苯并菲-柔性桥-苝二酰亚胺二元化合物及其制备方法与应用与流程
本发明涉及一种苯并菲-柔性桥-苝二酰亚胺二元化合物以及其制备方法及所述化合物在光学、电光学和电子学目的的应用。
背景技术:
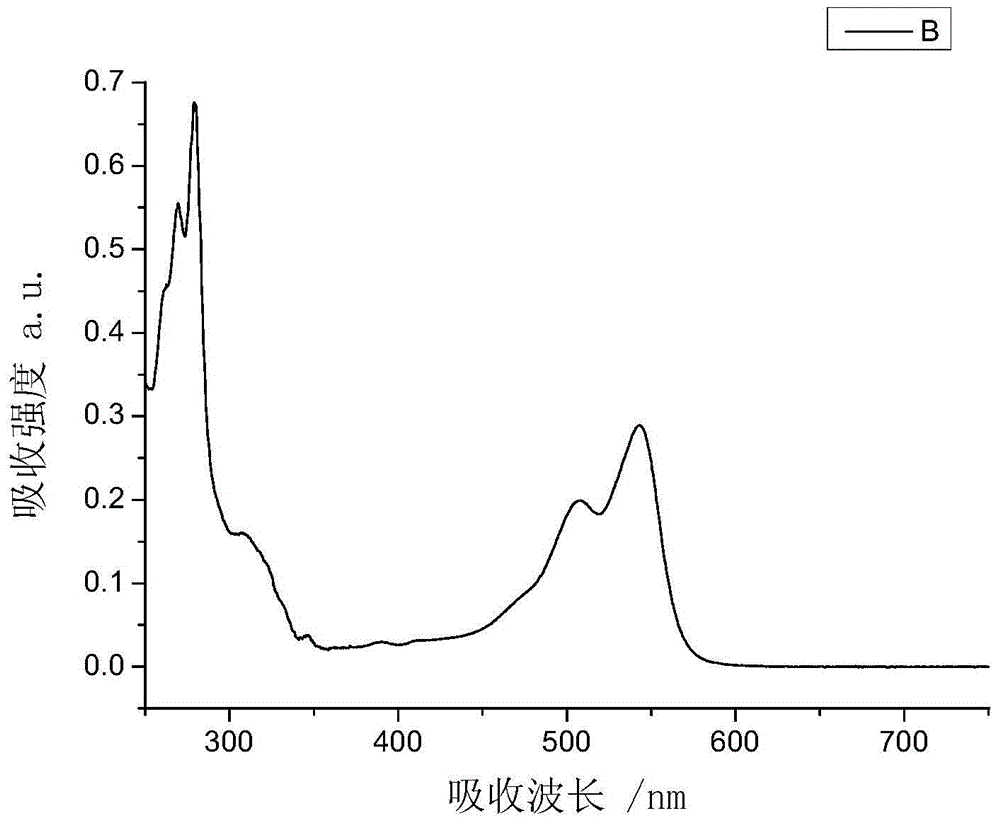
:柱状相盘状液晶材料处于液晶相时,由于盘状核之间较强的π-π电子相互作用,易自组装成有序的分子柱。从而使得沿分子柱的轴向具有较高的载流子迁移率,可以作为光电子器件的修饰层和活性层使用。苯并菲类液晶材料由于制备方法简单、产率较高、易于提纯,是目前研究地最为广泛的盘状液晶材料。Bacher等人报道苯并菲类盘状液晶可以作为有机发光器件的空穴传输层[参考文献]。Freudenmann等人报道了苯并菲二聚体可以作为有机发光器件的发光中心。Oukachmi等人报道苯并菲类盘状液晶可以作为有机光伏器件的空穴传输层。苝二酰亚胺衍生物是一类重要的有机半导体材料。苝二酰亚胺类盘状液晶易形成柱状相。通常不论是固体,还是在溶液中在可见光区均具有较强的光吸收和发射性能,因此可作为活性层用于制备有机光伏器件。Schmidt-Mende等人用苝二酰亚胺等材料制备的光伏器件开路电压为0.7伏。Halls等人用苝二酰亚胺等材料制备的光伏器件,开路电压为1伏,光电转换效率为6%。为了改善苝二酰亚胺类液晶材料的光电性能,专利CN102127015B中公开了苯并菲-苝二酰亚胺-苯并菲结构的三元液晶化合物。该三元化合物液晶材料易溶于普通有机溶剂,具有较高的熔点和清亮点。此外,还具有较高的摩尔消光系数,与P3HT混合后可作为有机光伏器件的活性层使用。但是在该三元化合物中苝二酰亚胺单元作为桥体连接两个苯并菲单元,由于该桥体上没有其它侧链,因此液晶的相转变温度及温度区间较难通过桥体和侧链调控。此外,聚酰亚胺类材料制作的配向膜已经广泛应用于液晶显示器。本发明提供了一种含有燕尾型苝二酰亚胺单元的盘状分子二元化合物,目的是更方便地调节液晶相转变温度及温度区间,同时使盘状液晶分子形成的柱状相更易于在基板表面形成垂面取向,从而提高载流子迁移率,并提太阳能电池的光电转换效率。技术实现要素:本发明的目的是提供一种具有盘状结构单元的燕尾型苝二酰亚胺单元的盘状分子二元化合物,所述二元化合物在液晶相时,具有柱状相结构的性质。本发明的另一目的是提供一种具有盘状结构单元的燕尾型苝二酰亚胺单元的盘状分子二 元化合物的制备方法。本发明的再一目的是提供一种具有盘状结构单元的燕尾型苝二酰亚胺单元的盘状分子二元化合物作为有机太阳能电池活性层的用途。为了达到上述发明目的,本发明提供一种具有通式(Ⅰ)的化合物,以及其包含的柱状相盘状液晶介质。其中:所述R11表示具有1个到20个碳原子的直链或支链烷基,其中一个或多个不相邻的CH2基团也可以彼此独立地被-O-代替,其前提是氧原子不直接彼此连接,其中一个或多个氢原子可被F、Cl或Br取代;所述R21或R22相同或不同,各自独立的表示-H、具有1个到20个碳原子的直链或支链烷基,其中一个或多个不相邻的CH2基团也可以彼此独立地被-O-代替,其前提是氧原子不直接彼此连接,其中一个或多个氢原子可被F、Cl或Br取代,R11和R12不同时为-H;并且所述L是选自由亚烷基、亚芳基、-CO-、-NH-、-O-、-S-及其组合组成的组的二价的连接基团。在本发明的的一些优选的实施方式中,所述R11独立地表示具有1个到12个碳原子的直链或支链烷基,其中一个或多个不相邻的CH2基团也可以彼此独立地被-O-代替,其前提是氧原子不直接彼此连接,其中一个或多个氢原子可被F、Cl或Br取代。在本发明的的一些优选实施方式中,所述R21或R22相同或不同,各自独立的表示-H、具有1个到12个碳原子的直链或支链烷基,其中一个或多个不相邻的CH2基团也可以彼此独立地被-O-代替,其前提是氧原子不直接彼此连接,其中一个或多个氢原子可被F、Cl或Br取代,R11和R12不同时为-H。在本发明的一些优选实施方式中,所述L含有彼此连接的至少两个选自由亚烷基与亚芳 基、-CO-、-NH-、-O-及-S-组成的组的基团。