影响、易受光/湿度影响〉、需通过控释给药〈例如:需精准调控药物血中浓度、仅能于特定部位吸收〉以及高使用剂量〈例如:每次投予剂量大于300毫克〉。较佳地,根据本发明部分实施例,药物为酸敏性,或需包覆功能性膜衣。质子泵抑制剂,像是omeprazole、lansoprazole、pantoprazole、rabeprazole以及ilaprazole皆适用于本发明,然而本发明的药物使用并不局限于此。
本说明书中「口服剂形式」是指一种通过口服的药剂形式,包含但不局限于药锭、胶囊、颗粒、粉末、颊膜片、舌下含片、口内膏、悬浮液、乳状液或糖浆。
本说明书中「赋型剂」是指一药剂中非具药效的医药级可接受的惰性物质。赋型剂包含但不局限于粘合剂、崩解剂、填充剂、成膜剂、香料、色料、润滑剂、助流剂、吸附剂、防腐剂、释放速率控制剂或甜味剂。
本说明书中「控释制剂」是指一改变药物释放时间以及/或速率的药剂形式。控释制剂的药物释放时间以及/或位置特性可被选择,以达成已知药剂例如一溶液或速释剂形式无法匹配的医疗效果。控释制剂包含延迟释剂以及长效型释剂。延迟释剂为一在服用之后药物不立即释放或不连续释放药物的配方。长效型释剂则让药物在服用后长时间内皆能持续释出药物。相较于速释药剂型式,长效型释剂可减少投药频率。
本说明书中「释放速率控制剂」是指一可控制药物释放表现的物质,其通常通过形成基质或是膜衣来调节药物释放,但不限于此种方式。此物质包含但不局限于不溶于酸或碱的物质、不溶于水或在水中溶解速率慢的物质、遇水后膨胀并阻碍药物释放的物质或遇水后形成胶质的物质。较佳地,可成胶物质、肠衣物质或非水蚀/不溶解的物质可用来当作释放速率控制物质。此物质还包含但不局限于聚乙烯醇、聚丙烯酸钠、羟丙基甲基纤维素〈hydroxypropylmethyl cellulose,HPMC〉、羟丙基纤维素〈hydroxypropyl cellulose〉、羟乙基纤维素〈hydroxyethyl cellulose〉、甲基纤维素〈methyl cellulose〉、羧乙基纤维素〈carboxyethyl cellulose〉、羧甲基羟乙基纤维素〈carboxymethyl hydroxyethyl cellulose〉、卡波树脂〈carbomer〉、羧甲基钠纤维素〈sodium carboxymethyl cellulose〉、聚乙烯吡咯烷酮〈polyvinylpyrrolidone,PVP〉、丙烯酸甲酯/甲基丙烯酸共聚物、醋酸琥珀酸纤维素〈cellulose acetate succinate〉、羟丙基甲基苯二甲酸纤维素〈hydroxypropylmethyl cellulose phthalate,HPMCP〉、羟丙基甲基醋酸琥珀酸纤维素〈hydroxypropylmethyl cellulose acetate succinate,HPMCAS〉、聚醋酸乙烯邻苯二甲酸酯〈polyvinyl acetate phthalate〉、甲基丙烯酸甲酯/甲基丙烯酸共聚物、海藻酸钠〈sodium alginate〉以及乙基纤维素〈ethylcellulose〉。
本说明书中「肠衣物质」是指一种物质在酸性环境较碱性环境稳定。举例来说,肠衣物质不会溶解于酸性胃液〈pH约为3〉当中,但是在肠道的碱性环境〈pH7-9〉可溶解。该物质包含但不局限于脂肪酸、蜡、虫胶、塑料、植物纤维以及聚合物等。
本说明书中「高纯度」一词是指由适当分析仪器测量后纯度至少等于或高于95%。
