含野菊花提取物的降血脂药物及野菊花提取物的制备方法与流程
本发明涉及降血脂药物技术领域,尤其涉及一种含野菊花提取物的降血脂药物,以及野菊花提取物的制备方法。
背景技术:
高脂血症可诱发动脉粥样硬化、高血压、糖尿病、冠心病或其它心脑血管疾病,是常见多发的关键性疾病,位列危害我国人民健康的十大疾病之一。高效、低毒、经济合理的降血脂药物的成功开发,可以减少一群疾病的发病率和由此导致的死亡率,如动脉粥样硬化、高血压、糖尿病和冠心病等。因此,降血脂药物的研发问题是我国社会发展中迫切需要解决的关键科技问题,具有重大的社会效益。
胆固醇水平是衡量高脂血症的重要指标之一,肝X受体(LXR)是维持机体胆固醇代谢平衡的关键感受器,能调控胆固醇的吸收、合成、代谢和转运。LXR与视黄醇类X受体(RXR)结合形成二聚体,而后激活LXR反应元件,从而调节一系列关键靶基因的转录表达,比如ABC转运蛋白(ABCA1,ABCG5和ABCG8)、载脂蛋白E(apoE)和胆固醇调节元件结合蛋白(SREBP-1c)。LXR是近年来研究降血脂药物的热门靶标,开发新型LXR激动剂作为治疗高血脂的潜在药物一直是韦氏、科华、辉瑞等龙头药企的重点方向。
现有LXR激动剂主要包括天然的氧化性甾醇和叔胺类化合物(T0901317和GW3965)的衍生物,分子量在300-650之间。然而,到目前 为止,这些化合物由于激动活性差、生物活性低而无法走进临床试验,或由于人体毒副作用而止步于临床试验。LXR激动剂治疗高脂血症的尝试仍未成功,而对现有LXR激动剂先导化合物进行结构改造的努力显得力不从心。
中药是中国劳动人民几千年来与疾病作斗争的实践经验总结,从中药资源宝库中寻找先导化合物是我国药物开发的优势之一。野菊花属于菊科菊花种野菊花亚种,野生于山坡草地、田边、路旁等野生地带,因具有抗炎、抗菌以及保护心血管的生物功效而被广泛用于中药处方、茶饮中。1987年于德泉从野菊花中鉴别出野菊花醇的结构(野菊花化学成分的研究.药学学报,1987,22(11):837-840.),2000年M.Yoshikawa鉴别出野菊花抑制NO生成的活性成分倍半萜烯(Chemical&pharmaceutical bulletin,2000,48(5):651-656.),2002年H.Matsuda鉴别出野菊花抑制醛酮还原酶的活性成分黄酮苷衍生物(Chemical&pharmaceutical bulletin,2002,50(7):972-975.),2005年W.Cheng报道了野菊花粗提物的抗炎活性(Journal of ethnopharmacology,2005,101(1-3):334-337.)。至此,野菊花的活性成分还远未被完全开发和利用。
技术实现要素:
本发明针对现有的LXR激动剂因激动活性差、生物活性低而无法走进临床试验,LXR激动剂治疗高脂血症的尝试仍未成功的问题,提供一种有效成分为野菊花提取物且活性优于LXR激动剂GW395的降血脂药物,以及降血脂药物中的有效成分野菊花提取物的制备方法。
为实现上述目的,本发明采用以下技术方案。
一种含野菊花提取物的降血脂药物,包括野菊花提取物和/或药学上可接受的载体,所述野菊花提取物中含有以下结构的倍半萜类化合物:
所述野菊花提取物中所含的倍半萜类化合物的结构式是(化合物1):
所述野菊花提取物中所含的倍半萜类化合物的结构式是(化合物2):
所述野菊花提取物中所含的倍半萜类化合物的结构式是(化合物3):
所述野菊花提取物中所含的倍半萜类化合物的结构式是(化合物4):
一种含有以上所述的倍半萜类化合物的野菊花提取物的制备方法,包括以下步骤:
S1提取:用乙醇浸泡干野菊花,得提取液,浓缩提取液后得到粗提取物浸膏。
S2萃取:向粗提取物浸膏中加入水,搅拌使粗提取物浸膏变成浑浊悬浮液,然后用乙酸乙酯萃取,得萃取液,浓缩萃取液后得到提取物浸膏。
S3柱层析:提取物浸膏用甲醇溶解后吸附于等重量的200-300目的硅胶中,硅胶干燥后得到层析样品,然后用200-300目硅胶对层析样品进行柱层析,依次用石油醚和氯仿进行梯度洗脱,收集氯仿流出液;浓缩氯仿流出液, 得到初层析物。
