苯酚衍生物的晶型及其制备方法与流程
本发明涉及了一种式(I)所示化合物及其异构体的晶型及其制备方法以及它们在医药领域的用途。
背景技术:
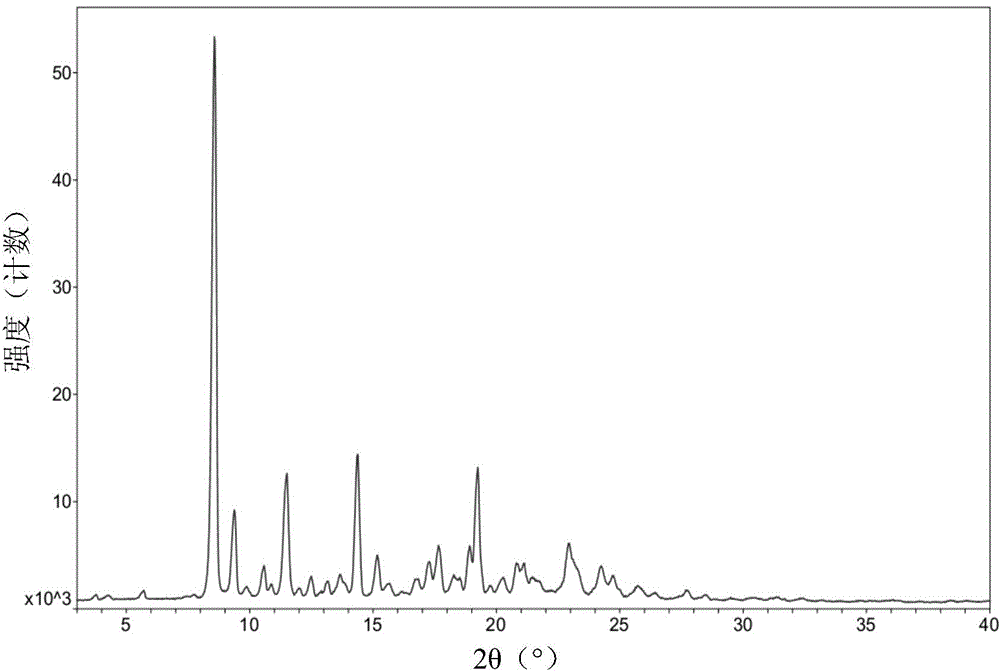
:丙泊酚可激活多种GABAA受体亚型,是一个临床上成熟的静脉麻醉药,广泛用于全身麻醉的诱导和维持。临床剂量相关的丙泊酚可直接激活哺乳动物神经元中的GABAA受体-氯离子通道复合体,增加氯离子传导,降低神经网络的兴奋性,进而引起全身麻醉(ManamiHara等(1993).Anesthesiology,79,781-788)。丙泊酚的显著药代动力学和药效学性质是起效快,维持时间短和快速可逆。静脉给药后,丙泊酚迅速从血液进入心、肺和肝等高灌注区,高脂溶性使丙泊酚容易跨越血脑屏障进入大脑发挥全身麻醉作用。2-[(1R)-1-环丙基乙基]-6-异丙基-苯酚为丙泊酚类似物,是一种高脂溶性物质,直接给予到血流中,可引起麻醉的迅速起效。[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-苯基乙基]氨基甲酸酯(式(I)化合物)是制备2-[(1R)-1-环丙基乙基]-6-异丙基苯酚的中间体,经过对其固体形态的研究,我们发现了具有明显粉末X-射线衍射图谱特征的固态形式。技术实现要素:本发明涉及一种式(I)化合物及其异构体式(II)化合物的晶型I。本发明涉及一种式(IV)化合物及其异构体式(III)化合物的晶型II。本发明涉及一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法。本发明提供式(I)化合物及其异构体式(II)化合物具有相同的晶型,即的晶型I,其粉末X-射线衍射图谱在d值为19.3±0.2、9.5±0.2、4.8±0.2和3.7±0.2处对应有特征衍射峰;优选在d值为19.3±0.2、9.5±0.2、8.7±0.2、4.8±0.2、4.5±0.2、4.1±0.2和3.7±0.2处对应有特征衍射峰;优选具有如图1所示的粉末X-射线衍射图 谱所代表的特征。进一步地,本发明所述的式(I)化合物的晶型I的特征性XRD峰,以°2θ和d值表示:序号°2θd值014.61619.128029.3949.4070310.1658.6950411.8407.4690515.6085.6730616.4595.3810717.0915.1840818.4134.8140919.7964.4811021.8874.0571123.8803.723进一步地,本发明所述的式(II)化合物的晶型I的特征性XRD峰,以°2θ和d值表示:序号°2θd值014.53519.467029.2579.5450310.0878.7620416.9945.2130517.4305.0840618.3944.8190719.7364.4950821.8104.0720923.7813.7381024.0173.7021124.2353.6691225.3393.5121325.7733.454本发明提供式(IV)化合物及其异构体式(III)化合物具有相同的晶型,即的晶型II,其粉末X-射线衍射图谱在d值为10.3±0.2、9.4±0.2、7.7±0.2、6.2±0.2和4.6±0.2处对应有特征衍射峰,优选在d值为10.3±0.2、9.4±0.2、8.4±0.2、7.7±0.2、6.2±0.2、5.8±0.2、5.0±0.2、4.6±0.2和3.9±0.2处对应有特征衍射峰;优选具有如图2所示的粉末X-射线衍射图谱所代表的特征;进一步地,本发明所述的式(IV)化合物的晶型II的特征性XRD峰,以°2θ和d值表示:序号°2θd值018.56610.314029.3759.4260310.5788.3560411.5057.6850514.3686.1600615.1765.8330717.2695.1310817.6455.0220918.9084.6901019.2424.6091120.8034.2661221.0994.2071322.9523.8721423.2683.8201524.2363.669进一步地,本发明所述的式(III)化合物的晶型II的特征性XRD峰,以°2θ和d值表示:序号°2θd值018.54510.339029.3549.4470310.5568.3740411.4477.7240514.3296.1760615.1385.8480717.2295.1420817.6245.0280918.8694.6991019.2024.6181120.8024.2671221.0784.2111322.9133.8781423.2293.8261524.1783.678在一具体实施方案中,本发明提供的制备的式(I)化合物的晶型I含量(质量含量)一般大于70%,优选大于80%,进一步最优选大于90%。