基于Sant‑75结构的化合物的制作方法与工艺
基于Sant-75结构的化合物技术领域本发明涉及蛋白抑制剂,特别是涉及一种基于Sant-75结构的化合物。
背景技术:
七次穿膜蛋白(Smoothened,Smo)是Hedgehog(Hh)信号通路中的一个重要组分。Hh信号通路参与正常的动物发育以及恶性肿瘤的发生,对后生动物的胚胎和成体组织的动态平衡也必不可少。Hh信号通路包括四个主要组分:Hh配体,十二次穿膜蛋白受体Patched1(Ptch1),七次穿膜蛋白受体Smo和Gli家族的转录因子。Hh配体通过结合Ptch1来激活Hh信号通路,在非结合的状态,Ptch1受体抑制了下游七次穿膜蛋白受体Smo的活性;而Hh配体和Ptch1受体的结合减轻了这种抑制作用,最终激活了Gli家族转录因子并移位到细胞核内,引起靶基因的表达从而导致细胞的增值和分化。Hh信号通路在正常的胚胎发生中调节细胞增殖和分化,由于其在调节细胞命运和增殖方面的关键作用,过度激活的Hh通路参与了一些恶性肿瘤和癌症的发生。基底细胞癌(Basalcellcarcinoma,BCC)是西方国家最为普遍的癌症之一,Ptch1和Smo的突变参与了其发生。在脑的发育中,Hh是小脑颗粒神经元祖细胞(cerebellargranuleneuronprogenitors)的有丝分裂原,并在出生后的发育中被下调。相比之下,大于30%的人成神经管细胞瘤显现出Gli1的高表达。在人成神经管细胞瘤中也发现Ptch1和Smo的突变。除了在胚胎发生中的作用,Hh信号通路也参与诸如血管发生和胰腺的器官发生。在斑马鱼和小鼠中的研究表明,Hh的缺陷会导致血管结构的丧失。而Shh缺陷小鼠会出现肺部脉管系统形成异常和支气管减少的表型。缺少Hh信号的斑马鱼胚胎表现出初级节间血管出芽缺陷。Shh似乎可以通过调节血管生成素1,-2和VEGF的mRNA的水平来促进血管成熟。Hh信号通路也在肿瘤相关血管发生中起重要作用。近年来的研究还表明,异常的Hh信号还和许多其它恶性肿瘤的发生相关,比如:小细胞肺癌、胰腺癌、乳腺癌和骨髓瘤等。因此发展有效的Hh信号通路的阻断剂是抗肿瘤药物研究的新策略和新思路。由于异常活化的Hh信号与人类疾病相关,而突变的Ptch1和Smo蛋白几乎都导致Hh信号的异常活化,所以人们已经花费了很多精力去寻找确定作用于Smo或其下游的治疗型抑制剂。如图1所示,环巴胺(cyclopamine)就是这样一个化合物,它通过与Smo直接相互作用从而特异性地阻断Hh信号通路。环巴胺是一种从植物中提取的甾体类生物碱,对Hh信号通路有抑制作用,当用其癌症治疗时,肿瘤发生中的血管结构会显著降低。Smo和Patched中的致癌突变似乎可以被环巴胺逆转。小规模的临床研究发现,局部施用环巴胺可以有效减小BCC病灶的大小。环巴胺在鼠类肿瘤的同种异体移植模型中也可以导致髓质母细胞瘤的衰退和凋亡。然而,环巴胺较差的水溶性(ca.5μg/mL)和酸不稳定性阻碍了其在临床上治疗肿瘤的应用。结合图1,SAG是Hh通路的激活剂,它与Smo蛋白有亲和力,但是却以一种与环巴胺作用相反的方式。由于SAG在更高浓度也抑制Hh通路,因此可以通过修饰其结构,开发出靶向Smo的抑制剂Sant-75,详请参见专利CN101597280A。但是,在对Sant-75的进一步研究中发现,Sant-75作为七次穿膜蛋白抑制剂的生物学活性较低,Sant-75的生物活性有待于进一步提高。
技术实现要素:
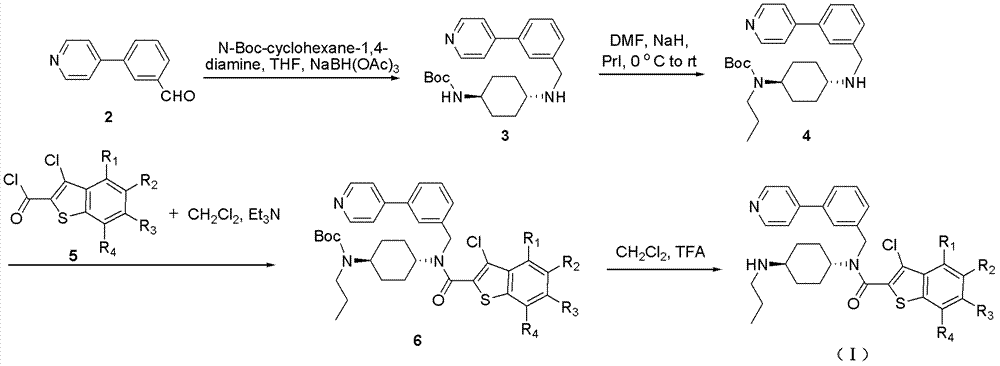
基于此,通过对Sant-75的结构进行修饰,提供一种作为七次穿膜蛋白抑制剂的生物学活性较高的基于Sant-75结构的化合物。一种基于Sant-75结构的化合物,具有如下结构式:其中,-R1为-H、-F或-Cl;-R2为-H、-F或-OH;-R3为-H、-F、-Cl、-OH、-NO2、-NH2、-Me、-SO2Me、-CO2Me、-CO2H、-NHAc或-NHSO2Me;-R4为-H、-F、-Cl或-SO2Me;并且-R1、-R2、-R3和-R4不同时为-H。在一个实施例中,上述基于Sant-75结构的化合物,具有如下化学式:一种基于Sant-75结构的化合物,具有如下结构式:一种基于Sant-75结构的化合物,具有如下结构式:其中,为一种基于Sant-75结构的化合物,具有如下结构式:其中,为一种基于Sant-75结构的化合物,具有如下结构式:其中,-R1为-H或-n-Pr;-R2为-CH3、-C2H5、-CH(CH3)2、-(CH2)2CH3、-n-Bu、-(CH2)4CH3、-(CH2)3CH3、-R3为-H或-Cl;-R4为-H或-Cl。在一个实施例中,上述基于Sant-75结构的化合物中,-R1为-H。在一个实施例中,上述基于Sant-75结构的化合物,具有如下结构式:在一个实施例中,上述基于Sant-75结构的化合物中,-R1为-n-Pr。在一个实施例中,上述基于Sant-75结构的化合物,具有如下结构式:上述基于Sant-75结构的化合物,通过对Sant-75进行系统的结构修饰,相对于Sant-75,其作为七次穿膜蛋白抑制剂的生物学活性较高。