S4二次柱层析:用200-300目硅胶对初层析物进行柱层析,洗脱液是石油醚与乙酸乙酯组成的复合洗脱液,复合洗脱液中石油醚的体积百分比是20-90%;收集流出液并浓缩,得到野菊花提取物。
优选的,步骤S4中,所述柱层析是用极性不同的复合洗脱液对初层析物进行梯度洗脱,复合洗脱液的极性依次是90:10、80:20、70:30、60:40、50:50、40:60、20:80,以复合洗脱液中石油醚与乙酸乙酯的体积比计;收集流出液并浓缩,得到野菊花提取物。
更优选的,步骤S4中,通过薄层色谱跟踪流出液,依次收集到流份A、流份B、流份C和流份D,分别浓缩各流份,对应得到提取物A、提取物B、提取物C和提取物D;所述提取物A、提取物B、提取物C和提取物D为野菊花提取物。
可分别对提取物A、提取物B、提取物C、提取物D作进一步分离提纯。
提取物A先通过使用MCI柱进行柱层析(梯度洗脱,甲醇/水,体积比为20:80至90:10)可得到提取物A1。提取物A1再通过使用MCI柱进行柱层析(梯度洗脱;石油醚/乙酸乙酯,体积比为90:10至70:30;氯仿/甲醇,体积比为95:5至80:20)可得到提取物A2。提取物A2再通过使用反相硅胶柱进行柱层析(梯度洗脱,甲醇/水,体积比为70:30和60:40)可得到化合物1。
提取物B先通过使用硅胶柱进行柱层析(梯度洗脱,氯仿/乙酸乙酯,体积比为100:0至70:30)可得到提取物B1。提取物B1再依次通过使用Sephadex LH-20(甲醇/氯仿,体积比为50:50)、Rp-18(梯度洗脱,甲醇/水, 体积比为70:30至100:0)、制备HPLC(甲醇/水,体积比为60:40)进行柱层析,可分离得到化合物2。
提取物C先通过MCI柱进行柱层析(梯度洗脱,甲醇/水,体积比为20:80至90:10)可得到提取物C1。提取物C1再依次通过使用硅胶柱(梯度洗脱,氯仿/甲醇,体积比为95:5至70:30)、Sephadex LH-20(甲醇)进行柱层析可得到化合物3。
提取物D先通过使用Sephadex LH-20柱进行柱层析(甲醇)可得到提取物D1,提取物D1通过使用MCI柱进行柱层析(梯度洗脱,甲醇/水,体积比为20:80至100:0)除去色素,得到提取物D1,提取物D1再依次通过使用硅胶(梯度洗脱,氯仿/乙酸乙酯,体积比为90:10至30:70)、Rp-18(梯度洗脱,甲醇/水,体积比为60:40至90:10)、Toyopearl HW-40(甲醇)、硅胶(梯度洗脱,氯仿/甲醇,体积比为100:0至80:20)、Sephadex LH-20(甲醇/氯仿,体积比为50:50)、Rp-8(梯度洗脱,甲醇/水,体积比为60:40至90:10)、制备HPLC(甲醇/水,体积比为65:35)进行柱层析,得到化合物4。
与现有技术相比,本发明的有益效果是:本发明的降血脂药物中的野菊花提取物含有以上公开的倍半萜类化合,这些倍半萜类化合物能显著抑制3T3-L1前脂肪细胞分化过程中甘油三酯和总脂肪的积累,抑制效果优于阳性对照化合物小檗碱;还能明显激动肝X受体α和β(LXRα和LXRβ),激动效果强于现有的LXR激动剂工具分子GW3965;尤其是化合物4,其激动效果明显优于GW3965。此外,这些倍半萜类化合物还能调节对胆固醇代谢至关重要的转录因子SREBP-1c、PPARγ和CEBPδ的mRNA表达量。因此,含有所公开的野菊花提取物的降血脂药物具有良好的降血脂效果,在 治疗高脂血症方面有良好的发展前景。本发明通过组合运用一系列具体的柱层析方法,使从野菊花中分离出的提取物含有所公开的倍半萜类化合物,从而使应用该野菊花提取物时可减少野菊花的其它成分产生的影响。
附图说明
图1为实施例1中从野菊花提取物中分离的化合物1、2、4抑制总脂肪积累的活性数据;
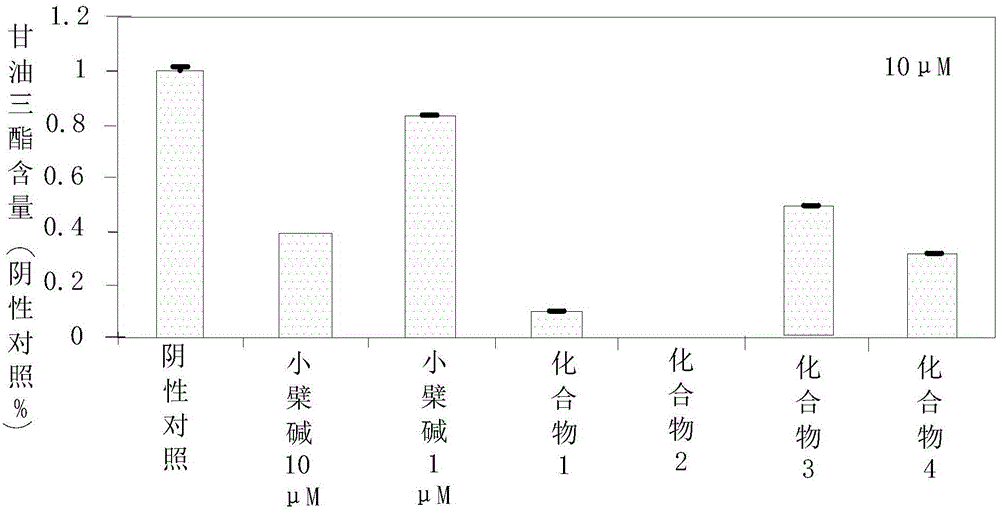