在一具体实施方案中,本发明提供的制备的式(II)化合物的晶型I含量(质量含量)一般大于70%,优选大于80%,进一步优选大于90%。在一具体实施方案中,本发明提供的制备的式(IV)化合物的晶型II含量(质量含量)一般大于70%,优选大于80%,进一步优选大于90%。在一具体实施方案中,本发明提供的制备的式(III)化合物的晶型II含量(质量含量)一般大于70%,优选大于80%,进一步优选大于90%。本发明提供一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法,包括:1.晶浆结晶法:式(I)化合物或其异构体与单一溶剂或组合溶剂混合后,室温下超声得到悬浊液,然后将超声得到的悬浊液在室温下晶浆3天。取晶浆后的浑浊液减压过滤,干燥后得到目标晶型。2.挥发结晶法:式(I)化合物或其异构体与单一溶剂混合后,室温下超声得到溶清液或接近溶清液,敞口放置在室温下自然挥发,再干燥后得到目标晶型。3.冷却结晶法:式(I)化合物或其异构体与组合溶剂混合后,室温下超声得到悬浊液,将悬浊液加热搅拌至溶清或接近溶清,放置于室温搅拌,有结晶后减压过滤,干燥后得到目标晶型。4.抗溶剂结晶法:式(I)化合物或其异构体中加入良溶剂,室温下超声得到溶清液或接近溶清液,然后采用正向添加的方式向样品溶清液中添加相应比例的抗溶剂并搅拌,搅拌至有结晶后减压过滤,干燥后得到目标晶型。本发明优选方案,一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法,该方法包括:式(I)化合物或其异构体与溶剂混合,室温下超声得到悬浊液,悬浊液在室温下晶浆3天,取晶浆后的浑浊液减压过滤,干燥;所述溶剂选自水与乙醇、异丙醇、丙酮或1,4-二氧六环中的一种或多种的组合;优选所述的溶剂选自水与乙醇、异丙醇、丙酮或1,4-二氧六环中的一种的组合,且两种溶剂之比为5:1~0:1;进一步优选两种溶剂之比为1:1;本发明优选方案,一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法,该方法包括:式(I)化合物或其异构体与溶剂混合,室温下超声得到溶清液或接近溶清液,敞口放置在室温下自然挥发,再干燥;所述溶剂选自乙醇、丙酮、1,4- 二氧六环、异丙醚、正己烷或正庚烷中的一种或多种的组合;优选所述的溶剂为两种的组合,两种溶剂之比为5:1~0:1;进一步优选两种溶剂之比为0:1。本发明优选方案,一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法,该方法包括:式(I)化合物或其异构体与溶剂混合,室温下超声得到悬浊液,将悬浊液加热搅拌至溶清或接近溶清,放置于室温搅拌,有结晶后减压过滤,干燥;所述溶剂选自水、正庚烷、正己烷、异丙醚、乙醇、1,4-二氧六环、丙酮或乙腈中的一种或多种的组合;优选所述的溶剂为两种的组合,两种溶剂之比为5:1~0:1;其中所述溶剂为两种的组合时,可以选自水、正庚烷、正己烷、异丙醚、乙醇、1,4-二氧六环、丙酮或乙腈中的任意两种;在本发明的一些优选实施方案中,所述溶剂为水、以及选自正庚烷、正己烷、异丙醚、乙醇、1,4-二氧六环、丙酮和乙腈中的任意一种的组合;进一步优选两种溶剂之比为3:1~1:2。本发明优选方案,一种制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型的方法,该方法包括:式(I)化合物或其异构体中加入良溶剂,室温下超声得到溶清液或接近溶清液,然后采用正向添加的方式向样品溶清液中添加相应比例的抗溶剂并搅拌,搅拌至有结晶后减压过滤,干燥;所述良溶剂选自乙醇、异丙醇、丙酮、1,4-二氧六环、乙腈、异丙醚中的一种或多种的组合,所述抗溶剂选自水或正庚烷中的一种或多种的组合;其中当抗溶剂为水时,良溶剂可以选自乙醇、异丙醇、丙酮、1,4-二氧六环、乙腈、异丙醚中的一种或多种的组合;其中当抗溶剂为正庚烷时,良溶剂可以选自乙醇、异丙醇、丙酮、1,4-二氧六环、乙腈、异丙醚中的一种或多种的组合;优选所述良溶剂选自乙醇、异丙醇、丙酮、1,4-二氧六环、乙腈、异丙醚,所述抗溶剂选自水或正庚烷,且抗溶剂与良溶剂之比为5:1~0:1;进一步优选抗溶剂与良溶剂之比为4:1~1:1。附图说明图1为式(I)化合物晶型I的XRD图谱。图2为式(IV)化合物晶型II的XRD图谱。图3为式(II)化合物晶型I的XRD图谱。图4为式(III)化合物晶型II的XRD图谱。图5为式(I)化合物晶型I的差示扫描量热法(DSC)曲线。图6为式(I)化合物晶型I的热重量分析(TGA)曲线。图7为式(IV)化合物晶型II的差示扫描量热法(DSC)曲线。图8为式(IV)化合物晶型II的热重量分析(TGA)曲线。具体实施方式以下通过具体实施例详细说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,不作为对本案可实施范围的限定。