附图说明图1为现有的几种Smo抑制剂和激活剂的结构对比图;图2为一实施例的具有结构式(I)的化合物的制备方法的示意图;图3为一实施例的具有化合物Hh-001的制备方法的示意图;图4为一实施例的具有结构式(II)的化合物的制备方法的示意图;图5为一实施例的具有结构式(III)的化合物的制备方法的示意图;图6为一实施例的具有结构式(IV)的化合物的制备方法的示意图。具体实施方式通过对Sant-75进行系统的结构修饰,并借助于斑马鱼胚胎筛选和细胞实验,发现如下具有更佳水溶性和药代动力学特性的Smo抑制剂。一实施方式的基于Sant-75结构的化合物,具有如下结构式:其中,-R1为-H、-F或-Cl;-R2为-H、-F或-OH;-R3为-H、-F、-Cl、-OH、-NO2、-NH2、-Me、-SO2Me、-CO2Me、-CO2H、-NHAc或-NHSO2Me;-R4为-H、-F、-Cl或-SO2Me;并且-R1、-R2、-R3和-R4不能同时为-H。结合图2,具有结构式(I)的化合物的制备方法如下:第一步骤——还原胺化(化合物2到化合物3):将单Boc保护的反式环己二胺加入到化合物2的四氢呋喃溶液,室温搅拌30分钟;然后在0℃加入三乙酰氧基硼氢化钠,并在室温中搅拌过夜;反应用饱和碳酸钠淬灭后,用二氯甲烷提取三次,有机层合并之后用无水硫酸镁干燥,真空浓缩后残渣由硅胶柱纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物3。第二步骤——烷基化反应(化合物3到化合物4):向化合物3的DMF溶液中加入氢化钠,反应在0℃搅拌1小时;然后滴加碘丙烷,再升至室温搅拌3小时,反应用饱和碳酸氢钠淬灭后,用二氯甲烷萃取三次;合并有机相,用无水硫酸钠干燥,有机相浓缩之后残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物4。第三步骤——酰基化反应(化合物4到化合物6):化合物4和三乙胺溶于的二氯甲烷溶液,加入化合物5,室温搅拌2小时后,移除溶剂,残渣由硅胶柱层析纯化(洗脱液:正己烷/乙酸乙酯=2/1)得到化合物6。第四步骤——脱Boc保护基(化合物6到具有结构式(I)的化合物):化合物6溶于的二氯甲烷溶液,在0℃下加入三氟乙酸,室温搅拌1小时,然后以饱和碳酸钠淬灭反应,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1),得到具有结构式(I)的化合物。在一个实施例中,上述具有结构式(I)的化合物为:一实施方式的基于Sant-75结构的化合物,具有如下结构式:结合图3,化合物Hh-001的制备方法如下:第一步骤——还原胺化(化合物2到化合物3):将单Boc保护的反式环己二胺加入到化合物2的四氢呋喃溶液,室温搅拌30分钟;然后在0℃加入三乙酰氧基硼氢化钠,并在室温中搅拌过夜;反应用饱和碳酸钠淬灭后,用二氯甲烷提取三次,有机层合并之后用无水硫酸镁干燥,真空浓缩后残渣由硅胶柱纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物3。第二步骤——烷基化反应(化合物3到化合物4):向化合物3的DMF溶液中加入氢化钠,反应在0℃搅拌1小时;然后滴加碘丙烷,再升至室温搅拌3小时,反应用饱和碳酸氢钠淬灭后,用二氯甲烷萃取三次;合并有机相,用无水硫酸钠干燥,有机相浓缩之后残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物4。第三步骤——酰基化反应(化合物4到化合物6’):化合物4和三乙胺溶于的二氯甲烷溶液,加入化合物5’,室温搅拌2小时后,移除溶剂,残渣由硅胶柱层析纯化(洗脱液:正己烷/乙酸乙酯=2/1)得到化合物6’。第四步骤——脱Boc保护基(化合物6’到化合物Hh-001):化合物6’溶于二氯甲烷溶液,在0℃下加入三氟乙酸,室温搅拌1小时,然后以饱和碳酸钠淬灭反应,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1),得到化合物Hh-001。一实施方式的基于Sant-75结构的化合物,具有如下结构式:其中,为上述具有结构式(II)的化合物,具体为:结合图4,具有结构式(II)的化合物的制备方法如下:第一步骤——还原胺化(化合物2a到化合物3a):将单Boc保护的反式环己二胺加入到化合物2a的四氢呋喃溶液,室温搅拌30分钟;然后在0℃加入三乙酰氧基硼氢化钠,并在室温中搅拌过夜;反应用饱和碳酸钠淬灭后,用二氯甲烷提取三次,有机层合并之后用无水硫酸镁干燥,真空浓缩后残渣由硅胶柱纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物3a。