一种制备手性二氢1,4-苯并噁嗪类化合物的方法与流程
本发明属于有机合成领域,具体涉及一种由邻氨基苯酚类化合物和炔丙基类化合物通过不对称[4+2]环加成反应合成手性二氢1,4-苯并噁嗪类化合物的方法。
背景技术:
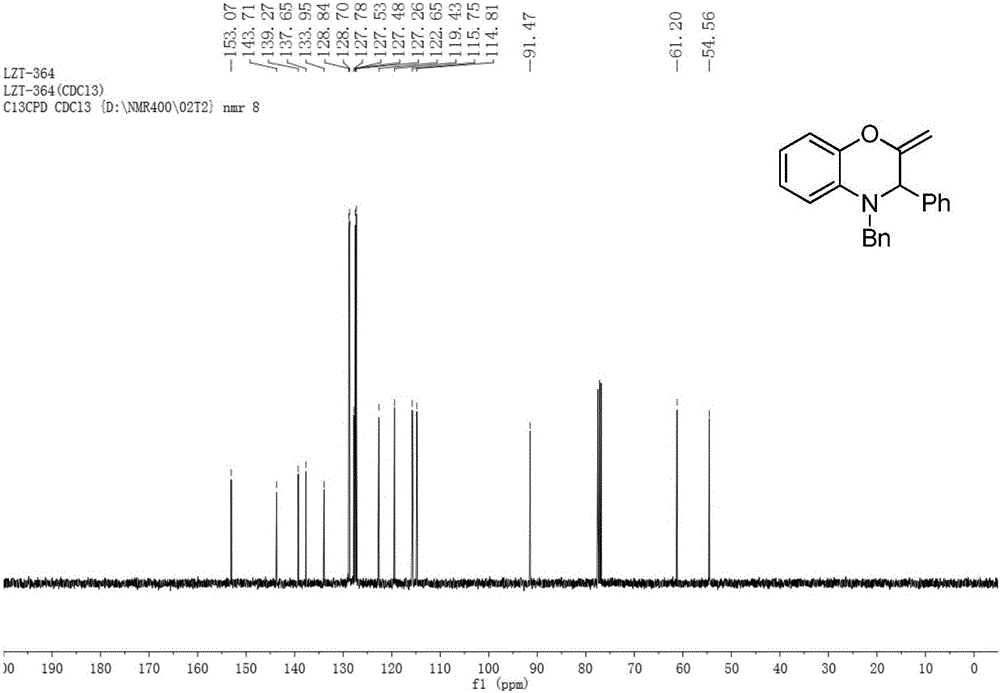
:许多1,4-苯并噁嗪类衍生物在临床医学和生物学上具有极其重要的生物活性,常见的有细胞内钙拮抗物、风湿药、钾通道活化剂、神经抗氧化剂、降压药、抗菌剂、凝血酶阻聚剂、抗炎药、抗生素C-1027等等[(a)Yamazaki,A.;Achiwa,I.;Achiwa,K.Tetrahedron:Asymmetry.1996,7,403.(b)J.;Jakopin,T.;Stegnar,M.;Kikelj,D.J.Med.Chem.2008,51,5617.(c)Otsuka,H.;Hirai,Y.Nagao,T.;Yamasaki,K.JournalofNaturalProducts1988,51,74.(d)Minami,Y.;Yoshida,K.;Azuma,R.;Saeki,M.;Otani,T.TetrahedronLett.1993,34,2633.(e)Touzeau,F.;Arrault,A.;Guillaumet,G.;Scalbert,E.;Pfeiffer,B.;Rettori,M.;Renard,P.;Mérour,J.-Y.J.Med.Chem.2003,46,1962.(f)Largeron,M.;Fleury,M.-B.TetrahedronLett.1998,39,8999.(g)Largeron,M.;Lockhart,B.;Pfeiffer,B.;Fleury,M.-B.J.Med.Chem.1999,42,5043.(h)Matsuoka,H.;Ohi,N.;Mihara,M.;Suzuki,H.;Miyamoto,K.;Maruyama,N.;Tsuji,K.;Kato,N.;Akimoto,T.;Takeda,Y.;Yano,K.;Kuroki,T.J.Med.Chem.1997,40,105–111.在这些具有药物活性的二氢1,4-苯并噁嗪类化合物中,通常含有手性碳中心,即为手性二氢1,4-苯并噁嗪类化合物。因此发展不对称合成方法来制备手性二氢1,4-苯并噁嗪类化合物具有重要的意义。二氢1,4-苯并噁嗪类化合物的制备,多以2-卤苯酚,2-氨基苯酚和2-硝基苯酚为起始底物,与合适底物进行环加成反应得到;也有通过环氧化物开环或者取代反应生成单取代产物,接着进行分子内环化反应得到目标产物。[(a)Albanese,D.;Landini,D.;Lupi,V.;Penso,M.Ind.Eng.Chem.Res.2003,42,680.(b)Kuroita,T.;Sakamori,M.;Kawakita,T.Chem.Pharm.Bull.1996,44,756.(c)Singh,S.K.;Bajpai,A.K.;Saini,R.TetrahedronLetters2013,54,7132.(d)Matsumoto,Y.;Tsuzuki,R.;Matsuhisa,A.;Takayama,K.;Yoden,T.;Uchida,W.;Asano,M.;Fujita,S.;Yanagisawa,I.;Fujikura,T.Chem.Pharm.Bull.1996,44,103.