化合物的结构是通过核磁共振(NMR)或(和)质谱(MS)来确定的。NMR位移(δ)以10-6(ppm)的单位给出。NMR的测定是用(BrukerAvanceIII400和BrukerAvance300)核磁仪,测定溶剂为氘代二甲基亚砜(DMSO-d6),氘代氯仿(CDCl3),氘代甲醇(CD3OD),内标为四甲基硅烷(TMS)。X-射线粉末衍射仪用所使用的仪器为BrukerD8Advancediffractometer,采用Cu-Ka辐射源,除非特别说明,样品在检测前未经研磨。差热分析扫描仪(DSC)数据采自于TAInstrumentsQ200DSC,样品以10℃/min的升温速度在40mL/min干燥N2的保护下将样品从0℃升温至200℃。热重分析仪(TGA)数据采自于TAInstrumentsQ500TGA,样品以10℃/min的升温速度加热到300℃。傅里叶红外(FT-IR)数据采自于BrukerTensor27。MS的测定用(Agilent6120B(ESI)和Agilent6120B(APCI))。HPLC的测定使用安捷伦1260DAD高压液相色谱仪(ZorbaxSB-C18100×4.6mm)。薄层层析硅胶板使用烟台黄海HSGF254或青岛GF254硅胶板,薄层色谱法(TLC)使用的硅胶板采用的规格是0.15mm~0.20mm,薄层层析分离纯化产品采用的 规格是0.4mm~0.5mm。柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。本发明的己知的起始原料可以采用或按照本领域已知的方法来合成,或可购买于泰坦科技、安耐吉化学、上海德默、成都科龙化工、韶远化学科技、百灵威科技等公司。氮气氛是指反应瓶连接一个约1L容积的氮气气球。氢气氛是指反应瓶连接一个约1L容积的氢气气球。氢化反应通常抽真空,充入氢气,反复操作3次。实施例中无特殊说明,反应在氮气氛下进行。实施例中无特殊说明,溶液是指水溶液。实施例中无特殊说明,反应的温度为室温。室温为最适宜的反应温度,为20℃~30℃。实施例1:2-[(1R)-1-环丙基乙基]-6-异丙基-苯酚(化合物1)2-[(1R)-1-cyclopropylethyl]-6-isopropyl-phenol第一步:1-[(E)-丁-2-烯氧基]-2-异丙基-苯(1B)1-[(E)-but-2-enoxy]-2-isopropyl-benzene10L反应器中加入2-异丙基苯酚1A(3000g,22.0mol)和N,N-二甲基甲酰胺(9.0L),冰水浴降温至10℃。分批加入固体氢氧化钠(969g,24.2mol),保持温度在10℃~15℃之间,加毕,滴加1-氯-2-丁烯(2595g,28.7mol)的N,N-二甲基甲酰胺(6.0L)溶液。滴加完毕,保持温度在10℃~15℃反应5小时。反应液倒入冰水中,用正己烷萃取,有机相用饱和氯化钠溶液洗涤后,无水硫酸钠干燥,过滤减压浓缩得到亮黄色液体的标题产物1-[(E)-丁-2-烯氧基]-2-异丙基-苯1B(3985g,产率95.08%)。1HNMR(400MHz,CDCl3)δ7.20(dd,1H),7.16~7.07(m,1H),6.90(t,1H),6.82(dd,1H),5.89~5.67(m,2H),4.50~4.39(m,2H),3.35(m,1H),1.79~1.69(m,3H),1.21(dd,6H)。第二步:(R,S)-2-异丙基-6-(1-甲基烯丙基)苯酚(1C)(R,S)-2-isopropyl-6-(1-methylallyl)phenol5L反应器中加入1-[(E)-丁-2-烯氧基]-2-异丙基-苯1B(3985g,20.5mol)和碳酸钾(28.3g,0.205mol),氮气保护,升温至200℃反应7小时。反应液冷却至室温,用乙二醇和甲醇的混合溶剂(v:v=1:1)溶解后,加正己烷萃取3次,分出下层。下层加入饱和氯化钠溶液,用正己烷萃取,合并正己烷层,用饱和氯化钠溶液洗涤后,无水硫酸钠干燥。过滤浓缩,残余物柱层析(洗脱剂为正己烷:乙酸乙酯(v:v)=100:1~20:1)后减压蒸馏得无色油状的标题产物(R,S)-2-异丙基-6-(1-甲基烯丙基)苯酚1C(1340g,产率33.63%,HPLC:98.22%)。1HNMR(400MHz,CDCl3)δ7.10(d,1H),6.98(d,1H),6.88(t,1H),6.07(m,1H),5.20(m,3H),3.69~3.56(m,1H),3.24(m,1H),1.40(d,3H),1.23(dd,6H)。第三步:(R,S)-2-(1-环丙基乙基)-6-异丙基-苯酚(1D)(R,S)-2-(1-cyclopropylethyl)-6-isopropyl-phenol氮气流保护下,100L玻璃反应釜中加入1.0M三乙基铝的正己烷溶液(64.00L,64.00mol),冷却降温至-5℃~5℃。