本说明书中「溶剂」是指水或有机溶剂,其中有机溶剂是指根据国际医药法规协和会〈ICH〉Q3C指南所列的第2类及第3类溶剂,包含但不局限于C4-C8直链烷、单环芳香烃、卤代烃、醇、酮、醚、酯、腈、酰胺。
本说明书中的溶解度是依据美国药典-国家处方集〈USP-NF〉定义。也就是说,「不溶」是指1单位溶质需要超过10,000单位溶剂。「微溶」是指1单位溶质需要1,000至10,000单位溶剂。「略溶」是指1单位溶质需要100至1,000单位溶剂。「稍溶」是指1单位溶质需要30至100单位溶剂。「可溶」是指1单位溶质需要10至30单位溶剂。「即溶」是指1单位溶质需要1至10单位溶剂。「完全相溶」是指1单位溶质仅需少于1单位溶剂。
根据本发明部分实施例,通过其制备药物凝集体方法所产生的药物凝集体外观大致呈球状。产生的药物凝集体平均颗粒直径大小约为0.05至2毫米,较佳地,可为0.1至1.5毫米,更佳地,可为0.1至1.0毫米。根据本发明部分实施例产生的药物凝集体包含高纯度〈高於95%(w/w)〉药物。
如图1所示,本发明提供一种制备药物凝集体的方法,包含步骤S110,将药物粒子添加至一第一溶剂中并使的悬浮,步骤S120,添加一第二溶剂以凝集药物粒子,并对药物凝集体外型加工使其圆融,接下来,步骤S130,搜集药物凝集体,可选择让其干燥以移除多余溶剂。已知技术需经过药物溶解-再结晶的步骤,本发明则无须再结晶,且凝集体的形成受控于一预定颗粒大小的原物料,因此,凝集体大小可被有效控制在一定范围内。
本发明还提供另一种制备药物凝集体的方法,包含将药物粒子溶解于有效溶剂形成一饱和溶液,接着,将该饱和溶液与第一溶剂混合,使得药物结晶沉淀,再添加第二溶剂以凝集药物结晶,并对药物凝集体外型加工使其圆融,接下来,搜集药物凝集体,可选择让其干燥以移除多余溶剂。
上述步骤S110与S120皆在机械搅拌的状态下进行。另外,将药物粒子溶解于有效溶剂形成饱和溶液,接着,将该饱和溶液与第一溶剂混合,使得药物结晶沉淀,再添加一第二溶剂以凝集药物结晶,并对药物凝集体外型加工使其圆融的步骤皆在机械搅拌的状态下进行。药物凝集体在搅拌的过程中形成球状凝集体。机械搅拌可通过桨叶器材搅拌,或其他合适的搅拌工具。为了使搅拌的 效果更好,可在反应槽内放置数片折流板。干燥步骤用来将凝集体与溶剂分离,任何合适的干燥手段皆可用于本发明。
相较于第二溶剂,第一溶剂对于药物具有较差的溶解度。更详细地说,药物粒子不溶于第一溶剂〈溶解度低于0.0001克药物/克溶剂〉。在上述将药物粒子溶解于有效溶剂形成饱和溶液之后,将该饱和溶液与第一溶剂混合,即使得药物结晶沉淀。
有效溶剂与第一溶剂互溶。另外,相较于第一溶剂,有效溶剂具有较高的药物粒子溶解度。更详细地说,药物在有效溶剂中为即溶或完全相溶〈高于0.03克药物/克溶剂〉。
第一溶剂可以包含但不局限于C4-C8直链烷〈如:戊烷、庚烷〉、单环芳香烃〈如:甲苯、二甲苯〉、乙酸乙酯、N,N-二甲基苯胺以及丁酮。有效溶剂包含但不局限于水、甲醇、乙醇、异丙醇、丁醇、乙腈、丙酮、四氢呋喃、三氯甲烷、1,4-二氧六环以及二甲基甲酰胺。
第二溶剂与第一溶剂不相溶。另外,第二溶剂对于药物的溶解度介于微溶与稍溶之间〈0.0001-0.03克药物/克溶剂〉。添加适量第二溶剂以促进凝集药物粒子/结晶,形成更大的粒子。除此之外,在步骤S110中,在添加药物粒子之前,第一溶剂可以与一部份第二溶剂混合,如此以来可促进药物在第一溶剂中的分布状态,并增进之后的凝集效果。第二溶剂包含但不局限于乙腈、甲醇、乙醇、水、二甲基甲酰胺及其组合。第二溶剂的选择取决于第一溶剂以及使用的药物。