图2为实施例1中从野菊花提取物中分离的化合物1-4抑制甘油三酯积累的活性数据;
图3为实施例1中从野菊花提取物中分离的化合物4激活LXRα和LXRβ的活性数据;
图4为实施例1中从野菊花提取物中分离的化合物3和化合物4上调胆固醇调节过程中关键转录因子的mRNA表达的活性数据(PPARγ);
图5为实施例1中从野菊花提取物中分离的化合物3和化合物4上调胆固醇调节过程中关键转录因子的mRNA表达的活性数据(SREBP-1c);
图6为实施例1中从野菊花提取物中分离的化合物3和化合物4上调胆固醇调节过程中关键转录因子的mRNA表达的活性数据(CEBPδ)。
具体实施方式
为了更充分理解本发明的技术内容,下面结合具体实施例对本发明的技术方案作进一步介绍和说明。
实施例1
本实施例提供一种野菊花提取物的制备方法,以及野菊花提取物中化合物1-4的生理活性分析。
野菊花提取物的制备方法包括以下步骤:
(1)提取
称取7.0Kg干野菊花,在常温常压下用10L的乙醇(95%)浸泡干野菊花24h,然后过滤,收集滤液。重复提取野菊花三次,合并三次提取的滤液,然后将滤液置于水浴中常压浓缩,得到2.0Kg提取物浸膏。
(2)萃取
将提取物浸膏放入5L水中,搅拌使提取物浸膏变成浑浊悬浮液,然后用乙酸乙酯萃取,得萃取液,浓缩萃取液得浓缩物,浓缩物干燥后得到425g萃取物。
(3)柱层析
萃取物用甲醇溶解后吸附于等重量的200-300目的硅胶中,硅胶在室温下干燥后得到层析样品,然后用2.0Kg的200-300目硅胶对层析样品进行柱层析,依次用石油醚、氯仿进行梯度洗脱,收集氯仿流出液;浓缩氯仿流出液,得到120g初层析物。
(4)二次层析
用200-300目硅胶对初层析物进行柱层析,用复合洗脱液进行梯度洗脱,复合洗脱液由石油醚和乙酸乙酯组成,所用的系列极性的复合洗脱液如下:90:10、80:20、70:30、60:40、50:50、40:60、20:80(石油醚与乙酸乙酯的体积比)。通过薄层色谱(TLC)跟踪流出液,根据TLC反映的层析情况,合并溶解有相同极性(在该TLC层析条件下)的物质的流出液,从而依次收集到极性由小到大的流份A、流份B、流份C和流份D。分别浓缩各流份,对应得到18g提取物A、29g提取物B、19g提取物C和15g提取物D。提 取物A、提取物B、提取物C和提取物D组成野菊花提取物。
分别对提取物A、提取物B、提取物C、提取物D作进一步分离提纯。经过进一步分离提纯,可得到如下结构通式的倍半萜类化合物:
具体的分离提纯步骤如下(5)-(8)所述。
(5)分离提纯化合物1
提取物A通过使用MCI柱进行柱层析,用甲醇/水进行梯度洗脱,所用的系列极性的甲醇/水洗脱液如下:20:80、30:70、40:60、50:50、60:40、70:30、80:20、90:10(甲醇与水的体积比),收集流出液,浓缩流出液后得到提取物A1。
提取物A1再通过使用MCI柱进行柱层析,依次用石油醚/乙酸乙酯、氯仿/甲醇进行梯度洗脱,所用的系列极性的石油醚/乙酸乙酯洗脱液如下:90:10、80:20、70:30(石油醚与乙酸乙酯的体积比),所用的系列极性的氯仿/甲醇洗脱液如下:95:5、80:20(氯仿与甲醇的体积比),收集流出液,浓缩流出液后得到提取物A2。
提取物A2再通过使用反相硅胶柱进行柱层析,用甲醇/水进行梯度洗脱,所用的甲醇/水洗脱液的体积比为70:30和60:40,收集流出液,浓缩流出液后可得到化合物1,化合物1为白色粉末。
分别测化合物1的质谱、核磁共振氢谱、核磁共振碳谱,由测试所得数 据可知化合物1的分子量是592.3,分子式是C35H44O8。化合物1的质谱、核磁共振氢谱、核磁共振碳谱与文献1中的化合物2的相应图谱一致,因此化合物1的结构式是:(文献1:Chrysanolide A,an unprecedented sesquiterpenoid trimer from the flowers of Chrysanthemum indicum L.RSC Advances,2013,3(26):10168.)