搅拌下滴加(R,S)-2-异丙基-6-(1-甲基烯丙基)-苯酚1C(4.06kg,21.34mol)的二氯甲烷(13.56kg)溶液,3小时加毕;滴加二碘甲烷(20.00kg,74.67mol),约40分钟加毕;加料完毕保持30℃~35℃反应48小时。搅拌下,将反应液滴加至10.0%的氢氧化钠水溶液中淬灭。淬灭液静置分层后,分出有机层;水层用正己烷萃取一次,分出碱水层后,合并有机层;有机层依次用去离子水、氯化钠溶液洗涤后,无水硫酸钠干燥。过滤浓缩后,减压蒸馏得到淡黄色油状标题产物(R,S)-2-(1-环丙基乙基)-6-异丙基-苯酚1D(3.52kg,产率80.73%,HPLC:96.59%)。1HNMR(400MHz,CDCl3)δ7.11(d,1H),7.06(d,1H),6.89(t,1H),4.92(s,1H),3.15(m,1H),2.57~2.39(m,1H),1.35~1.19(m,9H),1.04(m,1H),0.55(m,1H),0.51~0.38(m,1H),0.29~0.09(m,2H)。第四步:[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1R)-1-苯乙基]氨基甲酸酯(1E)[2-[(1R,1S)-1-cyclopropylethyl]-6-isopropyl-phenyl]N-[(1R)-1-phenylethyl]carbamate50L玻璃反应釜中依次投入正庚烷(25.00kg)、(R,S)-2-(1-环丙基乙基)-6-异丙基-苯酚1D(5.00kg,24.47mol)、(R)-(+)-1-苯乙基异氰酸酯(4.69kg,31.87mol)及三乙胺(1.24kg,12.26mol),28~33℃反应48小时。反应结束,反应料液过滤,抽滤至干,得湿品。将湿品用乙酸乙酯溶解,搅拌2小时后,加入正己烷,继续搅拌1小时。硅藻土过滤,收集滤液。滤液减压浓缩后,真空干燥得到类白色固状的标题产物[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1R)-1-苯乙基]氨基甲酸酯1E(2.68kg)。第五步:[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-苯基乙基]氨基甲酸酯 (1F)[2-[(1R)-1-cyclopropylethyl]-6-isopropyl-phenyl]N-[(1R)-1-phenylethyl]carbamate将[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1R)-1-苯乙基]氨基甲酸酯1E(2.68kg)用正庚烷重结晶3次,过滤,滤饼烘干得到类白色固体状的标题产物[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-苯基乙基]氨基甲酸酯1F,类白色固体(2.00kg,第四、五步两步总产率46.52%,HPLC:97.31%,chiral-HPLC:99.90%)。1HNMR(400MHz,CDCl3)δ7.45~7.01(m,8H),5.27(d,1H),4.91(m,1H),3.15~2.86(m,1H),2.08(s,1H),1.55(d,3H),1.33~0.86(m,10H),0.49(s,1H),0.31(s,1H),0.15~-0.04(m,2H)。第六步:2-[(1R)-1-环丙基乙基]-6-异丙基-苯酚(化合物1)2-[(1R)-1-cyclopropylethyl]-6-isopropyl-phenol50L玻璃反应釜,通入氮气流,投入1,4-二氧六环(10.17kg)和[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-苯基乙基]氨基甲酸酯1F(1.99kg),滴加6.0%氢氧化钠水溶液(10.17kg),加毕,升温至65℃~75℃反应2小时。将正己烷加入冷却的反应液中,搅拌后铺硅藻土过滤,收集滤液。滤液转入100L玻璃釜中,加入去离子水,搅拌静置分层后,分出水层,再用正己烷萃取一次,合并有机层;有机层依次用3.6%盐酸水溶液、去离子水、10.0%碳酸氢钠水溶液及氯化钠水溶液洗涤后,无水硫酸钠干燥。过滤浓缩后,减压蒸馏得到淡黄色油状标题产物2-[(1R)-1-环丙基乙基]-6-异丙基-苯酚化合物1(1.00kg,产率86.45%,HPLC:99.93%,chiral-HPLC:99.91%)。1HNMR(400MHz,CDCl3)δ7.11(d,2H),7.06(d,2H),6.89(t,1H),4.91(s,1H),3.15(m,1H),2.49(m,1H),1.35~1.19(m,9H),1.10~0.98(m,1H),0.55(m,1H),0.50~0.41(m,1H),0.19(m,2H)。