在本发明的一些更加优选的实施方式中,所述L含有彼此连接的至少两个选自由亚烷基、亚芳基、-CO-、-NH-和-O-组成的组的基团。在本发明的一些优选的实施方式中,所述亚烷基包括1到12个碳原子。在本发明的一些优选的实施方式中,所述亚芳基包括6到10个碳原子。在本发明的一些优选的实施方式中,所述L选自L1-L23中的L1;一种或多种:-O-Al-O-其中,L的右端连接在苝二酰亚胺核上,左端连接在苯并菲核上,并且其中,Al为亚烷基,Ar为亚芳基。在本发明的一些实施方式中,作为更加优选方案,所述L选自以下基团中的一种或多种:其中,L的右端连接在苝二酰亚胺核上,左端连接在苯并菲核上,并且其中,Al为1-12个碳原子的亚烷基,Ar为亚苯基。在本发明的一些实施方式中,特别优选方案,所述L为如下基团:-O-Al-O-L1;-O-Al-O-Ar-O-L2;其中,L的左端连接在苝二酰亚胺核上,右端连接在苯并菲核上,并且其中,Al为1-12个碳原子的亚烷基。本发明还提供了一种通式Ⅰ结构的盘状分子化合物的制备方法。合成路线如下所示:合成路线:以1-溴代烷为原料,与金属镁反应生成格氏试剂,再与甲酸乙酯反应生成仲醇(2);仲醇(2)再与邻苯二甲酰亚胺(3)发生Mitsunobu反应(光延反应)生成酰亚胺(4);酰亚胺与水合肼发生肼解反应生成仲胺(5);仲胺(5)再与苝四甲酸酐(6)反应生成苝二酰亚胺(7);苝二酰亚胺(7)再与硝酸铈铵反应生成1-硝基苝二酰亚胺(8);1-硝基苝二酰亚胺(8)与对苯二酚(9)在碱性条件下反应,生成1-对羟基苯氧基苝二酰亚胺(10);以邻苯二酚(11)为原料,与溴代烷发生Williamson(威廉姆逊合成)反应,生成二烷氧基苯(12)或邻烷氧基苯酚(13),化合物12和13在无水三氯化铁作用下发生Scholl反应(肖尔反应)生成单羟基烷基苯并菲(14)化合物14与ω-卤代醇发生Williamson反应;或是与α,ω-二醇发生Mitsunobu反应;或是与α,ω-二溴代烷发生Williamson反应,得产物15或16。进一步与化合物10,生成目标产物Ⅰ本发明的另一方面提供包含本发明的具有通式Ⅰ结构的化合物在有机太阳能电池活性层中的应用。有益效果:1.本发明通过大量实验筛选出具有通式Ⅰ化合物,与现有技术相比,本发明提供的苯并菲-柔性桥-苝二酰亚胺二元化合物,具有易于调节液晶相转变温度及温度区间的优点,使得该化合物具有优异的光学、电光学和电子学目的的用途,特别是作为有机太阳能电池活性层的用途。2.本发明通过大量实验筛选,优选得到通式Ⅰ化合物化合物的最佳制备工艺,可操作性强,易于工业化生产,生产效率高,制备得到的化合物纯度高。附图说明图1是化合物Ⅰ-1在152℃处于液晶态时在偏光显微镜下的织构图。图2是化合物Ⅰ-1在1×10-6mol·L-1的二氯甲烷溶液中的紫外-可见吸收光谱图。图3是化合物Ⅰ-2在159℃处于液晶态时在偏光显微镜下的织构图。图4是化合物Ⅰ-2在1×10-6mol·L-1的二氯甲烷溶液中的紫外-可见吸收光谱图。图5是化合物Ⅰ-3在148℃处于液晶态时在偏光显微镜下的织构图。图6是化合物Ⅰ-3在1×10-6mol·L-1的二氯甲烷溶液中的紫外-可见吸收光谱图。图7是化合物Ⅰ-4在214℃处于液晶态时在偏光显微镜下的织构图。图8是化合物Ⅰ-5在175℃处于液晶态时在偏光显微镜下的织构图。图9是化合物Ⅰ-5在1×10-6mol·L-1的二氯甲烷溶液中的紫外-可见吸收光谱图。图10是化合物Ⅰ-6在89℃处于液晶态时在偏光显微镜下的织构图。图11是化合物Ⅰ-1制备的有机太阳能电池的结构。具体实施方式以下将结合具体实施方案来说明本发明。需要说明的是,下面的实施例仅用来说明本发明,而不用来限制本发明。在不偏离本发明主旨或范围的情况下,可进行本发明构思内的其他各种组合和改良。在下述的实施例中,所有百分比表示重量百分比。下述的温度是摄氏度。1HNMR所用仪器为500M核磁共振仪,型号为AVANCE500,瑞士布鲁克公司生产;或者型号为Varian400MHz核磁共振仪,美国Varian生产;测试内标为三甲基硅烷(TMS),溶剂为CDCl3,有说明的除外。