一种利用短链引物阻遏提高DNA克隆及组装效率的方法与流程
本发明属于生物技术领域,具体涉及一种利用短链引物阻遏提高DNA克隆及组装效率的方法。
背景技术:
随着全基因组测序技术的普及以及DNA合成技术的发展,合成生物学得到飞速的发展。在合成生物学的很多研究领域,科学家们都需要合成大片段的DNA,而其中酿酒酵母基因组合成项目,甚至需要将DNA片段组装成染色体。因此探索DNA大片段的快速组装方法是当今的研究热点。
在过去的几十年中,酶切-连接是最常用的克隆方法。这种DNA重组技术始于限制性内切酶和DNA连接酶的发现,随后,多种多样的通过使用限制性内切酶和PCR等技术的连接DNA分子的方法被不断发展起来。然而,依赖于这些传统技术,待克隆或者拼接的序列本身需要存在限制性内切酶识别序列或者通过引入限制性内切酶识别序列来实现。这样不仅仅会增加操作的难度与限制,而且由于限制性内切酶识别序列的存在,会在新的序列中留下“疤痕”。因此,高效与高通量的克隆以及合成DNA的方法是当今合成生物学研究中的迫切需求。
不受序列限制的克隆的方法中包括不依赖于连接的克隆(LIC)、同时不依赖于序列和连接的克隆(SLIC)和Gibson assembly等。在近些年开发的DNA组装技术中,很多是利用DNA重组技术,比如利用酵母体内的同源重组系统进行DNA的体内组装。而Gibson assembly是一个非常经典利用体外重组的方法,该方法基于5’外切酶、DNA聚合酶以及DNA连接酶,可以将多个有重叠序列的DNA片段在一个等温反应中进行组装。
Golden gate shuffling则是一种基于IIs型限制性内切酶的快速DNA组装技术。IIs型限制性内切酶的酶切序列和识别序列不同,目标DNA片段末端可以引入精心设计的IIs型酶位点,通过酶切,酶的识别序列与DNA片段分离,留下粘性末端,用于DNA多片段的无痕组装。然而在组装长片段DNA的过程中,不能忽视组装的效率,即有多少比例的DNA片段可以正确连接成目的长片段。提高该比率是任何一个新方法都必须面临的问题。以Golden gate DNA shuffling为例,研究者们通过不断优化反应体系中的各种参数来实现高效组装。
技术实现要素:
本发明的第一个目的是提供一种阻遏环状质粒中的目的片段和骨架载体酶切后重新连接的方法。
本发明提供的阻遏环状质粒中的目的片段和骨架载体酶切后重新连接的方法包括如下步骤:
1)在环状质粒的目的片段两侧添加DNA片段甲和DNA片段乙,得到环状重组载体,所述环状重组载体从上游起依次包括所述DNA片段甲、所述目的片段、所述DNA片段乙和所述骨架载体;
所述DNA片段甲的两端分别为限制性内切酶A的识别切割序列和限制性内切酶B的识别切割序列,
所述DNA片段乙的两端分别为限制性内切酶C的识别切割序列和限制性内切酶D的识别切割序列;
用所述限制性内切酶A、所述限制性内切酶B、所述限制性内切酶C和所述限制性内切 酶D酶切所述环状重组载体后形成的黏性末端依次记作:骨架载体黏性末端1、DNA片段甲上游黏性末端、DNA片段甲下游黏性末端、目的片段上游黏性末端、目的片段下游黏性末端、DNA片段乙上游黏性末端、DNA片段乙下游黏性末端、骨架载体黏性末端2;
所述黏性末端满足如下条件:
所述目的片段上游黏性末端与所述骨架载体黏性末端1和所述骨架载体黏性末端2均不匹配,
所述目的片段下游黏性末端与所述骨架载体黏性末端1和所述骨架载体黏性末端2均不匹配,
所述目的片段上游黏性末端与所述目的片段下游黏性末端不同且不匹配;
2)用所述限制性内切酶A、所述限制性内切酶B、所述限制性内切酶C和所述限制性内切酶D酶切所述环状重组载体,得到酶切产物;
3)将所述酶切产物、短链引物A和短链引物B退火,即实现了阻遏所述目的片段和所述骨架载体酶切后重新连接;