第二步骤——烷基化反应(化合物3a到化合物4a):向化合物3a的DMF溶液中加入氢化钠,反应在0℃搅拌1小时;然后滴加碘丙烷,再升至室温搅拌3小时,反应用饱和碳酸氢钠淬灭后,用二氯甲烷萃取三次;合并有机相,用无水硫酸钠干燥,有机相浓缩之后残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物4a。第三步骤——酰基化反应(化合物4a到化合物6a):化合物4a和三乙胺溶于的二氯甲烷溶液,加入化合物5a,室温搅拌2小时后,移除溶剂,残渣由硅胶柱层析纯化(洗脱液:正己烷/乙酸乙酯=2/1)得到化合物6a。第四步骤——脱Boc保护基(化合物6a到具有结构式(II)的化合物):化合物6a溶于的二氯甲烷溶液,在0℃下加入三氟乙酸,室温搅拌1小时,然后以饱和碳酸钠淬灭反应,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1),得到具有结构式(II)的化合物。一种基于Sant-75结构的化合物,具有如下结构式:其中,为上述具有结构式(III)的化合物,具体为:结合图5,具有结构式(III)的化合物的制备方法如下:第一步骤——还原胺化(化合物2b到化合物3b):将单Boc保护的反式环己二胺加入到化合物2b的四氢呋喃溶液,室温搅拌30分钟;然后在0℃加入三乙酰氧基硼氢化钠,并在室温中搅拌过夜;反应用饱和碳酸钠淬灭后,用二氯甲烷提取三次,有机层合并之后用无水硫酸镁干燥,真空浓缩后残渣由硅胶柱纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物3b。第二步骤——烷基化反应(化合物3b到化合物4b):向化合物3b的DMF溶液中加入氢化钠,反应在0℃搅拌1小时;然后滴加碘丙烷,再升至室温搅拌3小时,反应用饱和碳酸氢钠淬灭后,用二氯甲烷萃取三次;合并有机相,用无水硫酸钠干燥,有机相浓缩之后残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物4b。第三步骤——酰基化反应(化合物4b到化合物6b):化合物4b和三乙胺溶于的二氯甲烷溶液,加入化合物5b,室温搅拌2小时后,移除溶剂,残渣由硅胶柱层析纯化(洗脱液:正己烷/乙酸乙酯=2/1)得到化合物6b。第四步骤——脱Boc保护基(化合物6b到具有结构式(II)的化合物):化合物6b溶于的二氯甲烷溶液,在0℃下加入三氟乙酸,室温搅拌1小时,然后以饱和碳酸钠淬灭反应,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1),得到具有结构式(III)的化合物。一种基于Sant-75结构的化合物,具有如下结构式:其中,-R1为-H或-n-Pr;-R2为-CH3、-C2H5、-CH(CH3)2、-(CH2)2CH3、-n-Bu、-(CH2)4CH3、-(CH2)3CH3、-R3为-H或-Cl;-R4为-H或-Cl。-n-Pr代表正丙基,-t-Bu代表叔丁基。在一个实施例中,上述的具有结构式(IV)的化合物中,-R1为-H,结构式为:在一个实施例中,上述的具有结构式(IV)的化合物中,-R1为-n-Pr,结构式为:结合图6,具有结构式(IV)的化合物的制备方法如下:第一步骤——还原胺化(化合物2c到化合物3c):将单Boc保护的反式环己二胺加入到化合物2c的四氢呋喃溶液,室温搅拌30分钟;然后在0℃加入三乙酰氧基硼氢化钠,并在室温中搅拌过夜;反应用饱和碳酸钠淬灭后,用二氯甲烷提取三次,有机层合并之后用无水硫酸镁干燥,真空浓缩后残渣由硅胶柱纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物3c。第二步骤——烷基化反应(化合物3c到化合物4c):向化合物3c的DMF溶液中加入氢化钠,反应在0℃搅拌1小时;然后滴加碘丙烷,再升至室温搅拌3小时,反应用饱和碳酸氢钠淬灭后,用二氯甲烷萃取三次;合并有机相,用无水硫酸钠干燥,有机相浓缩之后残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=10/1)得到化合物4c。第三步骤——酰基化反应/脱Boc保护基(化合物4c到化合物6c或化合物3c到化合物6c’):化合物4c或化合物3c和三乙胺溶于的二氯甲烷溶液,加入化合物5c,室温搅拌2小时后,移除溶剂,残渣溶于的二氯甲烷溶液,在0℃下加入三氟乙酸,室温搅拌1小时,然后以饱和碳酸钠淬灭反应,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1),得到化合物6c或6c’。第四步骤——酰基化反应(化合物6c到具有结构式(IV)的化合物或6c’到具有结构式(V)的化合物)化合物6c或6c’和三乙胺溶于的二氯甲烷溶液,加入化合物酰氯R2COCl,室温搅拌2小时后,移除溶剂,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=20/1)得到具有结构式(IV)或(V)的化合物。在一个实施例中,上述的具有结构式(V)的化合物为:在一个实施例中,上述的具有结构式(VI)的化合物为:上述基于Sant-75结构的化合物,通过对Sant-75进行系统的结构修饰,相对于Sant-75,其作为七次穿膜蛋白抑制剂的生物学活性较高。