缓释性医药组合物及其制备方法与流程
本发明涉及一种医药组合物,尤其涉及一种具有缓释性效果的医药组合物;本发明还涉及一种前述医药组合物的制备方法,尤其涉及一种无需针对氧化烯聚合物进行造粒化或整粒化处理即可获得具有缓释性效果的医药组合物的制备方法。
背景技术:
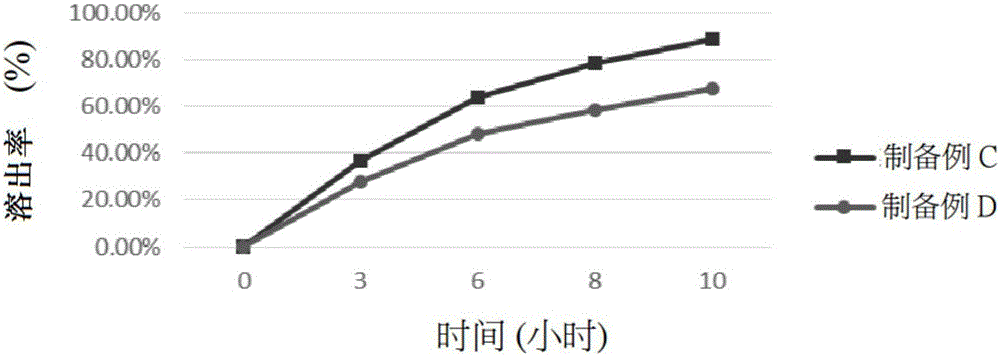
:口服持续释放剂型的药物释放常会受胃肠道食物的影响,使得制剂形态难以维持、释出速率不稳定,且常因制剂滞留消化道能力不佳,导致药物控释效果多集中于消化道上部(胃及小肠)的缺点,缩短药物吸收时间。中国台湾发明专利公告第393320号公开了一种口服用水胶体徐放性(缓释性)医药锭剂,其是以聚乙烯氧化物(polyethyleneoxide,PEO)作为该高分子物质水胶体形成基质,再搭配适当亲水基质[例如聚乙二醇(Polyethyleneglycol,PEG)]时,锭剂滞留于消化管上部的胃及小肠时几乎具有完全凝胶化能力,由于PEO凝胶化后胶体可塑性佳,且制剂表面侵蚀速率适中、不易受食物影响、能维持稳定的药物释出,因此当制剂向消化管下部移行时,仍能于结肠持续放出药物。在该案的制备方法中,多将药物、亲水性基质及水胶体形成基质等组份直接混合后进行打锭(直接压锭法),或将这些组份直接混合进行干式/湿式造粒后再打锭;在部分实施例中,甚至将活性成分连同部分亲水性基质及水胶体形成基质先溶于醇系溶剂或其与水的混合溶液后,进行喷雾干燥形成喷干品,最后将该喷干品与剩余的亲水性基质及水胶体形成基质混合打锭。前述所使用的直接压锭法和干式/湿式造粒法存在混合、造粒后粉体流动性差而不适合压锭的问题,且在制备活性成分含量低的医药组合物时,更会有明显的药物分布均匀性不佳的缺点;而先制得喷干品再混合压锭的工艺步骤又过于繁杂耗时。中国台湾发明专利公告第I307632号进一步针对中国台湾发明专利公告第393320号所提出的口服用水胶体徐放性(缓释性)医药锭剂及其工艺进行改良,发明第I307632号同样以PEO作为水胶体形成基质,为了改善中国台湾发明专利第393320号中颗粒大小不一、药物分布均一度不佳及混合造粒后粉体流动性差的问题, 其将药物与亲水性基质溶于水中,形成含药/亲水性基质的悬浮液,接着将该含药悬浮液对PEO进行喷雾干燥的整粒处理,此时的亲水性基质作为整粒剂。然而,虽然发明第I307632号的喷雾整粒处理,改善了药物分布均匀度不佳及造粒过后PEO粉体流动性差的缺点,但是,由于PEO本身物化性质的关系,其造粒/整粒条件难以掌控,研发过程需经大量尝试方能找到适合的造粒/整粒条件设定,此亦代表着当处于工业量产阶段时,针对PEO的造粒/整粒,其工艺势必需要较精细的调控。前述缺点从第I307632号发明的喷雾整粒工艺需调控众多因子可观察出:含药悬浮液的量、喷雾温度/时间、干燥温度/时间、振动频率等不仅工艺烦杂,加上其所使用的喷雾整粒装置(如:流动层造粒机)设备成本高,所需空间大,PEO的高黏度亦导致工艺后设备清洁更是不易,整体而言仍不利于工业大量制备。欧洲发明专利第1205190号(EP1205190)公开了以PEO作为缓释基质的制剂于光照下保存后,由PEO制成的基质溶解加快,造成药物从基质中释出的速率亦随之变快,进而影响药物释放特性。另外,目前已知聚氧化乙烯(PEO)由于分链上存在大量的醚键,因此很容易受到氧的攻击而发生降解,而某些重金属离子、氧化剂和紫外线都会加速其氧化降解的进程。由此可知,以PEO作为缓释基质的医药组合物,虽然能达到不错的控制释放效果,但由于PEO价格昂贵,且其易氧化和不安定的物理特性,为制剂稳定性及保存增添不少变数。技术实现要素:鉴于现有技术混合造粒后粉体流动性差、颗粒大小不一、药物分布均匀性不佳、制备过程繁杂耗时以及制备工艺后设备清洁不易的缺点,本发明的目的在于提供一种缓释性医药组合物的制备方法,其通过将药物与纤维素衍生物以低烷基醇溶剂进行造粒,进而氧化烯聚合物无需历经造粒或整粒,即可达到较佳均一度的效果。