添加第二溶剂时,较佳以渐进的方式逐步混合。然而,过长的添加时间却也可能造成凝集体大小不均的问题。本发明发现,控制第二溶剂的液滴大小将能有效影响凝集体的颗粒大小及分布情形。使用喷枪将第二溶剂以微细液珠及广域的方式喷洒,例如液滴大小介于0.1至1.0毫米之间,将特别适用于制备大批量凝集体。其他添加第二溶剂的方式,例如滴加于第一溶剂中或直接导入于第一溶剂也在本发明的保护范围内。
溶剂使用取决于药物粒子特性。举例来说,使用杜洛希酊当作药物粒子并欲制作其球状凝集体时,第一溶剂可为庚烷,第二溶剂可为乙腈、乙醇、水或其混合。
根据本发明部分实施例,有效溶剂与第一溶剂混合比例约为1:5至1:35〈v/v〉。第二溶剂与第一溶剂〈或第二溶剂相比于第一溶剂与有效溶剂的和〉混合比例约为1:20至1:70〈v/v〉。
由本发明的方法所产生的药物凝集体可进行下阶段加工程序,例如包覆功能性膜衣。功能性膜衣可增加凝集体其他特性,像是控制药物释放表现、增加安定性、保护凝集体等,使得凝集体在后续制程应用更多元。功能性膜衣可提供凝集体抗酸/碱、味道/气味遮蔽、光/湿度防护、调节释放表现等功能。
药物凝集体的大小与密度主要取决于原物料形状以及颗粒大小。较小的原物料可产生较小且外观圆滑的药物凝集体。举例来说,为了得到直径约为1.0毫米的球状凝集体,原物料的直径最好小于0.1毫米。根据本发明部分实施例,药物颗粒大小d90小于0.05毫米为较佳的起始大小。药物颗粒大小d90小于0.01毫米则可产生趋近0.1至1.0毫米的药物凝集体。0.1至1.0毫米的药物凝集体大小有利于后续包膜或成锭的程序。
以下实施例利用杜洛希酊氢氯化物〈duloxetine hydrochloride〉为原药物形成药物凝集体。杜洛希酊为一血清素与正肾上腺素再吸收抑制物,首见于欧盟专利第273658号,其可使用于重度忧郁症、泛焦虑症、糖尿病周边神经性疼痛以及肌纤维痛。其以盐类型式,杜洛希酊氢氯化物,呈现于产品中,目前由Eli Lilly以为名贩售于美国。欧盟专利第273658号揭露杜洛希酊氢氯化物粒子形成方式,其杜洛希酊氢氯化物粒子为针状结晶,且须经过其他加工手续才得以用于制备医药产品。美国专利第5508276号揭露杜洛希酊粒子需经过研磨到粒子小于50μm,方可通过粉末包覆程序将其覆盖到珠粒之上;或使杜洛希酊溶解或悬浮于溶液中,再将其喷洒至珠粒之上。由于结晶特性的限制,目前市面上的杜洛希酊几乎都使用与相同的配方,也就是将一层杜洛希酊覆盖于一惰性珠粒。然而,此种制程不仅耗时制作成本也较高。
在本发明下述的实施例一至四与八至九中,杜洛希酊氯化氢原物料的颗粒大小d10为7.9μm,d50为43.6μm,d90为119.5μm。实施例十与十三中,杜洛希酊氯化氢原物料的颗粒大小d10为0.6μm,d50为1.8μm,d90为5.2μm。
实施例一