由一般已知的方法确定物理、物化和电光参数,如同在小册子《MerckLiquidCrystals–PhysicalPropertiesOfLiquidCrystals--DescriptionoftheMeasurementMethods》中所描述的那样。例1制备化合物Ⅰ-1反应步骤如下所示:化合物2的合成化合物1(39.6g,0.22mol)溶于120mL的干燥的四氢呋喃中,该溶液在1小时内滴加到镁屑(4.8g,0.2mol)上,并保持轻微的沸腾,再回流3h。冷却到室温后,滴加甲酸乙酯 (6.66g,0.09mol),室温搅拌5h。再加入饱和的氯化铵水溶液50mL,减压蒸出大部分四氢呋喃,用乙酸乙酯(20mL)萃取三次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到一淡黄色的油状物,加入乙腈(150mL)重结晶,得到化合物2为无色粉末(19.1g),产率93%。1HNMR(500MHz,CDCl3)δ3.51(s,1H),1.39-1.22(m,24H),0.81(t,J=7.0Hz,6H).化合物4的合成在氮气保护下,偶氮二甲酸二乙酯(2.58g,0.01484mol)溶于20mL干燥的四氢呋喃中,该混合溶液滴加到化合物2(2.82g,12.37mmol)和PPh3(3.56g,13.61mmol)和化合物3(2.32g,15.8mmol)的四氢呋喃中,室温搅拌24h。减压蒸出大部分THF,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到淡黄色的油状物,粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚1:30-1:20),得到化合物4为无色固体3.23g,产率73%。化合物5的合成化合物4(3.23g,0.00905mol)和水合肼(14.5mL)在无水乙醇(49mL)中回流2h。减压蒸出大部分乙醇,加入二氯甲烷(DCM)(70mL)萃取三次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到化合物5为淡黄色的油状物(1.76g),产率86%。1HNMR(500MHz,CDCl3)δ2.68(s,1H),1.43-1.27(m,24H),0.89(t,J=7.0Hz,6H)。化合物7的合成化合物5(2g,0.00881mol),苝四甲酸酐(1.568g,0.004mol)和咪唑(7.13g)在130℃反应6h。加入DCM(60mL)萃取三次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为乙酸乙酯/石油醚1:30-1:20),得到化合物7为红色固体(2.66g),产率82%。1HNMR(400MHz,CDCl3)δ8.62-8.54(m,8H),5.26–5.12(m,2H),2.22-2.30(m,4H),1.84-1.90(m,4H),1.39–1.20(m,40H),0.84(t,J=6.8Hz,12H)。化合物8的合成化合物7(0.66g,0000.815mol),98%浓硫酸(0.399g,0.00407mol),硝酸铈胺(2.23g,0.00407mol)在1,2-二氯乙烷(30ml)中回流2h。冷却至室温,滤掉沉淀,用DCM(70ml)萃取三次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚3:10),得到化合物8为红色固体(0.54g),产率77%。1HNMR(500MHz,CDCl3)δ8.74-8.64(m,5H),8.59(d,J=8.0Hz,1H),8.24(d,J=8.0Hz,1H),5.20-5.08(m,2H),2.25-2.19(m,4H),1.84(dd,J=11.5Hz,7.6Hz,4H),1.32-1.19(m,40H),0.82(t,J=6.0Hz,12H)。化合物10的合成氮气保护下,化合物8(0.70g,0.00082mol),化合物9(0.45g,0.0041mol),无水碳酸钾(1.13g,0.0082mol),碘化钾(0.316g,0.000082mol)在NMP(30mL)中于70℃反应3h。