实施例2:2-[(1S)-1-环丙基乙基]-6-异丙基-苯酚(化合物2)2-[(1S)-1-cyclopropylethyl]-6-isopropyl-phenol第一步:[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1S)-1-苯乙基]氨基甲酸酯(2A)[2-[(1R,1S)-cyclopropylethyl)]-6-isopropyl-phenyl]N-[(1S)-1-phenylethyl]carbamate向反应瓶中加入(R,S)-2-(1-环丙基乙基)-6-异丙基苯酚1D(42.00g,205.57mmol)和四氢呋喃(200mL),滴加三乙胺(58.00g,573.18mmol),搅拌均匀后加入(S)-(-)-1-苯乙基异氰酸酯(45.00g,308.36mmol),加热至63℃搅拌6小时,减压浓缩,用乙酸乙酯溶解,减压抽滤,将滤液减压浓缩得到白色固体状的标题产物[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1S)-1-苯乙基]氨基甲酸酯2A(80.00g)。MSm/z(ESI):352.5[M+1]+。1HNMR(400MHz,CDCl3):δ7.38~7.11(m,8H),5.27~5.08(m,1H),4.94~4.87(m,1H),3.00~2.97(m,1H),2.08(s,1H),1.55(d,3H),1.23~1.13(m,9H),0.95(s,1H),0.49(s,1H),0.31(s,1H),0.05(s,1H)。第二步:[2-[(1S)-1-环丙基乙基]-6-异丙基苯基]N-[(1S)-苯基乙基]氨基甲酸酯(2B)[2-[(1S)-1-cyclopropylethyl]-6-isopropyl-phenyl]N-[(1S)-1-phenylethyl]carbamate将上一步所得到的[2-[(1R,1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1S)-1-苯乙基]氨基甲酸酯2A(80.00g)用正己烷重结晶4次,过滤,滤饼烘干得到白色粉末状的标题产物[2-[(1S)-1-环丙基乙基]-6-异丙基苯基]N-[(1S)-1-苯乙基]氨基甲酸酯2B(39g,产率:54.93%,HPLC:97.62%,chiral-HPLC:99.84%)。化合物1D含有1个手性中心,拆分后只能得到两个异构体,即化合物1和化合物2。化合物2B含有两个手性中心,其中1个手性中心由(S)-(-)-1-苯乙基异氰酸酯引入,环丙烷基所连接的手性碳原子与化合物2的手性一致。第三步:2-[(1S)-1-环丙基乙基]-6-异丙基-苯酚(化合物2)2-[(1S)-1-cyclopropylethyl]-6-isopropyl-phenol将[2-[(1S)-1-环丙基乙基]-6-异丙基苯基]N-[(1S)-1-苯乙基]氨基甲酸酯2B(39.00g,110.96mmol)溶于四氢呋喃(390mL)中,加入1.0M氢氧化钠水溶液(190mL,190mmol),氮气保护,加热至70℃反应4小时,静置分层,收集有机层,水层用1M盐酸调节pH为7,用乙酸乙酯萃取,合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=100:1)得到淡黄色液体状的标题产物2-[(1S)-1-环丙基乙基]-6-异丙基-苯酚化合物2(17.2g,产率:75.80%,HPLC:97.67%,chiral-HPLC:99.86%)。化合物1D为含一个手性中心,拆分后能且只能得到两个异构体,即化合物1和化合物2。MSm/z(ESI):203.1[M-1]-。1HNMR(400MHz,CDCl3):δ7.14(dd,1H),7.08(dd,1H),6.93(t,1H),4.93(s,1H),3.22~3.15(m,1H),2.55~2.48(m,1H),1.32(d,6H),1.28(d,3H),1.10~1.04(m,1H),0.60~0.58(m,1H),0.49~0.46(m,1H),0.25~0.18(m,2H)。实施例3:[2-[(1S)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1R)-1-苯乙基]氨基甲酸酯 (化合物3)[2-[(1S)-1-cyclopropylethyl]-6-isopropyl-phenyl]N-[(1R)-1-phenylethyl]carbamate向反应瓶中加入2-[(1S)-1-环丙基乙基]-6-异丙基-苯酚(化合物2)(5.00g,24.47mmol)和四氢呋喃(50mL),滴加三乙胺(4.95g,48.95mmol),搅拌均匀后加入(1R)-1-苯乙基异氰酸酯(5.40g,36.71mmol),加热至63℃搅拌过夜,减压浓缩,用乙酸乙酯(10mL)溶解,减压抽滤,将滤液减压浓缩,得到白色固体状的[2-[(1S)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-1-苯乙基]氨基甲酸酯(化合物3)(9.00g,HPLC:98.4%,chiral-HPLC:97.3%)。