以下为具体实施例部分。实施例13-氯-6-硝基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-nitro-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备结合图2,化合物4(84mg,0.2mmol)和三乙胺(42μL,0.30mmol)溶于4ml的二氯甲烷溶液,加入6-硝基-3-氯代苯并噻吩酰氯(66mg,0.24mmol),室温搅拌2小时后,移除溶剂,得到带有Boc保护基的Hh-0224粗产物。将该粗产物溶于4ml的二氯甲烷溶液,在0℃下加入1ml三氟乙酸,室温搅拌1小时,然后以4ml饱和碳酸钠淬灭反应,用二氯甲烷(3×20mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/丙酮/三乙胺=40/10/1)得到85mg目标产物Hh-0224,两步总收率为75%。产物经测定:1HNMR(300MHz,CDCl3)δ8.68(d,J=5.4Hz,2H),8.28-8.42(br,1H),7.90-8.11(br,1H),7.26-7.67(m,7H),4.63-4.82(br,2H),3.78-4.18(br,1H),2.68-2.90(br,m,3H),1.25-2.21(m,10H),0.89(t,J=7.2Hz,3H);HRMS(ESI)理论值C30H32ClN4O3S[M+]:563.1884,实测值:563.1898。实施例23-氯-6-甲氧羰基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-methoxylcarbonyl-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备结合图2,化合物4(84mg,0.2mmol)和三乙胺(42μL,0.30mmol)溶于4ml的二氯甲烷溶液,加入6-甲氧羰基-3-氯代苯并噻吩酰氯(69mg,0.24mmol),室温搅拌2小时后,移除溶剂,得到带有Boc保护基的Hh-006的粗产物。将该粗产物溶于4ml的二氯甲烷溶液,在0℃下加入1ml三氟乙酸,室温搅拌1小时,然后以4ml饱和碳酸钠淬灭反应,用二氯甲烷(3×20mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液用:二氯甲烷/丙酮/三乙胺=40/10/1)得到64mg目标产物Hh-006,两步总收率为56%。产物经测定:1HNMR(300MHz,CDCl3)δ8.57-8.66(m,3H),7.26-8.19(m,8H),4.84(s,2H),3.99(s,3H),3.76(m,1H),2.35-2.60(m,3H),1.28-2.04(m,10H),0.83(t,J=6.7Hz,3H);HRMS(ESI)理论值C32H35ClN3O3S[M+]:576.2088,实测值:576.2079。实施例33-氯-5,6-亚甲二氧基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-5,6-methylenedioxy-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备结合图3,化合物4(84mg,0.2mmol)和三乙胺(42μL,0.30mmol)溶于4ml的二氯甲烷溶液,加入5,6-亚甲二氧基-3-氯代苯并噻吩酰氯(66mg,0.24mmol),室温搅拌2小时后,移除溶剂,得到带有Boc保护基的Hh-001的粗产物。将该粗产物溶于4ml的二氯甲烷溶液,在0℃下加入1ml三氟乙酸,室温搅拌1小时,然后以4ml饱和碳酸钠淬灭反应,用二氯甲烷(3×20mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液用:二氯甲烷/丙酮/三乙胺=40/10/1)得到81mg目标产物Hh-001,两步总收率为72%。产物经测定:1HNMR(300MHz,CDCl3)δ8.65(d,J=4.5Hz,2H),7.30-7.68(m,6H),7.15-7.20(m,2H),6.07(s,2H),4.77(s,2H),3.85(m,1H),2.33-2.55(m,3H),1.83-1.97(m,5H),1.26(m,5H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C31H33ClN3O3S[M+]:562.1931,实测值:562.1979。