(e)Albanese,D.;Landini,D.;Lupi,V.;Penso,M.Ind.Eng.Chem.Res.2003,42,680.(f)Brahma,K.;Das,B.;Chowdhury,C.Tetrahedron2014,70,5863.(g)Rao,R.K.Naidu,A.B.;Sekar,G.Org.Lett.2009,11,1923.(h)Largeron,M.;Lockhart,B.;Pfeiffer,B.;Fleury,M.B.J.Med.Chem.1999,42,5043.(i)Ramesh,C.;Raju,B.R.;Kavala,V.;Kuo,C.-W.;Yao,C.-F.Tetrahedron2011,67,1187.(j)Sharifi,A.;Abaee,M.S.; Mokhtare,Z.;Mirzaei,M.EnvironmentalChemistryLetters2014,12,365.(k)Yu,G.;Zheng,Y.;Wu,J.;Dai,W.-M.Tetrahedron2013,69,10488.(l)Zidar,N.;Kikelj,D.Tetrahedron2008,64,5756.(m)Trost,B.M.;VanVranken,D.L.;Bingel,C.J.Am.Chem.Soc.1992,114,9327.(n)Feng,G.;Wu,J.;Dai,W.-M.Tetrahedron2006,62,4635.(o)Ramesh,C.;Raju,B.R.;Kavala,V.;Kuo,C.-W.;Yao,C.-F.Tetrahedron2011,67,1187.本专利利用手性铜催化剂催化邻氨基苯酚类化合物与炔丙基类化合物进行[4+2]不对称环加成反应,高收率和高对映选择性地得到了手性二氢1,4-苯并噁嗪类化合物,为手性二氢1,4-苯并噁嗪类化合物提供了一条操作简单,高收率,对映选择性高的合成新路线。技术实现要素:本发明的目的是提供一种铜催化的邻氨基苯酚类化合物与炔丙基类化合物通过[4+2]不对称环加成反应来合成手性二氢1,4-苯并噁嗪类化合物的方法。本发明具有原料易得,操作简单,反应条件温和,对映选择性高等特点。本发明提供了一种手性二氢1,4-苯并噁嗪类化合物的催化不对称合成方法,在碱添加剂的存在下,手性铜催化剂在反应介质中催化邻氨基苯酚类化合物与炔丙基类化合物通过[4+2]不对称环加成反应合成手性二氢1,4-苯并噁嗪类化合物,具体步骤为:(1)手性铜催化剂的制备:氮气保护下,将铜盐与手性P,N,N-配体按摩尔比1:0.1-1:10在反应介质中搅拌0.5-2小时制得手性铜催化剂;(2)手性二氢1,4-苯并噁嗪类化合物的制备:将邻氨基苯酚类化合物、炔丙基类化合物和碱添加剂溶于反应介质中,然后将该溶液在氮气保护下加入到上述步骤(1)中制得的手性铜催化剂的溶液中,室温搅拌反应1-24小时;反应完毕,减压旋蒸,柱层析分离,得到手性二氢1,4-苯并噁嗪类化合物;上述步骤(2)中所述手性铜催化剂与炔丙基类化合物的摩尔比为0.001:1-1:1;所述碱添加剂与炔丙基类化合物的摩尔比为0-10:1;所述邻氨基苯酚类化合物与炔丙基类化合物的摩尔比为1:1-2:1。所述手性二氢1,4-苯并噁嗪类化合物具有以下两种结构之一:I和II互为对映异构体,式中,R1,R2为C1-C40的烷基、C3-C12的环烷基、带有取代基的C3-C12环烷基、苯基、取代苯基、苄基、取代苄基、含一个或二个以上氧、硫、氮原子的五元或六元杂环芳香基团或酯基中的 一种或两种以上;上述C3-C12环烷基上的取代基、苯基上的取代基和苄基上的取代基为C1-C40烷基、C1-C40的烷氧基、卤素、硝基、酯基或氰基中的一种或两种以上。