为达到上述目的,本发明提供一种缓释性医药组合物的制备方法,将活性成分与浓度不小于80%(w/v)的低烷基醇类溶剂及纤维素衍生物进行混合造粒,以获得颗粒性粉体;将颗粒性粉体进行干燥,移除低烷基醇类溶剂;将已干燥的颗粒性粉体与氧化烯聚合物(polyalkyleneoxide)进行混合,形成均匀的混合物;将润滑剂加入混合物并进行压缩制锭,获得缓释性医药组合物。较佳的,所述的该活性成分包含良性前列腺肥大(benignprostatichyperplasia,BPH)治疗剂、泌尿道疾病治疗剂、抗高血压剂、精神疾病治疗剂、消炎剂、解热剂、镇痛剂、代谢疾病治疗剂、心血管疾病治疗剂、免疫疾病治疗剂、抗肿瘤剂、其他以徐放化(缓释)为目的的药物或其组合。较佳的,所述的该活性成份包括,但不限于他姆斯罗辛(Tamsulosin)、米拉贝隆(Mirabegron)、喹硫平(Quetiapine)或其医药学上所容许的盐。较佳的,所述的将活性成分与浓度不小于80%(w/v)的低烷基醇类溶剂及纤维素衍生物混合造粒的步骤中,是将活性成分先溶于浓度不小于80%(w/v)的低烷基醇类溶剂后,再与纤维素衍生物混合造粒。较佳的,所述的将活性成分与浓度不小于80%(w/v)的低烷基醇类溶剂及纤维素衍生物混合造粒的步骤中,活性成分是先与纤维素衍生物混合后,再与浓度不小于80%(w/v)的低烷基醇类溶剂混合造粒。较佳的,所述的将润滑剂加入混合物并进行压缩制锭的步骤后还包括将已压缩的锭剂涂覆膜衣。依据本发明,所述的“浓度不小于80%(w/v)的低烷基醇类溶剂”是指低烷基醇类溶剂的浓度是在80%(w/v)至100%(w/v)之间;较佳的,低烷基醇类溶剂的浓度是在90%(w/v)至99.5%(w/v)之间。此外,所述的低烷基醇类溶剂包括,但不限于甲醇、乙醇、丙醇、异丙醇、丁醇或其组合。于本发明制备方法中,其中将活性成分与浓度不小于80%(w/v)的低烷基醇类溶剂及纤维素衍生物混合造粒的步骤中,所使用低烷基醇类溶剂的量需依据欲造粒的粉体量去作适当调整;较佳的,使用的低烷基醇类溶剂与欲造粒的粉体重量比约在1:100至100:1之间;更佳的,低烷基醇类溶剂与欲造粒的粉体重量比约在1:10至10:1之间;最佳的,低烷基醇类溶剂与欲造粒的粉体重量比约在1:5至5:1之间。较佳的,所述的该纤维素衍生物包括,但不限于羟丙基纤维素(hydroxypropylcellulose,HPC)、羟丙基甲基纤维素(hypromellose,HPMC)、甲基纤维素(methylcellulose,MC)、羟乙基纤维素(hydroxyethylcellulose,HEC)、乙基纤维素(ethylcellulose,EC)或其组合。较佳的,所述的该纤维素衍生物是黏度介于50mPa·s至100,000mPa·s的羟丙基 甲基纤维素,黏度测试条件为1%w/v、20℃。较佳的,所述的该氧化烯聚合物包括,但不限于聚氧化乙烯(polyethyleneoxide,PEO)、聚氧化丙烯(polypropyleneoxide,PPO)、氧化乙烯氧化丙烯共聚物(poly(propyleneoxide-ethyleneoxide))或其组合。较佳的,所述的该氧化烯聚合物是分子量介于500,000Da至7,000,000Da的聚氧化乙烯。较佳的,所述的将活性成分与浓度不小于80%(w/v)的低烷基醇类溶剂及纤维素衍生物混合造粒的步骤中,还可添加助溶剂。