MSm/z(ESI):352.5[M+1]。1HNMR(400MHz,CDCl3):δ7.36~7.13(m,8H),5.26(d,1H),4.94~4.87(m,1H),3.01~2.98(m,1H),2.04~2.09(m,1H),1.55(d,3H),1.20~1.15(m,9H),0.96~0.93(m,1H),0.45~0.43(m,1H),0.37~0.30(m,1H),0.18~0.09(m,2H)。实施例4:[2-[(1R)-(1-环丙基乙基)]-6-异丙基-苯基]N-[(1S)-1-苯乙基]氨基甲酸酯(化合物4)[2-[(1R)-1-cyclopropylethyl]-6-isopropyl-phenyl]N-[(1S)-1-phenylethyl]carbamate向反应瓶中加入2-[(1R)-1-环丙基乙基]-6-异丙基-苯酚(化合物1)(5.00g,24.47 mmol)和四氢呋喃(50mL),滴加三乙胺(4.95g,48.95mmol),搅拌均匀后加入(1S)-1-苯乙基异氰酸酯(5.40g,36.71mmol),加热至63℃搅拌过夜,减压浓缩,用乙酸乙酯(10mL)溶解,减压抽滤,将滤液减压浓缩,得到白色固体状的[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1S)-1-苯乙基]氨基甲酸酯(化合物4)(9.50g,HPLC:99.4%,chiral-HPLC:99.5%)。MSm/z(ESI):352.5[M+1]。实施例5:[2-[(1R)-1-环丙基乙基]-6-异丙基苯基]N-[(1R)-苯基乙基]氨基甲酸酯(1F)的X-射线单晶衍射测试从实施例1的第五步制备得到的类白色固体中选取大小为0.30mm×0.20mm×0.20mm的无色片状单晶粘合于玻璃丝上,衍射实验用晶体为三斜晶系,空间群为P1,晶胞参数:a=5.3665(3),b=10.3493(11),α=97.598(9)°,β=96.660(7)°,γ=90.165(6)°,晶胞体积不对称单元数为Z=2。在Xcalibur四圆单晶衍射仪上于293.15K用MoKα射线(λ=0.7107,射线管管压:50kv,管流:40ma)收集衍射强度数据,晶体与CCD探测器间的距离D=45mm,扫描方式:2θ(6.32°<θ<52.744°),总共收集到8385个衍射点(-6≤h≤6,-12≤k≤12,-21≤l≤23),其中独立衍射点为5645个[Rint=0.0372,Rsigma=0.0588]。晶体衍射强度数据的采集和还原使用了衍射仪配套软件:CrysAlisPro,晶体结构解析使用了Olex2和SHElXS-13(直接法),对全部原子的坐标以及各向异性参数用SHElXL-13精修(偏最小二乘法)。最终获得的晶体结构其残差因子R1=0.0850,wR2=0.2088[I>=2σ(I)],R1=0.1115,wR2=0.2405[alldata],S=1.064,精修参数480个,约束条件3个。化合物1F的16位碳原子的绝对构型由已知的(R)-(+)-1-苯乙基异氰酸酯引入, 故化合物1F的16位碳原子的绝对构型为已知的R构型,根据X-射线单晶衍射图谱(图1)显示:C-7的绝对构型与16-C的绝对构型一致,所以也为R构型。由化合物1F的绝对构型从而确证了化合物1的C-7的绝对构型为R构型。实施例1:晶浆结晶法制备式(I)化合物及其异构体式(II)、(III)、(IV)晶型。取约50mg研究样品放置于10mL玻璃小瓶中,将5.0mL单一溶剂或溶剂A和溶剂B以不同比例形成的混合溶剂加入至上述放置样品的玻璃小瓶中,然后将放置样品和溶剂的玻璃小瓶置于室温下超声10min保证得到悬浊液,然后将超声得到的悬浊液在室温下晶浆3天。取晶浆后的浑浊液减压过滤,结晶物干燥后取样XRD、DSC和TGA表征。研究结果见表1-表8。表1式(I)化合物制得晶型结果编号温度溶剂A溶剂BA:B研制结果XRD结果1室温水乙醇1:1有结晶晶型I2室温水异丙醇1:1有结晶晶型I3室温水丙酮1:1有结晶晶型I4室温水1,4-二氧六环1:1有结晶晶型I式(I)化合物晶型研究结果显示结晶物均为同一种晶型I,未发现其他新晶型。如图5中DSC曲线所见,晶型I具有良好的热稳定性,DSC曲线显示晶型I吸热热流的起点在约112.6℃处(焓为102.2J/g)。