所述反应介质为甲醇、乙醇、甲苯、苯、二甲苯、二氯甲烷、二氯乙烷、乙醚、四氢呋喃或乙酸乙酯中的一种或两种以上。所述邻氨基苯酚类化合物具有以下结构:式中:R1为C1-C40的烷基、C3-C12的环烷基、带有取代基的C3-C12环烷基、苯基、取代苯基、苄基、取代苄基、含一个或二个以上氧、硫、氮原子的五元或六元杂环芳香基团或酯基中的一种或二种以上;上述C3-C12环烷基上的取代基、苯基上的取代基和苄基上的取代基为C1-C40烷基、C1-C40的烷氧基、卤素、硝基、酯基或氰基中的一种或两种以上。所述炔丙基类化合物具有以下结构:式中:R2为C1-C40的烷基、C3-C12的环烷基、带有取代基的C3-C12环烷基、苯基、取代苯基、苄基、取代苄基、含一个或二个以上氧、硫、氮原子的五元或六元杂环芳香基团或酯基中的一种或二种以上;上述C3-C12环烷基上的取代基、苯基上的取代基和苄基上的取代基为C1-C40烷基、C1-C40的烷氧基、卤素、硝基、酯基或氰基中的一种或两种以上;X为氟、氯、溴、碘、烷基羧酸酯、烷基碳酸酯、烷基磺酸酯、烷基磷酸酯、苯基、取代苯基羧酸酯、苯基、取代苯基碳酸酯、苯基、取代苯基磺酸酯、苯基或取代苯基磷酸酯中的一种或两种以上。所述铜盐为Cu(OAc)2·H2O、CuSO4·H2O、Cu(OAc)2、CuSO4、Cu(OTf)2、CuCl2、CuOAc、CuCl、CuI、CuClO4、CuOTf·1/2C6H6、Cu(CH3CN)4BF4或Cu(CH3CN)4ClO4中的一种或两种以上。所述手性P,N,N-配体具有以下结构特征:式中:R3,R4为H、C1~C10内的烷基、C3~C8内的环烷基、苯基、取代苯基、苄基或取代苄基中的一种或两种以上;R5,R6为H、卤素、烷基、环烷基、苯基、取代苯基、烷氧基、苯氧基、酰基或硝基中的一种或两种以上;R7为烷基、环烷基、苯基、取代苯基、萘基、取代萘基、含一个或以上氧、硫、氮原子的五元或六元杂环芳香基团中的一种或两种以上。所述碱添加剂为各种无机碱或有机碱,为iPr2NEt、NEt3、tBuOK、KOH、NaOH、K2CO3、Na2CO3、或NaHCO3中的一种或两种以上。所述步骤(2)催化反应条件优选为:温度为-40℃;反应介质为甲醇;压力为常压;时间为24小时。所述手性铜催化剂与炔丙基类化合物的摩尔比优选为0.001:1-1:1;所述碱添加剂与炔丙基类化合物的摩尔比优选为1.2:1;所述邻氨基苯酚类化合物与炔丙基类化合物的摩尔比优选为1:1;所述铜盐优选Cu(OAc)2·H2O、Cu(OTf)2、Cu(CH3CN)4BF4或Cu(CH3CN)4ClO4中的一种或两种以上;所述反应介质优选甲醇、二氯甲烷中的一种或两种。本发明的反应方程式为:本发明具有以下优点:1、起始原料廉价易得。2、手性配体合成简便,催化剂廉价易得,用量少。3、反应活性好、立体选择性高,反应条件易实现。4、与传统方法相比较,该方法可以更方便地合成各种取代的手性二氢1,4-苯并噁嗪类化合物。