在20℃环境下,使2.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率400rpm〉。渐进地滴加7毫升乙腈至该悬浮液。将溶液持续搅拌220分钟达到匀相平衡〈稳定大小以及表面平滑的凝集体形成〉,过滤产物并在40℃下干燥20小时。由上述手段可得到1.8克凝集体,且外观大致呈球状,平均颗粒直径大小介于1.0至1.8毫米之间。凝集体平均抗压强度大约是230.7克/珠粒。抗压强度测试方法已见于Braz.J.Pharm.Sci.vol.48No.4Oct/Dec.2012或Asian Journal of Biomedical and Pharmaceutical Sciences 3(18)2013,10-16。由示差扫描热分析可看到,产物在173.8℃出现高峰,与原物料特性类似,且热重分析显示溶剂并不存在于杜洛希酊凝集体。
实施例二
在20℃环境下,使2.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率400rpm〉。渐进地滴加6.6毫升第二溶剂〈乙腈:95%乙醇,约为19:1〉至该悬浮液。将溶液持续搅拌200分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥13小时。最后产物为总重1.8克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小介于0.8至1.2毫米。凝集体平均抗压强度大约是200.0克/珠粒。
实施例三
在20℃环境下,使9.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率1000rpm〉。渐进地滴加11毫升乙腈至该悬浮液。将溶液持续搅拌60分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥11小时。最后产物为总重8.7克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小介于0.3至0.7毫米。凝集体平均密度大约是0.88克/立方公分。
实施例四
在20℃环境下,使20.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率400rpm〉。渐进地滴加15毫升乙腈至该悬浮液。将溶液持续搅拌210分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥12小 时。最后产物为总重19.6克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小介于0.8至1.0毫米。凝集体平均密度大约是0.77克/立方公分。
实施例五〈杜洛希酊丸肠衣包膜〉
实施例四所产生的近球状杜洛希酊凝集体可继续进行肠衣包覆程序。近球状杜洛希酊凝集体通过筛号60的筛子筛选。准备一密封膜溶液,该密封膜溶液包含22.5克的HPMC、45.0克的蔗糖以及67.5克的滑石粉溶解于764.9克的纯水。将300克筛选过的近球状杜洛希酊凝集体放置于一流体化床,并将密封膜溶液洒在凝集体上。经过密封膜包覆后,重量将增加约30.7%,含水量约为1.2%。密封膜包覆程序后,其产物进入肠衣包覆。将密封膜包覆的300克凝集体包覆一肠衣层溶液,该肠衣层溶液包含19.0克HPMCAS,2.9克柠檬酸三乙酯以及8.1克硬酯酸溶解于711.2克的95%乙醇。在包膜程序之后,即产生肠衣聚合物包覆的杜洛希酊凝集体,重量增加约为7.0%,含水量约为1.2%。每一杜洛希酊丸包含约71.5%w/w杜洛希酊氯化氢。包膜环境请见以下表格一。
表格一