实施例43,6,7-三氯-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3,6,7-trichloro-N-((trans)-4-(propylamino)cyclohexyl)N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备结合图2,化合物4(84mg,0.2mmol)和三乙胺(42μL,0.30mmol)溶于4ml的二氯甲烷溶液,加入5,6-亚甲二氧基-3-氯代苯并噻吩酰氯(71mg,0.24mmol),室温搅拌2小时后,移除溶剂,得到带有Boc保护基的Hh-019的粗产物。将该粗产物溶于4ml的二氯甲烷溶液,在0℃下加入1ml三氟乙酸,室温搅拌1小时,然后以4ml饱和碳酸钠淬灭反应,用二氯甲烷(3×20mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液用:二氯甲烷/丙酮/三乙胺=40/10/1)得到92mg目标产物Hh-019,两步总收率为78%。产物经测定:1HNMR(300MHz,CDCl3)δ8.66(d,J=4.2Hz,2H),7.46-7.68(m,8H),4.69-4.85(br,2H),3.79(m,1H),2.42-2.72(m,3H),1.80-2.08(m,4H),1.24-1.68(m,6H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C30H31Cl3N3OS[M+]:586.1253,实测值:586.1232。实施例53-氯-6-氨基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-amino-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备实施例1制得的带有Boc保护基的Hh-0224(331mg,0.5mmol)溶于5ml的DMF和水(1∶1)的混合溶剂中,加入FeCl3(225mg,1.5mmol),锌粉(325mg,5mmol)。混合物在100℃反应1小时。当反应转化完全后,过滤混合物,滤液用10ml水稀释,接着加入饱和的Na2CO3溶液中和反应体系,并用二氯甲烷(3x10ml)萃取,无水硫酸钠干燥。浓缩后得到带有Boc保护基的Hh-0227的粗产物。将该粗产物溶于10ml的二氯甲烷溶液,在0℃下加入2ml三氟乙酸,室温搅拌1小时,然后以4ml饱和碳酸钠淬灭反应,用二氯甲烷(3×30mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣由硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=30/1)得到191mg目标产物Hh-0227,两步总收率为72%。产物经测定:1HNMR(300MHz,DMSO)δ8.63(s,2H),7.47-7.64(m,7H),6.83-7.02(m,2H),5.71(s,2H),4.75(s,2H),3.72(m,1H),2.7-2.89(m,3H),1.21-2.07(m,10H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C30H34ClN4OS[M+]:533.2142,实测值:533.2149。实施例63-氯-6-甲磺酰胺基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-(methylsulfonamido)-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将实施例5制得的带有Boc保护基的Hh-0227(63mg,0.1mmol)和三乙胺(20.8μl,0.15mmol)溶于10ml干燥的二氯甲烷,向其中加入MeSO2Cl(14mg,0.12mmol),然后室温搅拌反应4小时后浓缩。将浓缩后的残留物溶于2ml的二氯甲烷溶液,在0℃下加入0.5ml三氟乙酸,室温搅拌1小时,然后以2ml饱和碳酸钠淬灭反应,用二氯甲烷(3×10mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=40/1)得到48mg目标产物Hh-0215,两步总收率为78%。产物经测定:1HNMR(300MHz,DMSO)δ8.64(s,2H),7.29-7.80(m,9H),4.79(s,2H),2.98(s,3H),2.35-2.50(m,3H),1.21-2.07(m,10H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C31H36ClN4O3S2[M+]:611.1917,实测值:611.1922。实施例73-氯-6-乙酰氨基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-(actamido)-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将实施例5制得的带有Boc保护基的Hh-0227(63mg,0.1mmol)和三乙胺(20.8μl,0.15mmol)溶于5ml干燥的二氯甲烷,向其中加入乙酰氯(10mg,0.12mmol),然后室温搅拌反应3小时后浓缩。