具体的测试数据如下:
HRESI-MS m/z 591.2936[M-H]-。
1H-NMR(400MHz,CDCl3)δ:6.18(1H,dd,J=5.84,1.00Hz,H-3`),6.16(1H,dd,J=5.84,1.00Hz,H-3``),6.03(1H,d,J=3.64Hz,13`-Ha),5.30(1H,d,J=3.28Hz,13`-Hb),5.88(1H,d,J=5.48Hz,H-2`),5.55(1H,ddd,J=3.68,4.24,2.04Hz,H-8),5.49(1H,m,J=1.36Hz,H-3),4.08(1H,t,J=9.72Hz,H-6`),3.96(1H,t,J=9.80Hz,H-6),3.54(1H,t,J=8.65Hz,H-7),2.95(1H,dd,J=6.20,3.04Hz,H-7`),2.74(1H,m,J=7.96Hz,H-5),2.56(1H,dt,J=10.68,7.60Hz,H-1),2.36(1Hd,J=12.12Hz,H-13a),1.49(1H,s,H-13b),2.26(1H,d,J=4.54Hz,H-9a),1.97(1H,d,H-9b),2.18(1H,m,H-8`a),1.46(1H,m,H-8`b),2.24-2.04(2H,m,H-2),1.95(1H,m,H-5`),1.80(2H,m,H-9`),1.97(3H,d,J=7.36Hz,3``-CH3),1.89(3H,s,2``-CH3-),1.89(3H,s,H-15),1.46(3H,s,H-14`),1.29(3H,s,H-15`),1.21(3H,s,H-14)。
13C-NMR(101MHz,CDCl3)δ:54.6(C-1),33.5(C-2),125.8(C-3),144.8(C-4),54.9(C-5),79.0(C-6),47.8(C-7),70.1(C-8),38.3(C-9),73.6(C-10),59.0(C-11),178.3(C-12),37.4(C-13),33.7(C-14),18.5(C-15),64.8(C-1`),133.8(C-2`),140.6(C-3`),57.4(C-4`),65.9(C-5`),79.4(C-6`),43.1(C-7`),23.7(C-8`),34.9(C-9`),72.9(C-10`),141.3(C-11`),170.2(C-12`),118.4(C-13`),15.5(C-14`),29.9(C-15`),166.5(C-1``),126.6(C-2``),143.4(C-3``),16.3(C-4``),20.6(C-5``)。
(6)分离提纯化合物2
提取物B先通过使用300g的硅胶进行柱层析,用氯仿/乙酸乙酯进行梯度洗脱,所用系列极性的氯仿/乙酸乙酯洗脱液如下:100:0、90:10、80:20、70:30(氯仿与乙酸乙酯的体积比),收集流出液,浓缩流出液后得到提取物B1。提取物B1再依次通过使用Sephadex LH-20(甲醇/氯仿,体积比为50:50)、Rp-18(梯度洗脱,所用系列极性的甲醇/水洗脱液的体积比为:70:30、80:20、90:10、100:0)、制备HPLC(甲醇/水,体积比为60:40)进行柱层析,可分离得到化合物2,化合物2为白色固体。
分别测化合物2的质谱、核磁共振氢谱、核磁共振碳谱,由测试所得数据可知化合物2的分子量是306,分子式是C17H22O5。化合物2的质谱、核磁共振氢谱、核磁共振碳谱与文献2中的化合物12的相应图谱一致,因此化合物2的结构式是:(文献2:Cumambrin A in Chrysanthemum boreale Makino Preparation,X-ray Crystal Structure and 13C-and 1H-NMR Study of Cumambrin A[J]Kor.J.Pharmacogn,1996,27:207-211.)