先加入50mL10%盐酸,再加入10mL甲醇,趁热抽滤,的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚3:2),得到化合物10为红色固体(0.47g),产率61%。1HNMR(400MHz,CDCl3)δ9.60(d,J=8.4Hz,1H),8.67-8.57(m,5H),8.28(d,J=8.8Hz,1H),7.10(d,J=8.8Hz,2H),6.98(d,J=8.8Hz,2H),6.07(s,1H),5.29-5.01(m,2H),2.33-2.09(m,4H),1.86(m,6.9Hz,4H),1.34–1.15(m,40H),0.80(t,J=6.0Hz,12H)。化合物12的合成化合物11(20g,0.1816mol),1-溴代正己烷(89.9g,0.5448mol),无水碳酸钾(73.5g,0.5448mol)及碘化钾(6.0g)在无水乙醇(250mL)中回流48小时。冷却至室温后抽滤,滤饼用乙醇(150mL)洗涤,抽滤,重复两次。滤液旋蒸后,再减压蒸馏收集149-155℃(0.5mmHg)的馏分,得到化合物12为无色油状液体(48g),产率96%。化合物13的合成化合物11(50g,0.45mol),1-溴代正己烷(75g,0.45mol),无水碳酸钾(100g,0.725mol)及碘化钾(3.8g)在无水乙醇(500mL)中回流12小时。冷却至室温后抽滤,滤饼用乙醇(150mL)洗涤,抽滤,重复两次。滤液旋蒸后,在减压蒸馏收集140-143℃(0.5mmHg)的馏分,得到化合物13为无色油状液体(25.5g),产率29%。化合物14的合成三氯化铁(12.96g,0.0792mol)溶于二氯甲烷(80mL)和硝基甲烷(8mL)中。化合物12(4.44g,0.016mol)和化合物13(1.56g,0.008mol)溶于二氯甲烷(60mL),在0℃时,把该溶液滴加至三氯化铁溶液中,约40分钟滴加完毕。在0℃继续反应2.5小时后,加入甲醇(60mL)和水(30mL)终止反应。反应液用20mL稀盐酸(2mol/L)洗涤两次,饱和食盐水洗涤一次。分出有机层,用无水硫酸钠干燥,滤液旋干后得到的粗产物用硅胶柱提纯(淋洗液为乙酸乙酯/石油醚,体积比为1:30-1:20),得到化合物14为无色固体(2.14g),产率36%。1HNMR(400MHz,CDCl3)δ:7.96(s,1H),7.83(s,3H),7.82(s,1H),7.77(s,1H),5.91(s,1H),4.31-4.19(m,10H),1.98-1.90(m,10H),1.59-1.40(m,30H),0.96-0.92(m,15H)。化合物15的合成在氮气保护下,化合物14(0.94g,0.00126mol),6-氯-1-己醇(0.32g,0.00234mol),无水碳酸钾(0.43g,0.00311mol),碘化钾(0.36g,0.00217mol)加入N,N-二甲基甲酰胺(15mL)中,80℃下搅拌反应72h。冷却至室温后,加入乙酸乙酯(60mL)和水(15mL),分出有机层,用无水硫酸钠干燥,滤液旋干,用二氯甲烷(4ml)和甲醇(12ml)重结晶,滤饼再用硅胶柱提纯(淋洗液为二氯甲烷),得到化合物15为无色固体(0.64g),产率60%。1HNMR(500MHz,CDCl3)δ7.85(s,6H),4.24-4.28(m,12H),3.71(t,J=6.5Hz,2H),2.01-1.92(m,12H),1.70-1.49(m,16H),1.46-1.37(m,20H),0.95(t,J=7.0Hz,15H)。化合物Ⅰ-1的合成在氮气保护下,将偶氮二甲酸二异丙酯(0.105g,0.00052mol)和干燥的四氢呋喃(5mL)溶液滴加到化合物15(0.4g,0.000436mol),化合物10(0.58g,0.000686mol),三苯基磷(0.126g,0.00048mol)和四氢呋喃(10ml)的混合液中,室温搅拌反应72h。