将浓缩后的残留物溶于2ml的二氯甲烷溶液,在0℃下加入0.5ml三氟乙酸,室温搅拌1小时,然后以2ml饱和碳酸钠淬灭反应,用二氯甲烷(3×10mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,残渣用硅胶柱层析纯化(洗脱液用:二氯甲烷/甲醇=30/1)得到42mg目标产物Hh-0218,两步总收率为74%。产物经测定:1HNMR(300MHz,CDCl3)δ8.65(s,2H),8.25(s,1H),7.28-7.65(m,9H),4.78(br,2H),3.88-4.20(br,1H),2.70-2.88(m,3H),2.18(S,3H),1.92-2.10(m,4H),1.26-2.14(m,10H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C32H36ClN4O2S[M+]:575.2247,实测值:575.2246。实施例83-氯-5,6-二羟基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-5,6-dihydroxy-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将实施例3制得的化合物Hh-001(56mg,0.1mmol)溶于5ml干燥的二氯甲烷,冷却至-78℃,并用氮气保护反应体系,用注射器慢慢滴加三溴化硼(22.6μL,0.24mmol)。加完后,撤除低温浴,让反应体系自然升至室温并搅拌过夜。反应结束后,加入30ml乙酸乙酯稀释反应液,并连续用饱和食盐水(3x4mL)和水(2x4mL)洗涤。有机相用无水硫酸钠干燥,真空浓缩后,残渣用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=20/1)得到37mg目标产物Hh-002,收率为68%。产物经测定:1HNMR(300MHz,DMSO)δ9.79(br,2H),8.79-8.95(m,3H),7.18-8.26(m,7H),7.15-7.20(m,2H),4.80(s,2H),2.92-2.98(m,1H),2.65-2.75(m,2H),2.02-2.15(m,2H),1.56-1.78(m,6H),1.20(m,2H),0.87(t,J=6.7Hz,3H);HRMS(ESI)理论值C30H33ClN3O3S[M+]:550.1931,实测值:550.1947。实施例93-氯-6-羧基-氮-反式-4-丙胺基环己基-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-6-carboxyl-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将实施例2制得的化合物Hh-006(28mg,0.05mmol)溶于2ml甲醇和2mL0.25NKOH水溶液,混合物在40℃搅拌反应2小时。加入5ml的水,并搅拌至均相。水相用乙醚萃取(2X4ml)以除去小极性的杂志。然后用浓盐酸酸化,洗出固体,过滤出固体,水洗,干燥后得到18mg目标产物Hh-007,收率63%。产物经测定:1HNMR(300MHz,DMSO)δ8.58-8.66(m,3H),7.46-8.14(m,8H),4.83(s,2H),4.29(m,1H),2.66-2.91(m,3H),1.53-2.07(m,6H),1.15-1.21(m,4H),0.83(t,J=6.7Hz,3H);HRMS(ESI)理论值C31H33ClN3O3S[M+]:562.1931,实测值:562.1935。实施例10氮-(2,4’-联吡啶-6-基-甲基)-3-氯-氮-反式-4-丙胺基环己基苯并[b]噻吩-2-甲酰胺(N-(2,4′-bipyridin-6-ylmethyl)-3-chloro-N-((trans)-4-(propylamino)cyclohexyl)benzo[b]thiophene-2-carboxamide)的制备结合图2,以2,4′-联吡啶-6-甲醛为原料,采用通用的制备步骤,即:1)还原胺化;2)烷基化;3)酰胺化;4)脱Boc保护基;合成得到Hh-0273。产物经测定:1HNMR(300MHz,CDCl3)δ8.75(d,J=5.7Hz,2H),7.39-7.94(m,9H),4.76-4.93(br,2H),4.52(br,0.4H),3.82(br,0.6H),2.52-2.66(m,3H),1.43-2.33(m,10H),0.88(t,J=7.2Hz,3H);HRMS(ESI)理论值C29H32ClN4OS[M+]:519.19851,实测值:519.1986。