具体的测试数据如下:
EI-MSm/z=307.2([M+H]+)。
1H-NMR(400MHz,CDCl3)δ:6.18(1H,d,J=3.44Hz,H-13),5.50(1H,d,J=2.96Hz,H-13),5.50(1H,d,J=2.96Hz,H-3),5.16(1H,ddd,J=7.04,5.72,1.12Hz,H-8),3.99(1H,m,H-6),3.89(1H,tt,J=9.36,3.12Hz,H-7),2.76(1H,dd,J=10.84,4.12Hz,H-5),2.58(1H,dd,J=18.44,8.20Hz,H-1),2.31(1H,dd,J=16.68,5.80Hz,H-9),1.84(1H,d,J=16.68Hz,H-9),2.27-2.18(1H,m,H-2),2.13-2.03(1H,m,H-2),1.90(3H,s,H-14),1.24(3H,s,H-15),2.16(3H,s,OAc)。
13C-NMR(101MHz,CDCl3)δ:54.4(C-1),33.7(C-2),125.6(C-3),138.6(C-4),54.5(C-5),80.4(C-6),46.6(C-7),73.9(C-8),39.0(C-9),73.6(C-10),143.9(C-11),169.6(C-12),121.5(C-13),33.7(C-14),18.0(C-15),170.3(C-16),21.6(C-17)。
(7)分离提纯化合物3
提取物C先通过MCI柱进行柱层析,用甲醇/水进行梯度洗脱,所用系列极性的甲醇/水洗脱液如下:20:80、30:70、40:60、50:50、60:40、70:30、80:20、90:10(甲醇与水的体积比),收集流出液,浓缩流出液后得到提取物C1。提取物C1再依次通过使用硅胶柱(梯度洗脱,所用系列极性的氯仿/甲醇洗脱液的体积比为:95:5、85:15、70:30)、Sephadex LH-20(甲醇)进 行柱层析可得到化合物3,化合物3为白色固体。
分别测化合物3的质谱、核磁共振氢谱、核磁共振碳谱,由测试所得数据可知化合物3的分子量是246.1,分子式是C15H18O3。化合物3的质谱、核磁共振氢谱、核磁共振碳谱与文献3中的化合物1的相应图谱一致,因此化合物3的结构式是:(文献3:Conformational analysis of achillinand leukodin[J]J Nat Prod,1988,51(2):22-228.)
具体的测试数据如下:
EI-MSm/z=247.1([M+H]+)。
1H-NMR(400MHz,CDCl3)δ:6.14(1H,q,J=1.32,H-3),3.59(1H,td,J=11.20,10.08,1.36Hz,H-6),3.39(1H,d,J=10.04Hz,H-5),2.41(3H,s,H-14),2.41(1H,m,H-9a),2.24(1H,ddd,J=13.64,6.72,1.52Hz,H-9b),2.33(1H,dd,J=3.88,1.44Hz,H-11),2.27(3H,s,H-15),1.97(1H,m,H-8a),1.94(1H,m,J=12.44,2.72Hz,H-7),1.34(1H,dd,J=12.12,1.64Hz,H-8b),1.25(3H,dd,J=6.84,1.60Hz,H-13)。
13C-NMR(101MHz,CDCl3)δ:196.0(C-2),177.6(C-12),170.0(C-4),152.2(C-10),135.6(C-3),132.0(C-1),84.3(C-6),56.5(C-7),52.7(C-5),41.2(C-11),37.7(C-9),26.1(C-8),21.7(C-14),19.9(C-15),12.4(C-13)。
(8)分离提纯化合物4
提取物D先通过使用Sephadex LH-20柱进行柱层析(甲醇),收集流出 液,浓缩流出液后得到提取物D1。
提取物D1通过使用MCI柱进行柱层析,以甲醇/水进行梯度洗脱以除去色素,所用系列极性的甲醇/水洗脱液如下:20:80、30:70、40:60、50:50、60:40、70:30、80:20、90:10、100:0(甲醇与水的体积比),收集流出液,浓缩流出液后得到提取物D2。
提取物D2再依次通过使用硅胶(梯度洗脱,所用系列极性的氯仿/乙酸乙酯洗脱液的体积比为:90:10至30:70)、Rp-18(梯度洗脱,所用系列极性的甲醇/水洗脱液的体积比为60:40至90:10)、Toyopearl HW-40(甲醇)、硅胶(梯度洗脱,所用系列极性的氯仿/甲醇洗脱液的体积比为100:0、90:10、80:20)、Sephadex LH-20(甲醇/氯仿,体积比为50:50)、Rp-8(梯度洗脱,所用系列极性的甲醇/水洗脱液的体积比为60:40、70:30、80:20、90:10)、制备HPLC(甲醇/水,体积比为65:35)进行柱层析,得到化合物4,化合物4为白色固体。
分别测化合物4的质谱、核磁共振氢谱、核磁共振碳谱,由测试所得数据可知化合物4的分子量是798.4,分子式是C47H58O11。化合物4的质谱、核磁共振氢谱、核磁共振碳谱与文献1中的化合物3的相应图谱一致,因此化合物4的结构式是:(文献1:Chrysanolide A,an unprecedented sesquiterpenoid trimer from the flowers of Chrysanthemum indicum L.RSC Advances,2013,3(26):10168.)