DSC和TGA曲线(图6)数据说明晶型I中无混晶、溶剂和结晶水现象。表2式(I)化合物晶型I的特征粉末X-射线衍射(XRD)峰(图1)序号°2θd值014.61619.128029.3949.4070310.1658.6950411.8407.4690515.6085.6730616.4595.3810717.0915.1840818.4134.8140919.7964.4811021.8874.0571123.8803.723表3式(IV)化合物制得晶型结果编号温度溶剂A溶剂BA:B研制结果XRD结果1室温水乙醇1:1有结晶晶型II2室温水异丙醇1:1有结晶晶型II式(IV)化合物晶型研究结果显示结晶物均为同一种晶型II,未发现其他新晶型。如图7中DSC曲线所见,晶型II具有良好的热稳定性,DSC曲线显示晶型II吸热热流的起点在约76.2℃处(焓为56.8J/g)。DSC和TGA曲线(图8)数据显示晶型II中无混晶、溶剂和结晶水现象。表4式(IV)化合物晶型II的特征粉末X-射线衍射(XRD)峰(图2)序号°2θd值018.56610.314029.3759.4260310.5788.3560411.5057.6850514.3686.1600615.1765.8330717.2695.1310817.6455.0220918.9084.6901019.2424.6091120.8034.2661221.0994.2071322.9523.8721423.2683.8201524.2363.669表5式(II)化合物制得晶型结果编号温度溶剂A溶剂BA:B研制结果XRD结果1室温水乙醇1:1有结晶晶型I2室温水异丙醇1:1有结晶晶型I3室温水丙酮1:1有结晶晶型I4室温水1,4-二氧六环1:1有结晶晶型I式(II)化合物晶型研究结果显示结晶物均为同一种晶型I,未发现其他新晶型,DSC和TGA数据显示晶型I中无混晶、溶剂和结晶水现象。表6式(II)化合物晶型I的特征粉末X-射线衍射(XRD)峰(图3)序号°2θd值014.53519.467029.2579.5450310.0878.7620416.9945.2130517.4305.0840618.3944.8190719.7364.4950821.8104.0720923.7813.7381024.0173.7021124.2353.6691225.3393.5121325.7733.454表7式(III)化合物制得晶型结果编号温度溶剂A溶剂BA:B研制结果XRD结果1室温水乙醇1:1有结晶晶型II2室温水异丙醇1:1有结晶晶型II式(III)化合物晶型研究结果显示结晶物均为同一种晶型II,未发现其他新晶型,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。表8式(III)化合物晶型II的特征粉末X-射线衍射(XRD)峰(图4)序号°2θd值018.54510.339029.3549.4470310.5568.3740411.4477.7240514.3296.1760615.1385.8480717.2295.1420817.6245.0280918.8694.6991019.2024.6181120.8024.2671221.0784.2111322.9133.8781423.2293.8261524.1783.678实施例2:挥发结晶法制备式(I)化合物及其异构体式(II)、(III)、(IV)的晶型。取约25mg研究样品放置于10ml或25mL玻璃小瓶中,将0.25~20.0mL单一溶剂加入至上述放置样品的玻璃小瓶中,然后将放置样品和溶剂的玻璃小瓶置于室温下超声30min得到溶清液或接近溶清液(若超声后形成的是分层混悬液,则取溶清层),直接敞口放置在室温下自然挥发。所得结晶物干燥后取样XRD、DSC和TGA表征。结果见表9-12。表9式(I)化合物制得晶型结果编号温度溶剂B研制结果XRD结果1室温乙醇有结晶晶型I2室温丙酮有结晶晶型I3室温1,4-二氧六环有结晶晶型I4室温异丙醚有结晶晶型I5室温正己烷有结晶晶型I6室温正庚烷有结晶晶型I式(I)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显示晶型I中无混晶、溶剂和结晶水现象。表10式(IV)化合物制得晶型结果编号温度溶剂B研制结果XRD结果1室温乙醇有结晶晶型II2室温丙酮有结晶晶型II3室温1,4-二氧六环有结晶晶型II4室温异丙醚有结晶晶型II5室温正己烷有结晶晶型II6室温正庚烷有结晶晶型II式(IV)化合物晶型研究结果显示结晶物均为同一种晶型II,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。