压减蒸出大部分四氢呋喃,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:5-1:2),得到化合物I-1为红色固体(0.30g),产率39%。1HNMR(400MHz,CDCl3)δ9.61(d,J=8.4Hz,1H),8.75-8.56(m,5H),8.24(d,J=8.8Hz,1H),7.82(m,6H),7.14(d,J=8.8Hz,2H),7.01(d,J=8.8Hz,2H),5.15(m,2H),4.28-4.21(m,12H),4.04(t,J=6.4Hz,2H),2.29-2.23(m,4H),2.07-1.77(m,18H),1.74-1.55(m,14H),1.40(m,20H),1.24(m,38H),0.95-0.92(m,15H),0.85-0.77(m,12H)。图1是化合物Ⅰ-1在152℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-1在降温 过程中于152℃时拍摄的织构图,表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司生产。图2是将化合物Ⅰ-1溶解在二氯甲烷中,浓度为1×10-6mol/L,利用紫外可见吸收光谱仪测试该物质的紫外可见吸收光谱。该化合物在可见光区及紫外光区有较强的光吸收性能。所用的紫外可见光谱仪(型号为Lambda35)由美国PerkinElmer公司生产。实施例2制备化合物Ⅰ-2反应步骤如下所示:依照实施例1的方法合成化合物1-14。化合物16的合成在氮气保护下,将偶氮二甲酸二乙酯(0.24g,0.00138mol)和干燥的四氢呋喃(5mL)溶液滴加到化合物14(0.95g,0.00125mol),1,8-辛二醇(0.94g,0.00625mol),三苯基磷(0.36g,0.00138mol)和四氢呋喃(10mL)的混和物中,室温搅拌反应48h。减压蒸出 大部分THF,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,分出有机层,用无水硫酸钠干燥,滤液旋蒸后,加入甲醇(7mL),过滤除去剩余的1,8-辛二醇,滤饼用二氯甲烷(5mL)和甲醇(20mL)重结晶,得到的粗产物用硅胶柱提纯(淋洗液为乙酸乙酯/石油醚,体积比1:10-1:5),得到化合物16为无色固体(0.58g),产率53%。1HNMR(500MHz,CDCl3)δ7.86(s,6H),4.25(t,J=7.0Hz,12H),3.67(t,J=7.0Hz,2H),1.99-1.93(m,12H),1.66-1.55(m,16H),1.45–1.39(m,24H),0.95(d,J=7.0Hz,15H)。化合物Ⅰ-2的合成在氮气保护下,将偶氮二甲酸二异丙酯(0.115g,0.00057mol)和干燥的四氢呋喃(5mL)溶液滴加到化合物16(0.44g,0.000479mol),化合物10(0.50g,0.000573mol),三苯基磷(0.138g,0.000527mol)加入到干燥的四氢呋喃(10mL)中,室温搅拌反应72h。压减蒸出大部分四氢呋喃,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:5-1:2),得到化合物I-2为红色固体(0.55g),产率64.7%。1HNMR(400MHz,CDCl3)δ9.60(d,J=8.4Hz,1H),8.74-8.57(m,5H),8.24(d,J=8.4Hz,1H),7.81(m,6H),7.14(d,J=8.8Hz,2H),7.00(d,J=8.8Hz,2H),5.26-5.18(m,2H),4.25-4.21(m,12H),4.01(t,J=6.4Hz,2H),2.29-2.23(m,4H),2.01-1.76(m,18H),1.60-1.20(m,76H),0.95-0.92(m,15H),0.85-0.77(m,12H)。