具体的测试数据如下:
EI-MS m/z=799.40([M+H]+)。
1H-NMR(400MHz,CDCl3)δ:6.25(1H,d,J=5.36Hz,H-3`),6.06(1H,d,J=3.16Hz,H-3``),5.99(1H,d,J=5.24Hz,H-13``a),5.32(1H,d,J=5.60Hz,H-13``b),5.88(1H,d,J=5.40Hz,H-2``),5.85(1H,d,J=5.48Hz,H-2`),5.46(1H,s,H-8),5.34(1H,m,J=2.76Hz,H-3),4.14(1H,t,J=9.60Hz,H-6`),4.00(1H,t,J=9.60Hz,H-6``),3.88(1H,t,J=9.76Hz,H-6),3.48(1H,dd,J=18.28,9.08Hz,H-7),3.18(1H,d,J=18.84,9.56Hz,H-7`),2.81(1H,m,J=17.12,9.20Hz,H-5``),2.73(1H,m,J=17.68,8.20Hz,H-5),2.53(1H,dd,J=17.68,7.88Hz,H-5`),2.38(1H,d,J=11.80Hz,H-7``),2.11(2H,s,H-9),1.84(3H,s,H-OAc),1.84(3H,s,H-15),1.37(3H,s,H-14`),1.32(3H,s,H-15``),1.29(3H,s,H-15`),1.25(3H,s,H-14``),1.19(3H,s,H-14)。
13C-NMR(101MH,CDCl3)δ:179.4(C-12),178.7(C-12``),170.9(C-OAc),170.6(C-12`),144.1(C-4),141.3(C-11``),141.1(C-3`),139.9(C-3``),134.3(C-2`),133.6(C-2``),125.5(C-3),118.7(C-13``),80.1(C-6`),78.8(C-6``),77.4(C-6),73.4(C-10),73.1(C-10`),71.8(C-10``),70.9(C-8),66.3(C-5`),65.8(C-5``),64.5(C-1`),64.3(C-1``),60.4(C-11),57.7(C-11`),56.8(C-4`), 56.7(C-4``),54.2(C-1),54.0(C-5),47.1(C-7),43.9(C-7`),43.5(C-7``),38.4(C-9),36.8(C-13),36.8(C-13`),35.5(C-9`),35.0(C-9``),33.6(C-14),33.3(C-2),29.8(C-14`),29.4(C-14``),23.5(C-8`),22.3(C-8``),22.2(C-OAc),18.2(C-15),15.5(C-15`),15.3(C-15`)。
由以上对提取物A、提取物B、提取物C、提取物D的进一步分离提纯的结果可知,本实施例的野菊花提取物中含有上述的化合物1、化合物2、化合物3和化合物4。
化合物1-4的药效相关实验如下。
试验1:化合物1-4抑制甘油三酯积累、总脂肪积累的活性测定。
(1)细胞培养
3T3-L1(小鼠胚胎成纤维细胞,前脂肪细胞)采用完全培养基(DMEM培养基中添加10%胎牛血清、100U/mL青霉素、100μg/mL链霉素)在37℃、体积分数5%CO2的培养箱中培养。
(2)药物干预
待测化合物以DMSO配成10mM母液,测试时稀释成不同浓度工作液。为校正母液溶剂DMSO影响,空白对照添加0.1%DMSO。
3T3-L1接种于48孔板中培养至完全接触(第0天),将培养基换成分化培养基I或加入指定浓度待测化合物的分化培养基I培养3天(第3天)。分化培养基I配方:完全培养基中添加2μg/mL胰岛素,100ng/mL地塞米松,0.5mM 3-异丁基-1-甲基黄嘌呤和10ng/mL生物素。
分化培养基和待测化合物作用第3天,将培养基更换成分化培养基II或加入指定浓度待测化合物的分化培养基II继续培养3天(第6天)。分化 培养基II配方:完全培养基中添加2μg/mL胰岛素。