所述短链引物A与所述DNA片段甲互补配对,以阻遏所述目的片段和所述骨架载体酶切后重新连接;
所述短链引物B与所述DNA片段乙互补配对,以阻遏所述目的片段和所述骨架载体酶切后重新连接。
上述阻遏环状质粒中的目的片段和骨架载体酶切后重新连接的方法中,所述短链引物A或其部分片段与所述DNA片段甲或其部分片段(或DNA片段甲或其部分片段的互补链)互补;所述短链引物B或其部分片段与所述DNA片段乙或其部分片段(或DNA片段乙或其部分片段的互补链)互补。
上述方法中,所述匹配是指互补配对。
本发明的第二个目的是提供一种用于将m个待拼接DNA片段拼接到接收载体上的成套产品。
本发明提供的用于将m个待拼接DNA片段拼接到接收载体上的成套产品由m个环状重组载体、接收载体x、短链引物A和短链引物B组成;
所述m个环状重组载体按照待拼接DNA片段的拼接顺序依次命名为环状重组载体1、环状重组载体2、……、环状重组载体m;
所述m为大于等于2的整数;
所述环状重组载体1从上游起依次包括DNA片段甲1、待拼接DNA片段1、DNA片段乙1和骨架载体;
所述环状重组载体2从上游起依次包括DNA片段甲2、待拼接DNA片段2、DNA片段乙2和骨架载体;
依次类推,
所述环状重组载体m从上游起依次包括DNA片段甲m、待拼接DNA片段m、DNA片段乙m和骨架载体;
所述接收载体x从上游起依次包括DNA片段甲x、待替换DNA片段x、DNA片段乙x和骨架载体;
每个所述环状重组载体中的DNA片段甲的两端分别为限制性内切酶A的识别切割序列和限制性内切酶B的识别切割序列,
每个所述环状重组载体中的DNA片段乙的两端分别为限制性内切酶C的识别切割序列和限制性内切酶D的识别切割序列,
用所述限制性内切酶A、所述限制性内切酶B、所述限制性内切酶C和所述限制性内切酶D酶切每个所述环状重组载体后形成的黏性末端依次记作:骨架载体黏性末端1、DNA片段甲上游黏性末端、DNA片段甲下游黏性末端、待拼接DNA片段上游黏性末端、待拼接DNA片段下游黏性末端、DNA片段乙上游黏性末端、DNA片段乙下游黏性末端、骨架载体黏性末端2;
所述黏性末端满足如下条件:
每个所述待拼接DNA片段上游黏性末端与任一个环状重组载体的骨架载体黏性末端1和任一个环状重组载体的骨架载体黏性末端2均不匹配;
每个所述待拼接DNA片段下游黏性末端与任一个环状重组载体的骨架载体黏性末端1和任一个环状重组载体的骨架载体黏性末端2均不匹配;
对于某个所述环状重组载体来讲,其产生的待拼接DNA片段上游黏性末端与该环状重组载体中的骨架载体黏性末端1和骨架载体黏性末端2不匹配,并且还与除接收载体x外的其它环状重组载体中的骨架载体黏性末端1和骨架载体黏性末端2也不匹配;对于某个所述环状重组载体来讲,其产生的待拼接DNA片段下游黏性末端与该环状重组载体中的骨架载体黏性末端1和骨架载体黏性末端2不匹配,并且还与除接收载体x外的其它环状重组载体中的骨架载体黏性末端1和骨架载体黏性末端2也不匹配;
每个所述待拼接DNA片段上游黏性末端与所述待拼接DNA片段下游黏性末端不同且不匹配;
待拼接DNA片段1下游黏性末端与待拼接DNA片段2上游黏性末端匹配;
待拼接DNA片段2下游黏性末端与待拼接DNA片段3上游黏性末端匹配;
依次类推;
待拼接DNA片段m-1下游黏性末端与待拼接DNA片段m上游黏性末端匹配;
所述接收载体x经限制性内切酶酶切产生的载体骨架上游黏性末端与待拼接DNA片段1上游黏性末端匹配;
所述接收载体x经限制性内切酶酶切产生的载体骨架下游黏性末端与待拼接DNA片段m下游黏性末端匹配;
所述短链引物A与所述DNA片段甲互补配对,以阻遏待拼接DNA片段和所述重组环状载体的骨架载体酶切后重新连接;
所述短链引物B与所述DNA片段乙互补配对,以阻遏待拼接DNA片段和所述重组环状载体的骨架载体酶切后重新连接。