图3是化合物Ⅰ-2在159℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-2在降温过程中于159℃时拍摄的织构图,表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司生产。图4是将化合物Ⅰ-2溶解在二氯甲烷中,浓度为1×10-6mol/L,利用紫外可见光谱仪测试该物质的紫外可见吸收光谱。该化合物在可见光区及紫外光区有较强的光吸收性能。所用的紫外可见光谱仪(型号为Lambda35)由美国PerkinElmer公司生产。实施例3制备化合物Ⅰ-3反应步骤如下所示:依照实施例1的方法合成化合物1-14。化合物17的合成在氮气保护下,化合物14(1.23g,0.00165mol),1,10-二溴癸烷(2.97g,0.0099mol),氢氧化钾(0.43g,0.0066mol),四丁基溴化铵(0.16g,0.0005mol)加入到二氯甲烷(10mL)和水(7mL)中,室温下搅拌反应24小时。静置,分出水层,向有机层中加入甲醇(40mL)。过滤出沉淀,用硅胶柱提纯(淋洗液为乙酸乙酯/石油醚=1:30-1:20),得到化合物17为无色固体(1.41g),产率89%。1HNMR(400MHz,CDCl3)δ7.84(s,6H),4.23(t,J=7.0Hz,12H),3.40(t,J=7.0Hz,2H),1.95(m,12H),1.84(m,2H),1.58(m,12H),1.38(m,30H),0.94(t,J=7.0Hz,15H)。化合物Ⅰ-3的合成在氮气保护下,化合物10(0.30g,0.000327mol),化合物17(0.45g,0.00049mol),无水碳酸钾(0.09g,0.000654mol),碘化钾(0.01g,0.00006mol)在DMF(15mL)中加热到80℃,保温反应24h。冷却至室温,加入乙酸乙酯(60mL)和水(15mL),分出有机层,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:2-1:1),得到化合物I-3为红色固体(0.29g),产率49%。1HNMR(400MHz,CDCl3)δ9.60(d,J=8.4Hz,1H),8.74-8.57(m,5H),8.26(d,J=8.4Hz,1H),7.81(s,6H),7.16(d,J=8.8Hz,2H),7.02(d,J=8.8Hz,2H),5.25-5.17(m,2H),4.24(t,J=6.4Hz,12H),4.03(t,J=6.4Hz,2H),2.31-2.23(m,4H),2.02-1.84(m,18H),1.61-1.24(m,80H),0.96-0.92(m,15H),,0.88-0.81(m,12H)。图5是化合物Ⅰ-3在148℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-3在降温过程中于148℃时拍摄的织构图,表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司生产。图6是将化合物Ⅰ-3溶解在二氯甲烷中,浓度为1×10-6mol/L,利用紫外可见光谱仪测试该物质的紫外可见吸收光谱。该化合物在可见光区及紫外光区有较强的光吸收性能。所用的紫外可见光谱仪(型号为Lambda35)由美国PerkinElmer公司生产。实施例4制备化合物Ⅰ-4反应步骤如下所示:依照实施例3的方法合成化合物1-17化合物19的合成取异辛胺(1g,7.75mmol),苝酐(1.39g,3.52mmol),咪唑(10.35g)在氮气保护下于130℃反应24小时,冷却用温水抽滤。得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷),得到化合物19为红色固体(1.1g),产率70%化合物20的合成化合物19(1g,1.629mmol),98%浓硫酸(0.82g,8.367mmol),硝酸铈铵(4.5g,8.212mmol)在1,2-二氯乙烷(40ml)于85℃反应3个小时结束。