第6天,收获细胞进行甘油三酯或油红O染色分析。
(3)甘油三酯测试
收获处理后的3T3-L1细胞,冰PBS(0.2M NaCl,10mM Na2HPO4,3mM KCl,2mM KH2PO4,pH 7.4)洗涤两次,超声破碎细胞,使用甘油三酯试剂盒和多功能酶标仪检测546nm处吸光度,从而计算细胞破碎液中甘油三酯含量。结果以空白对照细胞甘油三酯含量百分比的形式表示。
(4)油红O染色
3T3-L1细胞以10%(V/V)福尔马林溶液室温固定1小时,以新制备的油红O工作溶液60℃染色30分钟,蒸馏水洗两次,则总脂肪被油红O染成红色,以装有CCD数码相机的倒置显微镜观察并拍照。拍摄过程中放大倍数、光强度不变。
(5)结果处理
根据甘油三酯试剂盒和多功能酶标仪检测546nm处吸光度,减去空白对照组的本底吸光度值,比上阴性对照组细胞吸光度值,结果以阴性对照组细胞甘油三酯含量百分比的形式表示。所有数据均表示为平均值±标准方差。
测试结果如图1和图2所示。由图2可知,化合物1-4均能不同程度降低3T3-L1细胞内甘油三酯积累,其中CI-15处理6天后的3T3-L1细胞内几乎没有油滴积累,甘油三酯含量为零。由图1可知,降甘油三酯效果最好的化合物1、化合物2和化合物4明显降低3T3-L1细胞内总脂肪滴含量。
试验2:化合物4激动LXR的活性测定。
(1)细胞培养
HEK293T(人源胚胎肾细胞)采用完全培养基(DMEM培养基中添加10%胎牛血清、100U/mL青霉素、100μg/mL链霉素)在37℃、体积分数5%CO2的培养箱中培养。
(2)药物干预
HEK293T细胞以每孔2×104个细胞的密度平铺在96孔板中,培养至细胞生长到约90%覆盖率,按照脂质体Lipofectamine TM2000试剂说明书,将6.5μg pGL3/(DR-4)-c-fos-FF-luc(萤火虫荧光素酶报告基因)质粒、1.3μg pSG5/hLXRα(或pSG5/hLXRβ,肝X受体α或β)质粒、1.3μg pSG5/hRXRα(视黄酸X受体)质粒和0.13μgpCMV/Renilla-luc(海肾荧光素酶报告基因)质粒与10μLLipofectamineTM 2000脂质体试剂充分混匀,室温静置30分钟,将脂质体-DNA溶液加入到96孔板中,每孔5μL。
培养5小时后换成完全培养基,继续培养5小时。
加入待测化合物母液使工作浓度为10μM,与细胞共培养20小时。使用多功能酶标仪工作站Flex Station 3检测萤火虫荧光素酶活性和海肾荧光素酶活性。
(3)结果处理
结果分析用海肾荧光素酶活性校正萤火虫荧光素酶活性,故校正酶活性=萤火虫荧光素酶活性值/海肾荧光素酶活性值。相对荧光素酶活性值=实验组校正酶活性值/阴性对照组校正酶活性值。
测试结果如图3所示,化合物4激动LXRα的活性强于现有的LXRα激动剂工具分子GW3965,化合物4激动LXRβ的活性与现有LXRβ激动剂工具分子GW3965相当。
试验3:化合物3和化合物4上调LXR通路上关键转录因子的mRNA表达量的活性测定。
(1)细胞培养
3T3-L1(小鼠胚胎成纤维细胞,前脂肪细胞)采用完全培养基(DMEM培养基中添加10%胎牛血清、100U/mL青霉素、100μg/mL链霉素)在37℃、体积分数5%CO2的培养箱中培养。
(2)药物干预
待测化合物以DMSO配成10mM母液,测试时稀释成不同浓度工作液。为校正母液溶剂DMSO影响,空白对照添加0.1%DMSO。
3T3-L1接种于48孔板中培养至完全接触(第0天),将培养基换成分化培养基I或加入指定浓度待测化合物的分化培养基I培养24小时(第1天)。收获细胞进行荧光定量PCR扩增。
(3)荧光定量PCR