附图说明图1为实施例1制备的手性二氢1,4-苯并噁嗪类环加成产物Ⅱ-1的核磁共振氢谱;图2为实施例1制备的手性二氢1,4-苯并噁嗪类环加成产物Ⅱ-1的核磁共振碳谱;图3为实施例11制备的产物Ⅱ-2的核磁共振氢谱;图4为实施例11制备的产物Ⅱ-2的核磁共振碳谱;图5为实施例12制备的产物Ⅱ-3的核磁共振氢谱;图6为实施例12制备的产物Ⅱ-3的核磁共振碳谱;图7为实施例13制备的产物Ⅱ-4的核磁共振氢谱;图8为实施例13制备的产物Ⅱ-4的核磁共振碳谱;图9为实施例14制备的产物Ⅱ-5的核磁共振氢谱;图10为实施例14制备的产物Ⅱ-5的核磁共振碳谱;图11为实施例15制备的产物Ⅱ-6的核磁共振氢谱;图12为实施例15制备的产物Ⅱ-6的核磁共振碳谱;图13为实施例16制备的产物Ⅱ-7的核磁共振氢谱;图14为实施例16制备的产物Ⅱ-7的核磁共振碳谱。具体实施方式下面的实施例将对本发明予以进一步的说明,但并不因此而限制本发明。核磁共振是通过Bruker400核磁共振仪测定,高效液相色谱(HPLC)是通过Agilent1100系列高效液相色谱测定。实施例1:Cu(OTf)2和L-2-1络合作为催化剂催化反应,生成手性二氢1,4-苯并噁嗪类环加成产物Ⅱ-1。在反应瓶中加入金属前体Cu(OTf)2(0.015mmol,5mol%)及手性配体L-2-1(0.0165mmol,5.5mol%),氮气保护下加入1.0mL无水甲醇,室温搅拌1小时。然后将反应管移至-40℃的恒温反应冷冻机中,将炔丙醇酯Ⅳ-1(0.3mmol,1equiv),N-苄基邻氨基苯酚Ⅲ-1(0.3mmol,1equiv)和K2CO3(0.36mmol,1.2equiv)溶于2.0mL无水甲醇中,然后将该溶液在氮气保护下加入到上述搅拌好的催化剂的溶液中,-40℃搅拌反应24h。反应完毕,减压浓缩至无挥发性溶剂,硅胶柱层析分离(石油醚:乙酸乙酯=100:1),减压浓缩至无挥发性溶剂,真空干燥,得到化合物Ⅱ-1,无色油状物,98%收率,95%ee.产物Ⅱ-1的核磁共振氢谱和核磁共振碳谱分别如图1、图2所示:1HNMR(400MHz,CDCl3)δ7.23–7.17(m,4H),7.16–7.04(m,6H),6.81–6.78(m,1H),6.75–6.71(m,1H),6.65–6.60(m,2H),4.71(s,1H),4.62(d,J=1.1Hz,1H),4.42(d,J=15.7Hz,1H),4.20(d,J=1.4Hz,1H),4.12(d,J=15.7Hz,1H).13CNMR(101MHz,CDCl3)δ153.1,143.7,139.3,137.7,134.0,128.8,128.7,127.8,127.5,127.5,127.3,122.7,119.4,115.8,114.8,91.5,61.2,54.6.HPLC(ChiralcelAD-H,n-hexane/i-PrOH=98/2,0.8mL/min,254nm,40℃):tR(major)=7.0min,tR(minor)=8.0min。Ⅲ-1,Ⅳ-1,Ⅱ-1,L-2-1的结构式如下:实施例2:L-1-1作为配体反应生成产物Ⅱ-1将实施例1中的配体L-2-1用配体L-1-1代替,Cu(OTf)2用Cu(OAc)2·H2O代替,反应温度为0℃,其余同实施例1。反应得到化合物Ⅱ-1,96%收率,69%ee。L-1-1的结构式如下:实施例3:L-2-2作为配体反应生成产物Ⅱ-1。将实施例1中的配体L-2-1用配体L-2-2代替,Cu(OTf)2用Cu(CH3CN)4BF4代替,反应温度为0℃,其余同实施例1。反应得到化合物Ⅱ-1,95%收率,80%ee。