上述用于将m个待拼接DNA片段拼接到接收载体上的成套产品中,
所述短链引物A或其部分片段与所述DNA片段甲或其部分片段(或DNA片段甲或其部分片段的互补链)互补;
所述短链引物B或其部分片段与所述DNA片段乙或其部分片段(或DNA片段乙或其部分片段的互补链)互补。
上述用于将m个待拼接DNA片段拼接到接收载体上的成套产品中,
所述m为5时,所述成套产品由环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4、环状重组载体5、接收载体x、短链引物A和短链引物B组成;
所述m为6时,所述成套产品由环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4、环状重组载体6、环状重组载体7、接收载体x、短链引物A和短链引物B组成;
所述环状重组载体1中的DNA片段甲1为序列11中的第23-55位、DNA片段乙1为序列11中的第395-426位;
所述环状重组载体2中的DNA片段甲2为序列13中的第23-55位、DNA片段乙2为序列13中的第1139-1170位;
所述环状重组载体3中的DNA片段甲3为序列14中的第23-55位、DNA片段乙3为序列14中的第1472-1503位;
所述环状重组载体4中的DNA片段甲4为序列15中的第23-55位、DNA片段乙4为序列15中的第1640-1671位;
所述环状重组载体5中的DNA片段甲5为序列17中的第23-55位、DNA片段乙5为序列17中的第815-846位;
所述环状重组载体6中的DNA片段甲6为序列19中的第23-55位、DNA片段乙6为序列19中的第1235-1266位;
所述环状重组载体7中的DNA片段甲7为序列20中的第23-55位、DNA片段乙7为序列20中的第1211-1242位;
接收载体x中的DNA片段甲x为序列10中的第23-55位、待替换DNA片段x的核苷酸序列为序列10中的第56-743位、DNA片段乙x为序列10中的第744-775位;
所述短链引物A的核苷酸序列为序列表中序列21;
所述短链引物B的核苷酸序列为序列表中序列22。
本发明的第三个目的是提供一种用于将m个待拼接DNA片段拼接成全长DNA片段的成套产品。
本发明提供的用于将m个待拼接DNA片段拼接成全长DNA片段的成套产品由m个DNA片段、短链引物C和短链引物D组成;
所述m个DNA片段按照待拼接DNA片段的拼接顺序依次命名为DNA片段1、DNA片段2、……、DNA片段m;
所述m为大于等于2的整数;
所述DNA片段1从上游起依次包括DNA片段甲1、待拼接DNA片段1和DNA片段乙1;
所述DNA片段2从上游起依次包括DNA片段甲2、待拼接DNA片段2和DNA片段乙2;
依次类推,
所述DNA片段m从上游起依次包括由DNA片段甲m、待拼接DNA片段m和DNA片段乙m;
每个所述DNA片段中的DNA片段甲的两端分别为限制性内切酶A的识别切割序列和限制性内切酶B的识别切割序列,
每个所述DNA片段中的DNA片段乙的两端分别为限制性内切酶C的识别切割序列和限制性内切酶D的识别切割序列,
用所述限制性内切酶A、所述限制性内切酶B、所述限制性内切酶C和所述限制性内切 酶D酶切每个DNA片段后形成的黏性末端依次记作:DNA片段甲上游黏性末端、DNA片段甲下游黏性末端、待拼接DNA片段上游黏性末端、待拼接DNA片段下游黏性末端、DNA片段乙上游黏性末端、DNA片段乙下游黏性末端;
所述黏性末端满足如下条件:
待拼接DNA片段1下游黏性末端与待拼接DNA片段2上游黏性末端匹配;
待拼接DNA片段2下游黏性末端与待拼接DNA片段3上游黏性末端匹配;
依次类推;