使用RNAiso Plus提取3T3-L1细胞的总RNA,通过Oligo dT18将1μg总RNA逆转录为cDNA,通过定量PCR技术,使用Toyobo公司ThunderBird SYBR qPCR Mix试剂按照说明书对引物进行PCR扩增。每组样品3个复孔,以保证实验数据的有效性。使用β-actin为内参,检测目的基因相对于内参基因的表达变化来定量。采用2-ΔΔCt方法分析数据(即实验组目的基因相对于对照组的表达量的变化倍数)。
所用引物为:
β-actin(sense:5’-TGGAATCCTGTGGCATCCATGAAA-3’;antisense:5’-TAAAACGCAGCTCAGTAACAGTCC-3’)、
SREBP-1c(sense:5’-CAGCTCAGAGCCGTGGTGA-3’;antisense:5’-TGTGTGCACTTCGTAGGGTC-3’)、
PPAR-γ(sense:5’-TGCTGTTATGGGTGAAACTCTG-3’;antisense:5’-GAAATCAACTGTGGTAAAGGGC-3’)、
CEBPδ(sense:5’-AGCCCAACTTGGACGCCAG-3’;antisense:5’-TCGTCGTACATGGCAGGAGT-3’)。
(4)结果处理
检测目的基因相对于内参基因的表达变化来定量。采用2-ΔΔCt方法分析数据(即实验组目的基因相对于对照组的表达量的变化倍数)。
测试结果如图4-6所示,10μM浓度下的化合物4能显著增加3T3-L1细胞内SREBP-1c的mRNA表达量(化合物4处理过的细胞SREBP-1c的mRNA表达量为空白对照组的8倍),同时增加PPARγ和CEBPδ的mRNA表达量(均为3倍)。化合物3能显著增加PPARγ和CEBPδ的mRNA表达量。
实施例2
本实施例提供一种含野菊花提取物的降血脂药物,含有实施例1中所述的野菊花提取物和/或药学上可接受的载体,有效成分是野菊花提取物中所含的倍半萜类化合物,即实施例1中所述的化合物1、化合物2、化合物3和化合物4。
由实施例1中的测试结果可知,本实施例的含野菊花提取物的降血脂药物具有减少3T3-L1前脂肪细胞分化过程中油滴积累及甘油三酯积累的效果,因此具有良好的降血脂效果,在治疗高脂血症方面有良好的发展前景。
实施例3
本实施例提供一种含野菊花提取物的降血脂药物,含有实施例1中所述的化合物1和/或药学上可接受的载体,有效成分是化合物1。
由实施例1中的测试结果可知,本实施例的含野菊花提取物的降血脂药物具有减少3T3-L1前脂肪细胞分化过程中油滴积累及甘油三酯积累的效果,因此具有良好的降血脂效果,在治疗高脂血症方面有良好的发展前景。
实施例4
本实施例提供一种含野菊花提取物的降血脂药物,含有实施例1中所述的化合物2和/或药学上可接受的载体,有效成分是化合物2。
由实施例1中的测试结果可知,本实施例的含野菊花提取物的降血脂药物具有减少3T3-L1前脂肪细胞分化过程中油滴积累及甘油三酯积累的效果,因此具有良好的降血脂效果,在治疗高脂血症方面有良好的发展前景。
实施例5
本实施例提供一种含野菊花提取物的降血脂药物,含有实施例1中所述的化合物3和/或药学上可接受的载体,有效成分是化合物3。
由实施例1中的测试结果可知,本实施例的含野菊花提取物的降血脂药物具有减少3T3-L1前脂肪细胞分化过程中油滴积累及甘油三酯积累的效果,及提高PPARγ、SREBP-1c、CEBPδ的mRNA表达量的效果,因此具有良好的降血脂效果,在治疗高脂血症方面有良好的发展前景。
实施例6
本实施例提供一种含野菊花提取物的降血脂药物,含有实施例1中所述的化合物4和/或药学上可接受的载体,有效成分是化合物4。
由实施例1中的测试结果可知,本实施例的含野菊花提取物的降血脂药物具有减少3T3-L1前脂肪细胞分化过程中油滴积累及甘油三酯积累的效果,及提高PPARγ、SREBP-1c、CEBPδ的mRNA表达量的效果,因此具有良好的降血脂效果,在治疗高脂血症方面有良好的发展前景。
以上所述仅以实施例来进一步说明本发明的技术内容,以便于读者更容易理解,但不代表本发明的实施方式仅限于此,任何依本发明所做的技术延伸或再创造,均受本发明的保护。
- 还没有人留言评论。精彩留言会获得点赞!