冷却至室温,滤掉沉淀,用DCM(70ml)萃取三次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,粗产物用硅胶柱提纯(淋洗液为二氯甲烷/乙酸乙酯20:1),得到化合物20为红色固体(0.83g),产率77%.1HNMR(500MHz,CDCl3)δ8.77(d,J=8.0Hz,1H),8.74-8.64(m,4H),8.58(d,J=8.0Hz,1H),8.20(d,J=8.0Hz, 1H),4.20-4.10(m,4H),1.96-1.95(m,2H),1.42-1.33(m,16H),0.97(t,J=6.0Hz,6H),0.92(t,J=6.0Hz,6H)。化合物21的合成氮气保护下,化合物20(0.43g,0.7mmol),化合物9(0.385g,4.1mol),无水碳酸钾(0.97g,7mmol),碘化钾(0.316g)在NMP(30mL)中于70℃反应3h。先加入50mL10%盐酸,再加入10mL甲醇,趁热抽滤,的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚3:2),得到化合物21为红色固体(0.31g),产率65%。化合物Ⅰ-4的合成在氮气保护下,将偶氮二甲酸二异丙酯(0.105g,0.00052mol)和干燥的四氢呋喃(5mL)溶液滴加到化合物21(0.32g,0.000436mol),化合物15(0.58g,0.000686mol),三苯基磷(0.126g,0.00048mol)和四氢呋喃(10ml)的混合液中,室温搅拌反应72h。压减蒸出大部分四氢呋喃,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,合并有机相,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:5-1:2),得到化合物Ⅰ-4为红色固体(0.28g),产率41%。1HNMR(400MHz,CDCl3)δ9.51(d,J=8.4Hz,1H),8.59-8.46(m,5H),8.16(s,1H),7.71-7.69(m,6H),7.17(d,J=8.8Hz,2H),7.03(d,J=8.8Hz,2H),4.23-4.02(m,18H),2.01-1.94(m,16H),1.66-1.47(m,14H),1.40-1.30(m,34H),0.96-0.87(m,27H)。图7是化合物Ⅰ-4在214℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-4在降温过程中于214℃时拍摄的织构图,表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司 生产。实施例5制备化合物Ⅰ-5反应步骤如下所示:依照实施例4的方法合成化合物1-21化合物Ⅰ-5的合成在氮气保护下,化合物21(0.15g,0.0002mol),化合物17(0.6g,0.00065mol),无水碳酸钾(1.02g,0.0074mol),碘化钾(0.01g,0.00006mol)在DMF(10mL)中加热到80℃,保温反应24h。冷却至室温,加入乙酸乙酯(60mL)和水(15mL),分出有机层,用无水硫酸钠干燥,滤液旋蒸后,得到的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:2-1:1),得到化合物Ⅰ-5为红色固体(0.2g),产率58.8%。1HNMR(400MHz,CDCl3)δ9.45(d,J=8.4Hz,1H),8.54-8.36(m,5H),8.12(s,1H),7.64-7.59(m,6H),7.17(d,J=8.8Hz,2H),7.03(d,J=8.8Hz,2H),4.21-4.03(m,18H),1.97-1.86(m,16H),1.66-1.47(m,14H),1.40-1.30 (m,42H),0.96-0.87(m,27H)。图8是化合物Ⅰ-5在175℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-5在降温过程中于175℃时拍摄的织构图,表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司生产。