实施例113-氯-氮-反式-4-丙胺基环己基-氮-((2-(吡啶-4-基)-嘧啶)-4-基)甲基)苯并[b]噻吩-2-甲酰胺(3-chloro-N-((trans)-4-(propylamino)cyclohexyl)-N-((2-(pyridin-4-yl)pyrimidin-4-yl)methyl)benzo[b]thiophene-2-carboxamide)的制备结合图2,以2-(吡啶-4-基)嘧啶-4-甲醛为原料,采用通用的制备步骤,即:1)还原胺化;2)烷基化;3)酰胺化;4)脱Boc保护基;合成得到Hh-0530。产物经测定:1HNMR(300MHz,CDCl3)δ8.78-8.83(m,3H),7.46-7.86(m,7H),4.82(br,2H),4.52(m,0.4H),3.92(m,0.6H),2.73-2.90(m,3H),1.66-2.21(m,10H),0.88(t,J=7.2Hz,3H);HRMS(ESI)理论值C28H31ClN5OS[M+]:520.1938,实测值:520.1943。实施例123-氯-氮-(反式)-4-丙胺基环己基-氮-(3-(嘧啶-5-基)苄基)苯并[b]噻吩-2-甲酰胺(3-chloro-N-((trans)-4-(propylamino)cyclohexyl)-N-(3-(pyrimidin-5-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备结合图5,以3-(嘧啶-5-基)苯甲醛为原料,采用通用的制备步骤,即:1)还原胺化;2)烷基化;3)酰胺化;4)脱Boc保护基;合成得到Hh-0266。产物经测定:1HNMR(300MHz,CDCl3)δ9.21(s,1H),8.79-8.98(br,2H),7.49-7.84(m,8H),4.68-4.82(br,2H),3.80-4.28(m,1H),2.45-2.62(m3H),1.25-2.06(m,10H),0.87(t,J=7.2Hz,3H);HRMS(ESI)理论值C29H32ClN4OS[M+]:519.1985,实测值:519.1981。实施例133-氯-氮-(3-吗啡啉苄基)-氮-((反式)-4-丙胺基环己基)苯并[b]噻吩-2-甲酰胺(3-chloro-N-(3-morpholinobenzyl)-N-((1r,4r)-4-(propylamino)cyclohexyl)benzo[b]thiophene-2-carboxamide)的制备结合图5,以3-(吗啡啉)苯甲醛为原料,采用通用的制备步骤,即:1)还原胺化;2)烷基化;3)酰胺化;4)脱Boc保护基;合成得到Hh-0566。产物经测定:1HNMR(300MHz,CDCl3)δ7.78-7.81(m,2H),7.43-7.52(m,2H),7.00-7.22(m,1H)6.71-6.79(m,3H),4.55-4.68(m,2H),3.83-4.10(m,1H),3.83(s,4H),3.21-3.25(m,4H),2.68-2.86(m,3H),2.57-2.60(br,4H),1.79-2.23(m,10H),0.91(t,J=7.2Hz,3H);HRMS(ESI)理论值C29H37ClN3O2S[M+]:526.2295,实测值:526.2296。实施例143-氯-氮-((反式)-4-(氮-丙基环丁烷甲酰胺基)环己基)-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-N-((trans)-4-(N-propylcyclobutanecarboxamido)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将化合物Sant-75(52mg,0.1mmol)和三乙胺(20.8μl,0.15mmol)溶于5ml干燥的二氯甲烷,向其中加入环丁烷甲酰氯(14mg,0.12mmol),然后室温搅拌反应3小时后浓缩。将浓缩后的残留物用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=30/1)得到51mg目标产物Hh-01047,收率为86%。产物经测定:1HNMR(300MHz,CDCl3)δ8.67(br,2H),7.27-7.88(m,10H),4.65-4.86(m,2H),3.82-3.98(m,1H),2.90-3.18(m,4H),1.28-2.32(m,16H),0.85(t,J=7.2Hz,3H);HRMS(ESI)理论值C35H39ClN3O2S[M+]:600.2452,实测值:600.2437。实施例153-氯-氮-((反式)-4-(氮-丙基尼古丁甲酰胺基)环己基)-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺(3-chloro-N-((trans)-4-(N-propylnicotinamido)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide)的制备将化合物Sant-75(52mg,0.1mmol)和三乙胺(20.8μl,0.15mmol)溶于5ml干燥的二氯甲烷,向其中加入尼古丁甲酰氯(17mg,0.12mmol),然后室温搅拌反应3小时后浓缩。