更佳的,所述的助溶剂包括,但不限于聚乙二醇(polyethyleneglycol,PEG)、多库酯钠(docusatesodium)、十二烷基硫酸钠(sodiumlaurylsulfate)、中链三酸甘油酯(mediumchaintriglyceride)、聚乙烯吡咯烷酮(polyvinylpyrrolidone,PVP)、木糖醇(xylitol)、D-果糖(D-fructose)、尿素(urea)、乙酰胺(acetylamine)、聚羟40氢化蓖麻油(polyoxyl40hydrogenatedcastoroil)、聚羟60氢化蓖麻油(polyoxyl60hydrogenatedcastoroil)、泊洛沙姆407(poloxamer407)、安息香酸(benzoicacid)、安息香酸钠(sodiumbenzoate)、水杨酸(salicylicacid)、水杨酸钠(sodiumsalicylate)或其组合。本发明提供一种如前述的制备方法所制得的缓释性医药组合物。本发明更提供一种缓释性医药组合物,其是由至少:活性成分;黏度介于50mPa·s至100,000mPa·s的纤维素衍生物,其是难溶于浓度不小于80%(w/v)的低烷基醇类溶剂;重量百分含量少于5%的浓度不小于80%(w/v)的低烷基醇类溶剂,其中低烷基醇类溶剂是用于与活性成份及纤维素醚衍生物混合造粒,以使造粒物形成不结块的粉体;平均分子量500,000Da以上的氧化烯聚合物;以及,润滑剂;所组成的组分均匀混合打锭而成。较佳的,所述的缓释性医药组合物,其是以总重量(wt%)为基准,其包括:0.01wt%至25wt%的活性成分、1wt%至50wt%且黏度介于50mPa·s至100,000mPa·s的纤维素衍生物、0wt%至5wt%且浓度不小于80%(w/v)的低烷基醇类溶剂、30wt%至95wt%且平均分子量500,000Da以上的氧化烯聚合物以及0.1wt%至5wt%的润滑剂;其中纤维素醚衍生物是难溶于浓度不小于80%(v/v)的低烷基醇类溶剂;其中低烷基醇类溶剂是用于与活性成份及纤维素醚衍生物混合造粒,以使造粒物形成不结块的粉体。更佳的,所述的缓释性医药组合物中,其浓度不小于80%(w/v)的低烷基醇类溶剂的含量是在0wt%至2wt%之间;最佳的,其含量是在0wt%至1wt%之间。较佳的,所述的氧化烯聚合物是指未经造粒化或整粒化处理的氧化烯聚合物。较佳的,所述的缓释性医药组合物还包括含量为0wt%至10wt%的助溶剂。较佳的,所述的纤维素衍生物与氧化烯聚合物于医药组合物中的含量比例介于1:100至100:1之间;更佳的,其含量比例介于1:10至10:1之间;最佳的,其含量比例介于1:5至5:1之间。本发明相较于现有技术的优点在于:现有技术将活性成分溶于醇类溶剂,若需和HPMC进行造粒步骤时,由于HPMC已知难溶于高浓度的醇类溶剂,因此大多会选择使用喷雾造粒的方式进行造粒,而不会如本发明直接将含药醇类溶液加入HPMC粉体进行混合造粒,本发明所述的制备方法可节省喷雾造粒所需的时间、空间及金钱成本。此外,本发明通过特定黏度范围的纤维素衍生物,使所得的缓释性医药组合物中具有较佳均一度的优点。附图说明图1为本发明工艺方法所得的制备例A、B、E及F锭剂分别于0小时、3小时、6小时、8小时、10小时的溶出率。图2为本发明工艺方法所得的制备例C及D锭剂分别于0小时、3小时、6小时、8小时、10小时的溶出率。图3为本发明工艺方法所得的制备例A及C锭剂分别于0小时、3小时、6小时、8小时、10小时的溶出率。图4为本发明的制备例A锭剂历经0个月或3个月后置放后于0小时至24小时的溶出率。图5为本发明的制备例E锭剂历经0个月或3个月后置放后于0小时至24小时的溶出率。具体实施方式以下配合图式及本发明的较佳实施例,进一步阐述本发明为达到预定发明目的所采取的技术手段。制备例1,000单位缓释制剂的制备表1配方组成[单位:克(g)]如表1中的所示,将他姆斯罗辛盐酸(TamsulosinHCl)及聚乙二醇(PEG6000)溶于适量的95%乙醇中,调制成含药乙醇溶液。