L-2-2的结构式如下:实施例4:L-3作为配体反应生成产物Ⅰ-1将实施例1中的配体L-2-1用配体L-3代替,反应温度为0℃,其余同实施例1。反应得到化合物Ⅰ-1,<5%收率。实施例5:Cu(OAc)2·H2O和L-2-1催化反应生成产物Ⅱ-1将实施例1中Cu(OTf)2用Cu(OAc)2·H2O代替,反应温度为0℃,其余同实施例1。得到化合物Ⅱ-1,88%收率,79%ee。实施例6:CuI和L-2-1催化反应生成产物Ⅱ-1将实施例1中的Cu(OTf)2用CuI代替,反应温度为0℃,其余同实施例1,得到化合物Ⅱ-1,79%收率,82%ee。实施例7:没有碱添加剂,无产物Ⅱ-1将实施例1中的K2CO3去掉,没有得到产物Ⅱ-1。实施例8:N,N-iPr2NEt作为碱添加剂反应生成产物Ⅱ-1将实施例1中的K2CO3替换为N,N-iPr2NEt,温度为0℃,其余同实施例1,得到化合物Ⅱ-1,75%收率,83%ee。实施例9:二氯甲烷作为溶剂反应生成产物Ⅱ-1将实施例1中的甲醇溶剂替换为二氯甲烷,其余同实施例1,得到化合物Ⅱ-1,48%收率,35%ee。实施例10:甲苯作为溶剂反应生成产物Ⅱ-1将实施例1中的甲醇溶剂替换为甲苯,反应温度为0℃,其余同实施例1,得到化合物Ⅱ-1,26%收率,48%ee。实施例11:Ⅲ-2作为底物反应生成产物Ⅱ-2将实施例1中的N-苄基邻氨基苯酚Ⅲ-1替换为Ⅲ-2,其余同实施例1,得到化合物Ⅱ-2,94%收率,96%ee。产物Ⅱ-2的核磁共振氢谱和核磁共振碳谱分别如图3、图4所示:1HNMR(400MHz,CDCl3)δ7.35–7.31(m,2H),7.28–7.26(m,3H),7.25–7.21(m,3H),7.19–7.16(m,2H),6.86–6.79(m,2H),6.75–6.73(m,1H),4.81(s,1H),4.70(d,J=0.9Hz,1H),4.54(d,J=15.7Hz,1H),4.34(d,J=1.4Hz,1H),4.18(d,J=15.7Hz,1H).13CNMR(101MHz,CDCl3)δ152.5,142.5,138.8,136.7,135.2,128.9,128.8,128.0,127.7,127.5,127.0,121.6,117.0,116.4,114.9,91.8,60.6,53.7.HPLC(ChiralcelOJ-H,n-hexane/i-PrOH=95/5,0.8mL/min,254nm,40℃):tR(minor)=10.2min,tR(major)=15.5min.Ⅲ-2,Ⅱ-2的结构式如下:实施例12:Ⅲ-3作为底物反应生成产物Ⅱ-3将实施例1中的N-苄基邻氨基苯酚Ⅲ-1替换为Ⅲ-3,其余同实施例1,得到化合物Ⅱ-3,99%收率,97%ee。产物Ⅱ-3的核磁共振氢谱和核磁共振碳谱分别如图5、图6所示:1HNMR(400MHz,CDCl3)δ7.35–7.31(m,2H),7.29–7.20(m,6H),7.19–7.16(m,2H),6.81–6.79(m,1H),6.72–6.71(m,1H),6.68–6.65(m,1H),4.82(s,1H),4.70(d,J=1.3Hz,1H),4.54(d,J=15.8Hz,1H),4.34(d,J=1.6Hz,1H),4.19(d,J=15.8Hz,1H).13CNMR(101MHz,CDCl3)δ152.6,141.9,138.8,136.7,134.8,128.9,128.8,127.9,127.7,127.4,127.4,127.0,118.6,116.5,113.6,91.7,60.7,53.7.HPLC(ChiralcelOJ-H,n-hexane/i-PrOH=95/5,0.8mL/min,254nm,40℃):tR(minor)=10.5min,tR(major)=15.0min.