表11式(II)化合物制得晶型结果编号温度溶剂B研制结果XRD结果1室温乙醇有结晶晶型I2室温丙酮有结晶晶型I3室温1,4-二氧六环有结晶晶型I4室温异丙醚有结晶晶型I5室温正己烷有结晶晶型I6室温正庚烷有结晶晶型I式(II)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显 示晶型I中无混晶、溶剂和结晶水现象。表12式(III)化合物制得晶型结果编号温度溶剂B研制结果XRD结果1室温乙醇有结晶晶型II2室温丙酮有结晶晶型II3室温1,4-二氧六环有结晶晶型II4室温异丙醚有结晶晶型II5室温正己烷有结晶晶型II6室温正庚烷有结晶晶型II式(III)化合物晶型研究结果显示结晶物均为同一种晶型II,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。实施例3:冷却结晶法制备式(I)化合物及其异构体(II)、(III)、(IV)的晶型。取约50mg研究样品放置于10mL或25ml玻璃小瓶中,将2.0~10.0mL溶剂A和溶剂B以不同比例形成的混合溶剂加入至上述放置样品的玻璃小瓶中,然后将放置样品和溶剂的玻璃小瓶置于室温下超声30min得到悬浊液,将装有悬浊液的玻璃小瓶直接放入加热至60℃的水浴中并搅拌至溶清或接近溶清,放置于室温搅拌。有结晶后减压过滤,干燥后取样XRD、DSC和TGA表征。结果见表13-表16。表13式(I)化合物制得晶型结果编号溶剂A溶剂BA:B研制结果XRD结果1/正庚烷/有结晶晶型I2/正己烷/有结晶晶型I3/异丙醚/有结晶晶型I4水乙醇1:2有结晶晶型I5水1,4-二氧六环3:1有结晶晶型I6水丙酮1:1有结晶晶型I7水乙腈3:2有结晶晶型I式(I)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显 示晶型I中无混晶、溶剂和结晶水现象。表14式(IV)化合物制得晶型结果编号溶剂A溶剂BA:B研制结果XRD结果1/正庚烷/有结晶晶型II4水乙醇1:2有结晶晶型II式(IV)化合物晶型研究结果显示结晶物均为同一种晶型II,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。表15式(II)化合物制得晶型结果编号溶剂A溶剂BA:B研制结果XRD结果1/正庚烷/有结晶晶型I2/正己烷/有结晶晶型I3/异丙醚/有结晶晶型I4水乙醇1:2有结晶晶型I5水1,4-二氧六环3:1有结晶晶型I6水丙酮1:1有结晶晶型I7水乙腈3:2有结晶晶型I式(II)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显示晶型I中无混晶、溶剂和结晶水现象。表16式(III)化合物制得晶型结果编号溶剂A溶剂BA:B研制结果XRD结果1/正庚烷/有结晶晶型II2/正己烷/有结晶晶型II4水乙醇1:2有结晶晶型II5水1,4-二氧六环3:1有结晶晶型II式(III)化合物晶型研究结果显示结晶物均为同一种晶型II,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。实施例4:抗溶剂结晶法制备式(I)化合物及其异构体(II)、(III)晶型。取约50mg研究样品放置于5mL或10ml玻璃小瓶中,将1.0mL良溶剂加入至上述放置样品的玻璃小瓶中,然后将放置样品和良溶剂的玻璃小瓶置于室温下超声30min得到溶清液或接近溶清液,然后采用正向添加的方式向样品溶清液中添加相应比例的抗溶剂并搅拌,搅拌至有结晶后减压过滤,干燥后取样XRD、DSC和TGA表征。结果见17-表19。表17式(I)化合物制得晶型结果编号抗溶剂良溶剂抗溶剂:良溶剂研制结果XRD结果1水乙醇4:1有结晶晶型I2水异丙醇2:1有结晶晶型I3水丙酮4:1有结晶晶型I4水1,4-二氧六环4:1有结晶晶型I5水乙腈4:1有结晶晶型I6正庚烷异丙醚1:1有结晶晶型I式(I)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显示晶型I中无混晶、溶剂和结晶水现象。表18式(II)化合物制得晶型结果编号抗溶剂良溶剂抗溶剂:良溶剂研制结果XRD结果1水乙醇4:1有结晶晶型I2水异丙醇2:1有结晶晶型I3水丙酮4:1有结晶晶型I4水1,4-二氧六环4:1有结晶晶型I5水乙腈4:1有结晶晶型I6正庚烷异丙醚1:1有结晶晶型I式(II)化合物晶型研究结果显示结晶物均为同一种晶型I,DSC和TGA数据显示晶型I中无混晶、溶剂和结晶水现象。表19式(III)化合物制得晶型结果编号抗溶剂良溶剂抗溶剂:良溶剂研制结果XRD结果1水乙醇4:1有结晶晶型II2水异丙醇2:1有结晶晶型II3水丙酮4:1有结晶晶型II式(III)化合物晶型研究结果显示结晶物均为同一种晶型II,DSC和TGA数据显示晶型II中无混晶、溶剂和结晶水现象。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1