将该含药乙醇溶液分别与黏度为50mPa·s、4,000mPa·s或100,000mPa·s的羟丙基甲基纤维素,依序为Hypromellose50(厂商:Shin-EtsuChemicalCo.,Ltd.,型号:Metolose60SHGrades60SH-50)、Hypromellose4000(厂商:TheDowChemicalCompany,型号:MethocelK4mPremiumHydroxypropylMethylcelluloseID34693)及Hypromellose100000(厂商:Shin-EtsuChemicalCo.,Ltd.,型号:Metolose90SHGrades90SH-100000SR)混合,并以造粒机造粒,制得颗粒性粉体。将前述颗粒性粉体于65℃烘箱中进行烘干,移除乙醇,随后将干燥的颗粒性粉体通过适当的筛网。将PolyOXWSR303(厂商:TheDowChemicalCompany,型号:POLYOXWSR303-NFGrade)、硬脂酸镁以及过筛完的干燥颗粒性粉体均匀混合形成最终混合物。再将最终混合物于打锭机中压制成每锭为195mg或245mg的裸锭锭剂(半成品)。在较佳的实施例中,可视情况将获得的裸锭锭剂进行膜衣涂覆。实施例1缓释剂溶出速率测试将制备例A、B、C、D、E及F打锭后所获得的锭剂于500毫升0.1N的氯化氢溶液中,以溶出试验机USPApparatusII装置以每分钟转速100rpm下测试各锭剂分别于0小时、3小时、6小时、8小时、10小时的溶出率,其结果如表2与图1、图2及图3所示。表2缓释剂溶出率时间(小时)制备例A制备例B制备例C制备例D制备例E制备例F00.00%0.00%0.00%0.00%0.00%0.00%330.97%39.27%36.70%27.86%26.13%35.44%659.72%66.08%63.77%48.27%47.24%58.27%875.35%79.53%78.68%58.27%60.81%70.93%1088.58%90.51%88.54%67.83%71.42%81.26%图1显示本发明制备例A及E在各组份比例相同的情况下,由于制备例A使用的Hypromellose50黏度小于制备例E所使用的Hypromellose100000,因此于相同时间下制备例A的溶出率大于制备例E的溶出率。同样地,比对制备例B及F、制备例C及D(如图2所示)的溶出速率,观察到相同情形。由前述结果可知,若使用的纤维素衍生物黏度愈高者,其溶出速率则愈慢,即用户可视溶出速率的需求选择高黏度或低黏度的纤维素衍生物。另外,由图1亦可知,在本发明制备例A的溶出速率小于制备例B,是因制备例A的PolyOXWSR303含量较制备例B高的缘故,即各组份含量相同的情况下,氧化烯聚合物含量愈高者,其溶出速率愈慢。然而,若再进一步观察图3的制备例C可发现,若将其单元锭剂锭总重和制备例A一样固定为245mg,并使用不同比例的纤维素衍生物及聚乙烯氧化物,即将Hypromellose50含量提高且PolyOXWSR303含量降低,亦可使制备例C的溶出速率与制备例A趋近相同。由该结果可知,本发明的组合物,可通过提高纤维素衍生物比例来降低氧化烯聚合物的使用量,进而减少氧化烯聚合物所衍生的不安定因素及工艺不便性。实施例2粉体粒径分布及流动性分析1.粒径分布:将制备例A及制备例E于制备过程中获得的最终混合物分别随机取样定量后,置于数字式摇筛机(厂牌:Endecotts,型号:OctagonDigita),于频率3600次/分、振幅模式5的条件下振动20分钟,观察制备例A及制备例E于不同筛目筛网的滞留情形。2.安息角:使用粉体特性测试仪(厂牌:TsutsuiScientific,型号:ABD-100),将制备例A及制备例E定量的最终混合物分别以连续振动方式使粉体自然下落至测定用平台,测量粉体于平台形成圆锥状堆积层的自由表面在静平衡状态下与水平面形成的最大角度。安息角较佳为40°以下,安息角愈小,摩擦力愈小,流动性愈佳。3.松装密度:使用粉体特性测试仪,将制备例A及制备例E定量的最终混合物分别通过连续振动方式使粉体自然下落至内容积100毫升(ml)的容器中,测量粉体自然充满容器状态后的密度。