实施例六〈肠衣包覆的杜洛希酊丸溶离试验〉
由实施例五获得的膜覆杜洛希酊丸进一步经过溶离试验。该溶离试验根据美国药典的缓冲液替换手段,在一第一型溶离机中进行〈2小时在0.1N氯化氢,接着替换为pH6.8的PBS〉。膜覆杜洛希酊丸包含相当于30毫克的杜洛希酊自由碱基被置入3号胶囊中进行测试。结果显示其能降低药物在酸性环境的释放,而在中性环境始能立即释出药物,达到药物延迟释放的效果。
表格二
实施例七〈近球状杜洛希酊凝集体溶离试验〉
由实施例一以及二获得的膜覆杜洛希酊丸进一步经过溶离试验。该溶离试验根据美国药典在一第一型溶离机中、500毫升的pH6.8的PBS、100rpm以及37℃下进行。以下结果是对照于杜洛希酊原物料。由测试结果表格三来看,近球状药物凝集体释放表现与原物料十分类似。
表格三
实施例八
在20℃环境下,使2.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率400rpm〉。渐进地滴加0.8毫升纯水于该悬浮液。将溶液持续搅拌360分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥17小时。最后产物为总重1.9克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小介于1.2至1.6毫米。凝集体平均抗压强度大约是98.2克/珠粒。
实施例九
在20℃环境下,使2.0克杜洛希酊氯化氢在300毫升的庚烷中形成悬浮液,且持续搅拌〈桨叶速率400rpm〉。渐进地添加4.6毫升浓度95%乙醇于该悬浮液。将溶液持续搅拌180分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥12小时。最后产物为总重1.8克的凝集体,凝集体具有近球状的外观,平均颗粒直径大小介于1.7至1.9毫米。凝集体平均抗压强度大约是298.8克/珠粒。
实施例十
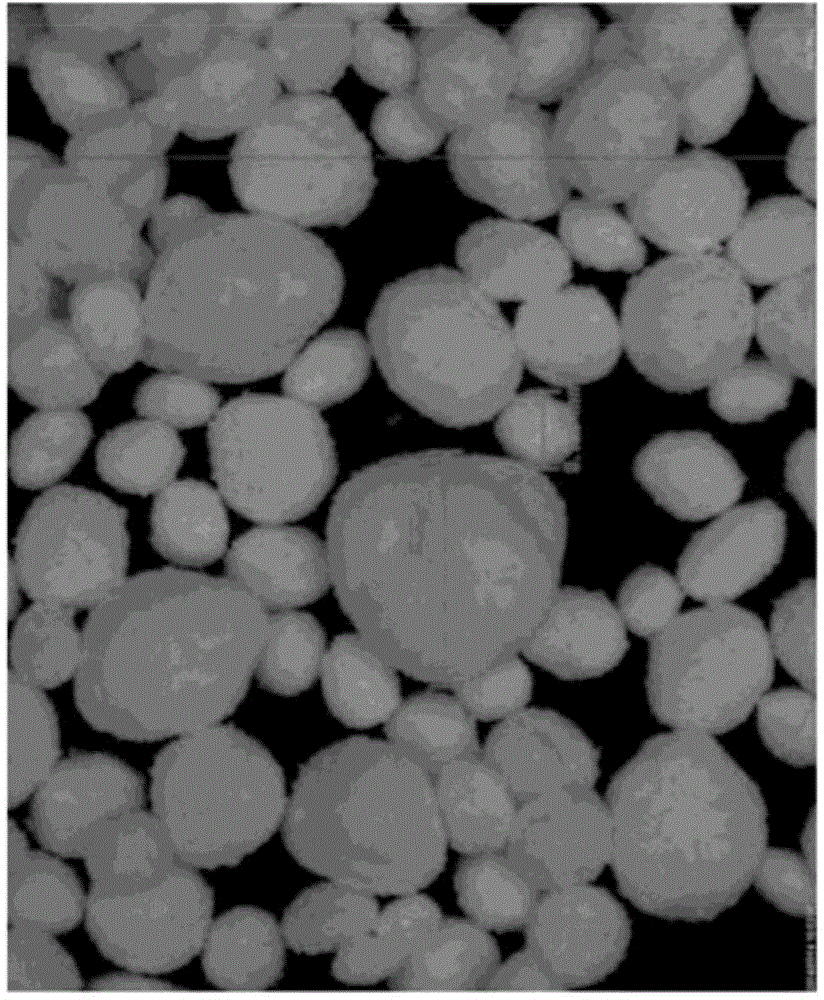
在20℃环境下,使35克杜洛希酊氯化氢在300毫升的庚烷以及5毫升的乙腈中形成悬浮液,且持续搅拌〈桨叶速率800rpm〉。在流速20rpm〈蠕动泵〉以及喷雾压力0.04巴渐进地通过0.5毫米孔径喷枪〈Mod.951S20〉喷洒27毫升乙腈于该悬浮液。将溶液持续搅拌180分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥11小时。最后产物为总重34.7克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小介于0.3至0.5毫米。凝集体平均抗压强度大约是43.0克/珠粒。图2为数码显微镜截图,显示在加入乙腈之后,不同时间点得到的凝集体。图2A为10分钟,图2B为40分钟,图2C为120分钟,图2D为180分钟。
实施例十一〈肠衣聚合物包覆的杜洛希酊丸〉
由实施例十获得的近球状杜洛希酊凝集体又经过密封程序,由一包含HPMC的材质包覆,且被一包含HPMCAS的肠衣层包覆〈参考实施例五的步骤〉。密封层增加总重量的30.7%,肠衣层增加7.0%。每一药丸包含约71.5%w/w杜洛希酊氯化氢。接下来,产物经过筛号16与20的筛子筛选,得到直径大小介于0.8至1.2毫米的颗粒。
实施例十二〈制备填料粒子与锭剂〉