图9是将化合物Ⅰ-5在1×10-6mol·L-1的二氯甲烷溶液中的紫外-可见吸收光谱图。化合物Ⅰ-5溶解在二氯甲烷中,浓度为1×10-6mol/L,利用紫外可见光谱仪测试该物质的紫外可见吸收光谱。该化合物在可见光区及紫外光区有较强的光吸收性能。所用的紫外可见光谱仪(型号为Lambda35)由美国PerkinElmer公司生产。实施例6制备化合物Ⅰ-6反应步骤如下所示:依照实施例1的方法合成化合物1-14。化合物24的合成在氮气保护下,将偶氮二甲酸二乙酯(0.24g,0.00138mol)和干燥的四氢呋喃(5mL)溶液滴加到化合物14(0.95g,1.25mmol),三甘醇(0.94g,6.3mmol),三苯基磷(0.36g,0.00138mol)和四氢呋 喃(10mL)的混和物中,室温搅拌反应48h。减压蒸出大部分THF,加入DCM(60mL),然后用水(15mL)洗涤两次,饱和食盐水洗一次,分出有机层,用无水硫酸钠干燥,滤液旋蒸后,加入甲醇(5mL),过滤除去剩余的三甘醇,滤饼用二氯甲烷(5mL)和甲醇(20mL)重结晶,得到的粗产物用硅胶柱提纯(淋洗液为乙酸乙酯/石油醚,体积比1:10-1:2),得到化合物24为淡色色固体(0.61g),产率55%。化合物Ⅰ-6的合成氮气保护下,化合物8(0.70g,0.00082mol),化合物24(0.79g,0.0009mol),无水碳酸钾(1.13g,0.0082mol),碘化钾(0.136g,0.00082mol)在NMP(30mL)中于70℃反应24h。先加入50mL10%盐酸,再加入10mL甲醇,趁热抽滤,的粗产物用硅胶柱提纯(淋洗液为二氯甲烷/石油醚,体积比1:3-1:1),得到化合物Ⅰ-6为红色固体(0.55g),产率40%。1HNMR(400MHz,CDCl3)δ9.49(d,J=8.0Hz,1H),8.51-8.23(m,6H),7.68(s,1H),7.55(s,1H),7.47-7.33(m,4H),5.17(s,2H),4.50(s,2H),4.43(s,2H),4.10(s,2H),2.01-1.94(m,16H),1.66-1.47(m,14H),1.40-1.30(m,34H),0.96-0.87(m,27H)。图10是化合物Ⅰ-6在89℃处于液晶态时在偏光显微镜下的织构图。化合物Ⅰ-6在降温过程中于89℃时拍摄的织构图,典型的焦锥状表明该液晶相为柱状相。所用偏光显微镜(型号为LeicaDMRX)由德国Leica公司生产。加热台(型号为THMSE600)由英国LinkamTHMSE公司生产。实施例7选取上述实施例制备得到的化合物Ⅰ-1、Ⅰ-2、Ⅰ-3和Ⅰ-4进行性能检测,各化合物的DSC数据如下表1所示表1编码升温过程的相变温度℃[焓变J/g]降温过程的相变温度℃[焓变J/g]Ⅰ-1Cr91.80[1.212]LC148.83[15.56]II142.23[15.19]CrⅠ-2Cr80.77[1.056]LC159.44[7.089]II143.42[8.095]LC29.02[0.135]CrⅠ-3Cr64.50[1.890]LC151.57[8.396]II155.23[6.995]CrⅠ-4Cr55.95[1.281]LC209.12[7.901]II205.21[7.230]Cr注:Cr表示晶体,LC表示液晶,I表示各向同性的液体通过以上表1所示的化合物Ⅰ-1、Ⅰ-2、Ⅰ-3及Ⅰ-4的DSC数据表明,本发明提供制备得到的化合物,具有易于调节液晶相转变温度及温度区间的优点。实施例8应用实例在有机太阳能电池中,用氧化铟锡(ITO)透明电极为正极,金属铝为负极,铝负极用真空蒸镀的方式镀在器件上。盘状液晶二元化合物Ⅰ-3为活性层,用真空蒸镀的方式或是旋涂的方式制备。修饰层用真空蒸镀的方式或是旋涂的方式制备。图11是应用化合物Ⅰ-3制备的有机太阳能电池的结构。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1