4.振实密度:使用粉体特性测试仪,将制备例A及制备例E定量的最终混合物分别盛装至内容积100ml的容器中,并以连续振动该容器以破坏粉体间的空隙后,测量粉体处于紧密填充状态的密度。5.压缩度:基于前述试验获得的粉体松装密度及振实密度,利用式(I)计算压缩度。压缩度较佳为等于或小于20%,压缩度愈小,粉体流动性愈佳。压缩度(%)=(振实密度-松装密度)/振实密度×100式(I)表3流动性测试结果由表3结果显示,本发明制备例A及制备例E约有80%的最终混合物的颗粒粒径分布于75微米(μm)至425μm区间,其粒径小于75μm的微粉含量皆≦20%,可知制备例A及制备例E于打锭过程中皆不易有附着或飞散的情形。此外,从表3可得知本发明制备例A及制备例E的最终混合物的安息角皆≦37°,压缩度皆小于20%,可得知本发明制备例A及制备例E的最终混合物具良好的粉体流动性。另外,从表3亦可观察到,虽制备例A及制备例E所使用的纤维素衍生物黏度不同,其最终混合物的粉体流动性却未因此而随之下降,其原因在于本发明工艺是利用高浓度醇溶剂与纤维素衍生物进行造粒,由于该纤维素衍生物难溶于高浓度醇溶剂,于造粒过程便不会产生用水造粒所产生的黏稠的团块,反而形成蓬松的粉体,因此即便使用高黏度纤维素衍生物仍能维持其极佳流动性。实施例3均一度试验将制备例A及制备例E于制备过程中获得的最终混合物进行随机取样,分别取得245mg等重样品进行药物(活性成分)含量分析,用以了解本发明工艺中,工艺中间体及半成品的药物分布均一度。以单元锭剂应有药物含量为基准,这些样品分析结果如表4所示。表4各样品及其裸锭的均一度结果表4显示,随机取样的制备例A及制备例E最终混合物(中间体)及裸锭(半成品),其药物分布均一度皆显著地优于USP标准的85%至115%,RSD≦6%;可推知本发明所使用的工艺,即便未对高黏度、流动性不佳氧化烯聚合物进行整粒或造粒,亦能使药物均匀分布,并可解决现有技术“粉体流动性差、不适合压锭”或“药物分布均匀度不佳”的问题。实施例4安定性试验1.活性成份安定性试验将制备例A、制备例E分别打锭后所获得的锭剂分别置于温度为40±2℃,湿度为75±5%的恒湿恒温箱中历经0个月、1个月、2个月、3个月,并测定锭剂中药物(活性成分)含量,以试验本发明所得的剂型于条件环境下药物的降解程度,其结果如表5所示。表5安定性试验结果如表5结果显示,本发明制备例A、制备例E经打锭后所获得的锭剂于高温、高湿度的环境下分别放置1个月至3个月后,活性成分并无显著的降解,其有效成分含量仍于标准95%至105%范围内,可推知本发明所述的剂型具高度稳定、安定性。2.溶出安定性试验将制备例A、制备例E分别打锭后所获得的锭剂置于温度为40±2℃,湿度为75±5%的恒湿恒温箱中历经0个月或3个月后,使这些锭剂于500毫升0.1N的氯化氢溶液中,以溶出试验机USPApparatusII装置以每分钟转速100rpm下测试各锭剂分别于0小时、1小时、2小时、3小时、6小时、8小时、10小时、16小时、20小时、24小时的溶出率,其结果如表6与图4及图5所示。表6溶出试验结果由表6所列出制备例A、制备例E的锭剂于各个时间点的溶出率可知,本发明制备例A及制备例E的锭剂可持续释放达16小时至24小时。通过图4及图5更可清楚观察到,即便制备例A、制备例E的锭剂历经长达3个月高温、高湿度的环境后,其溶出速率和未经处理的锭剂并无明显差异,仍能有效维持原有的释放效果,可见本发明剂型具高度稳定、安定性。以上所述仅是本发明的较佳实施例而已,并非对本发明做任何形式上的限制,虽然本发明已以较佳实施例公开如上,然而并非用以限定本发明,任何本领域的技术人员,在不脱离本发明技术方案的范围内,当可利用上述公开了的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1