制备控释锭剂,该锭剂通过压缩肠衣包覆的杜洛希酊丸以及填料粒子所形成。
填料粒子制备方法包含在造粒机中将480.0克微晶纤维素以及30.7克PVP,且加入150.0克纯水进行造粒。通过筛号16以及20的筛子筛选粒子。
将171克填料粒子、42克由上述实施例十一产生的肠衣包覆杜洛希酊丸、57克微晶纤维素、24克交联羧甲基纤维素钠、3克二氧化硅以及3克甘油山嵛酸酯混合。产生的混合物经过已知锭剂制备程序,制作成最终约为300毫克的锭剂,每锭包含30毫克杜洛希酊氯化氢。
实施例十三
在20℃环境下,使70克杜洛希酊氯化氢在600毫升的庚烷以及10毫升的乙腈中形成悬浮液,且持续搅拌〈桨叶速率800rpm〉。在流速20rpm以及喷雾压力0.04巴的环境,渐进地通过喷枪喷洒54毫升乙腈于该悬浮液。将溶液持续搅拌300分钟达到匀相平衡,再将产物滤出并在50℃的环境下干燥11小时。最后产物为总重69.6克的凝集体,且凝集体具有近球状的外观,平均颗粒直径大小约为0.4毫米。近球状凝集体成分经过气相色层分析,庚烷约为26ppm,且未侦测到乙腈。
实施例十四
表格六列举具有控释效果的杜洛希酊丸A以及杜洛希酊丸B进行胶囊充填。杜洛希酊丸A以及杜洛希酊丸B平均颗粒大小约为400微米。
表格四
实施例十五〈流动性比较〉
近球状杜洛希酊凝集体具有平均颗粒直径大小约1.6毫米,与杜洛希酊原物料颗粒一并经过Carr指标测试其流动性。近球状杜洛希酊凝集体的Carr指标值约为8.33%〈流动性佳〉。相较之下,杜洛希酊原物料颗粒平均颗粒直径大小约为6.7微米,Carr指标值约为46.25%,其流动性非常低。图3为数码显微镜截图,图3A为根据本发明的制备方法产生的近球状杜洛希酊凝集体,图3B为杜洛希酊原物料。
实施例十六〈肠衣聚合物包覆杜洛希酊丸〉
表格五
膜覆杜洛希酊凝集体进一步经过溶离试验。该溶离试验是根据美国药典的规范,在第一型溶离机中进行,首先,2小时置于一酸性介质内〈0.1N氯化氢,1000毫升,100rpm,37℃〉,接着置于一缓冲介质内〈pH6.8PBS,1000毫升,100rpm,37℃〉。
表格六
实施例十七
不同大小的近球状杜洛希酊凝集体的相容性可通过测量残余含量〈%assay〉得知。杜洛希酊原物料以及通过已知粒化手段得到的粒化杜洛希酊也进行相容性测试。粒化杜洛希酊由与制备凝集体相同的第二溶剂〈例如:乙腈〉进行造粒,且在50℃下干燥直到含水量小于1%。易造成杜洛希酊降解的肠衣物质HPMCP被用来进行实验。
根据实验结果,近球状杜洛希酊凝集体在固定时间内,相较于杜洛希酊原物料具有较佳的残余含量,可见本发明的药物凝集体制备方法能有效改善相容性。除此之外,近球状杜洛希酊凝集体相较于通过已知手段所得相同大小的粒化杜洛希酊具有较佳的相容性,由此可见相容性的改善并非仅归因于颗粒本身大小。图4是杜洛希酊不同态样数码显微镜截图。图4A为20-30筛产生的凝集体,图4B为40-60筛产生的凝集体,图4C为20-30筛产生的粒化杜洛希酊,图4D为40-60筛产生的粒化杜洛希酊。
表格七
*C:原物料;SA:近球状凝集体;G:粒化颗粒
虽然本发明已以实施方式揭露如上,然其并非用以限定本发明,任何熟习此技艺者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰,因此本发明的保护范围当视后附的权利要求书所界定的范围为准。
- 还没有人留言评论。精彩留言会获得点赞!