Ⅲ-3,Ⅱ-3的结构式如下:实施例13:Ⅲ-4作为底物反应生成产物Ⅱ-4将实施例1中的N-苄基邻氨基苯酚Ⅲ-1替换为Ⅲ-4,其余同实施例1,得到化合物Ⅱ-4,92%收率,92%ee。产物Ⅱ-4的核磁共振氢谱和核磁共振碳谱分别如图7、图8所示:1HNMR(400MHz,CDCl3)δ7.25–7.22(m,4H),7.19–7.10(m,6H), 6.70–6.68(m,1H),6.51–6.44(m,2H),4.68(s,1H),4.61(d,J=0.9Hz,1H),4.45(d,J=15.6Hz,1H),4.20(d,J=1.3Hz,1H),4.12(d,J=15.6Hz,1H),2.12(s,3H).13CNMR(101MHz,CDCl3)δ153.0,141.7,139.3,137.7,133.6,131.9,128.7,128.6,127.7,127.6,127.4,127.2,120.0,115.4,115.4,91.1,60.9,54.5,21.2.HPLC(ChiralcelOJ-H,n-hexane/i-PrOH=95/5,0.8mL/min,254nm,40℃):tR(major)=9.6min,tR(minor)=12.9min.Ⅲ-4,Ⅱ-4的结构式如下:实施例14:Ⅳ-2作为底物反应生成产物Ⅱ-5将实施例1中的炔丙醇酯Ⅳ-1替换为Ⅳ-2,其余同实施例1,得到化合物Ⅱ-5,91%收率,96%ee。产物Ⅱ-5的核磁共振氢谱和核磁共振碳谱分别如图9、图10所示:1HNMR(400MHz,CDCl3)δ7.23–7.20(m,4H),7.18–7.14(m,2H),7.07–7.01(m,3H),6.80–6.63(m,4H),4.67–4.64(m,2H),4.40(d,J=15.5Hz,1H),4.22(d,J=1.3Hz,1H),4.14(d,J=15.5Hz,1H).13CNMR(101MHz,CDCl3)δ151.9,143.9,141.3,137.3,134.5,133.5,129.9,128.9,127.9,127.7,127.6,127.5,125.3,122.8,120.1,115.9,115.9,92.2,60.5,55.4.HPLC(ChiralcelAD-H,n-hexane/i-PrOH=98/2,0.8mL/min,254nm,40℃):tR(major)=12.3min,tR(minor)=14.0min.Ⅳ-2,Ⅱ-5的结构式如下:实施例15:Ⅳ-3作为底物反应生成产物Ⅱ-6将实施例1中的炔丙醇酯Ⅳ-1替换为Ⅳ-3,其余同实施例1,得到化合物Ⅱ-6,98%收率,93%ee。