将浓缩后的残留物用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=20/1)得到45mg目标产物,收率为73%。产物经测定:1HNMR(300MHz,CDCl3)δ8.67(br,4H),7.19-7.91(m,12H),4.71-4.88(m,2H),2.95-3.91(m,4H),1.44-2.01(m,10H),0.90(br,3H);HRMS(ESI)理论值C36H36ClN4O2S[M+]:623.2247,实测值:623.2254。实施例163-氯-氮-((反式)-4-(2-苯氧乙酰胺基)环己基)-氮-(3-(4-吡啶基))苄基苯并[b]噻吩-2-甲酰胺3-chloro-N-((trans)-4-(2-phenoxyacetamido)cyclohexyl)-N-(3-(pyridin-4-yl)benzyl)benzo[b]thiophene-2-carboxamide的制备结合图6,化合物3c(190mg,0.5mmol)和三乙胺(104μL,0.75mmol)溶于8ml的二氯甲烷溶液,加入3-氯代苯并噻吩酰氯(138mg,0.6mmol),室温搅拌2小时后,移除溶剂,所得残留物溶于8ml的二氯甲烷溶液,在0℃下加入2ml三氟乙酸,室温搅拌1小时,然后以8ml饱和碳酸钠淬灭反应,用二氯甲烷(3×30mL)萃取,合并有机相并用无水硫酸钠干燥;真空浓缩后,所得中间体溶于20ml干燥的二氯甲烷,向其中加入三乙胺(104μl,0.75mmol)和2-苯氧乙酰氯(102mg,0.6mmol),然后室温搅拌反应3小时后浓缩。将浓缩后的残留物用硅胶柱层析纯化(洗脱液:二氯甲烷/甲醇=20/1)得到198mg目标产物,收率为65%。产物经测定:1HNMR(300MHz,CDCl3)δ8.66(d,J=4.8Hz,2H),6.28-7.86(m,15H),4.70-4.85(m,2H),4.40(s,2H),3.71-3.85(m2H),1.65-1.95(m,8H);HRMS(ESI)理论值C35H33ClN3O3S[M+]:610.1931,实测值:610.1943。性能测试一分别选取具有代表性的化合物进行性能测定,具体化合物见表1。将上述化合物作用于斑马鱼的FLK胚胎,通过显微镜观察胚胎发育型态的变化,可实现在整个活体胚胎中在体内水平上检测Hh的活性:Hh通路被抑制的胚胎会有U型体节(正常为V型),并且身体弯曲,眼睛发育异常。各化合物对FLK胚胎表型的最低作用浓度实验结果如表1所示:表1:代表化合物的转基因斑马鱼胚胎抑制试验化合物最低作用浓度Sant-755μMHh-02415μMHh-0115μMHh-027350μMHh-05665μMHh-010471μMHh-010342.5μMHh-0112120μMHh-0114910μMHh-0113720μMHh-010315μMHh-011570.6μMHh-011602.5μMHh-0118620μMHh-011932.5μM性能测试二上述化合物采用Shh-Light2细胞系来进一步检测对Hh通路的活性,用只有N端活性部分的合成的Shh蛋白作为信号分子,在培养基中加入上述待测的化合物以及荧光素酶报告质粒。通过从1×10-2到1×104nM浓度梯度的检测,发现化合物对Hh信号传导也有明显的抑制作用,结果见表2。对比文献(Taipale,J.,Chen,J.K.,Cooper,M.K.,Wang,B.,Mann.R.K.,Milenkovic,L.,Scott,M.P.,andBeachy,P.A.2000.EffectsofoncogenicmutationsinSmoothenedandPatchedcanbereversedbycyclopamine.Nature406:1005-1009.)中已经报道的Cyclopamine的浓度要低(IC50=300nM)。表2:代表化合物的Shh-light2细胞试验化合物NormalizedIC50withShh-light2Sant-759nMHh-02412.5nMHh-01117nMHh-027311nMHh-0566136nMHh-0104720.6nMHh-0103415.2nMHh-011219.8nMHh-0114911.4nMHh-0113711.6nMHh-0103123.8nMHh-0115720.2nMHh-011606.9nMHh-011865.6nMHh-0119313.1nM由性能测试一和性能测试二可以看出,这种基于Sant-75结构的化合物,通过对Sant-75进行系统的结构修饰,相对于Sant-75,其作为七次穿膜蛋白抑制剂的生物学活性较高。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1