产物Ⅱ-6的核磁共振氢谱和核磁共振碳谱分别如图11、图12所示:1HNMR(400MHz,CDCl3)δ7.24–7.16(m,5H),7.03–7.01(m,2H),6.96–6.94(m,2H),6.81–6.79(m,1H),6.76–6.72(m,1H),6.65–6.63(m,2H),4.70(s,1H),4.61(s,1H),4.43(d,J=15.7Hz,1H),4.19(s,1H),4.12(d,J=15.7Hz,1H),2.17(s,3H).13CNMR(101MHz,CDCl3)δ153.4,143.6,137.7,137.4,136.2,134.0,129.4,128.8,127.5,127.4,127.1,122.5,119.3,115.7,114.6,91.2,60.9,54.3,21.2.HPLC(ChiralcelAD-H,n-hexane/i-PrOH=98/2,0.8mL/min,254nm,40℃):tR(major)=6.5min,tR(minor)=8.1min.Ⅳ-3,Ⅱ-6的结构式如下:实施例16:Ⅳ-4作为底物反应生成产物Ⅱ-7将实施例1中的炔丙醇酯Ⅳ-1替换为Ⅳ-4,其余同实施例1,得到化合物Ⅱ-7,96%收率,91%ee。产物Ⅱ-7的核磁共振氢谱和核磁共振碳谱分别如图13、图14所示:1HNMR(400MHz,CDCl3)δ7.35–7.22(m,7H),7.12–7.10(m,2H),6.90–6.84(m,2H),6.81–6.74(m,2H),4.75(d,J=1.3Hz,1H),4.72(s,1H),4.49(d,J=15.4Hz,1H),4.30(d,J=1.4Hz,1H),4.23(d,J=15.4Hz,1H).13CNMR(101MHz,CDCl3)δ152.1,143.9,138.1,137.3,133.5,131.7,128.9,128.8,127.6,127.6,122.7,121.6,120.1,115.8,115.8,91.9,60.2,55.3.HPLC(ChiralcelAD-H,n-hexane/i-PrOH=98/2,0.8mL/min,254nm,40℃):tR(major)=8.7min,tR(minor)=11.4min.Ⅳ-4,Ⅱ-7的结构式如下:实施例17-30:反应底物适用性本发明具有广泛的底物适用性,按照实施例1中的反应条件,许多底物能参与该反应,高收率、高立体选择性地获得手性二氢1,4-苯并噁嗪类化合物,反应式为:表1实施例R1R2产率(%)对映体过量(%)1HPh9794173-MePh9790185-MePh9094196-MePh9797205-ClPh9795214-NO2Ph9885224-SO2NH2Ph9791234-tBuPh737124H2-ClPh958025H4-ClPh969126H4-FPh979027H4-CF3Ph768428H2-萘基709329H甲基968030H苄基9194实施例里17~30中,当R1和R2分别被替换,其产率和对映体过量值如上表1所示。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种制备手性二氢1,4-苯并噁嗪类化合物的方法与流程
- 一种手性螺环羟吲哚‑苯并吡喃‑酮并‑3,4‑二氢‑吡喃化合物的合成方法与流程
- 基于噻吩并异苯并吡喃不对称给电子单元的D‑A型聚合物光伏材料及其应用的制作方法与工艺
- 2‑芳氧基羧酸(二氢苯并呋喃‑7‑氧基)烷基酯的制作方法与工艺
- 一种具有6H‑二苯并吡喃结构化合物及其制备方法与流程
- 一种4H‑苯并吡喃类化合物的制备方法与流程
- 一种6H‑苯并[C]苯并吡喃及其衍生物的制备方法与流程
- 一种具有苯并吡喃结构的FSH拮抗剂的制备方法与流程
- 一种6H‑苯并[C]苯并吡喃类化合物的合成方法与流程
- 一种3,4‑二氢吡喃[3,2‑c]色烯‑5‑酮类化合物的合成方法及其应用与流程