待拼接DNA片段m-1下游黏性末端与待拼接DNA片段m上游黏性末端匹配;
所述短链引物C与所述DNA片段甲互补配对;
所述短链引物D与所述DNA片段乙互补配对。
上述方法或上述成套产品中,
所述DNA片段甲和所述DNA片段乙不匹配;
所述短链引物A和所述短链引物B不匹配;
所述短链引物C和所述短链引物D不匹配;
所述DNA片段甲和所述DNA片段乙的大小均为15-45bp;
所述短链引物A、所述短链引物B、所述短链引物C和所述短链引物D的大小均为15-45bp;
所述限制性内切酶A、所述限制性内切酶B、所述限制性内切酶C和所述限制性内切酶D为同一种IIs型限制性内切酶或不同种限制性内切酶。
上述用于将m个待拼接DNA片段拼接成全长DNA片段的成套产品中,
所述短链引物C或其部分片段与所述DNA片段甲或其部分片段(或DNA片段甲或其部分片段的互补链)互补;
所述短链引物D或其部分片段与所述DNA片段乙或其部分片段(或DNA片段乙或其部分片段的互补链)互补。
上述用于将m个待拼接DNA片段拼接成全长DNA片段的成套产品中,
所述m为3时,所述成套产品由DNA片段1、DNA片段2、DNA片段3、短链引物C和短链引物D组成;
所述m为5时,所述成套产品由DNA片段1、DNA片段2、DNA片段3、DNA片段4、DNA片段5、短链引物C和短链引物D组成;
所述m为7时,所述成套产品由DNA片段1、DNA片段2、DNA片段3、DNA片段4、DNA片段5、DNA片段6、DNA片段7、短链引物C和短链引物D组成;
所述DNA片段1中的DNA片段甲1为序列25中第20-57位核苷酸分子、DNA片段乙1为序列25中的第253-288位;
所述DNA片段2中的DNA片段甲2为序列26中第20-57位核苷酸分子、DNA片段乙2为序列26中的第251-286位;
所述DNA片段3中的DNA片段甲3为序列27中第20-57位核苷酸分子、DNA片段乙3为序列27中的第295-330位;
所述DNA片段4中的DNA片段甲4为序列28中第20-57位核苷酸分子、DNA片段乙4为序列28中的第295-330位;
所述DNA片段5中的DNA片段甲5为序列29中第20-57位核苷酸分子、DNA片段乙 5为序列29中的第295-330位;
所述DNA片段6中的DNA片段甲6为序列30中第20-57位核苷酸分子、DNA片段乙6为序列30中的第301-336位;
所述DNA片段7中的DNA片段甲7为序列31中第20-57位核苷酸分子、DNA片段乙7为序列31中的第250-285位;
所述短链引物C的核苷酸序列为序列23;
所述短链引物D的核苷酸序列为序列24。
本发明的第四个目的是提供一种将m个待拼接DNA片段拼接到接收载体上的方法。
本发明提供的将m个待拼接DNA片段拼接到接收载体上的方法包括如下步骤:
1)用限制性内切酶酶切上述m个所述环状重组载体和所述接收载体x,得到酶切产物;
2)将所述酶切产物、上述短链引物A和上述短链引物B退火,得到退火产物;
3)连接酶连接所述退火产物,使拼接后的长DNA片段替换所述接收载体x上的待替换DNA片段x,实现m个待拼接DNA片段拼接到接收载体上。
上述将m个待拼接DNA片段拼接到接收载体上的方法中,所述限制性内切酶为BsmBI。
本发明的第五个目的是提供一种将m个待拼接DNA片段拼接成全长DNA片段的方法。
本发明提供的将m个待拼接DNA片段拼接成全长DNA片段的方法包括如下步骤:
1)用限制性内切酶酶切上述m个DNA片段,得到酶切产物;
2)将所述酶切产物、上述短链引物C和上述短链引物D退火,得到退火产物;
3)连接酶连接所述退火产物,得到全长DNA片段,实现m个目的DNA片段的拼接。
上述将m个待拼接DNA片段拼接成全长DNA片段的方法中,所述限制性内切酶为BsaI。
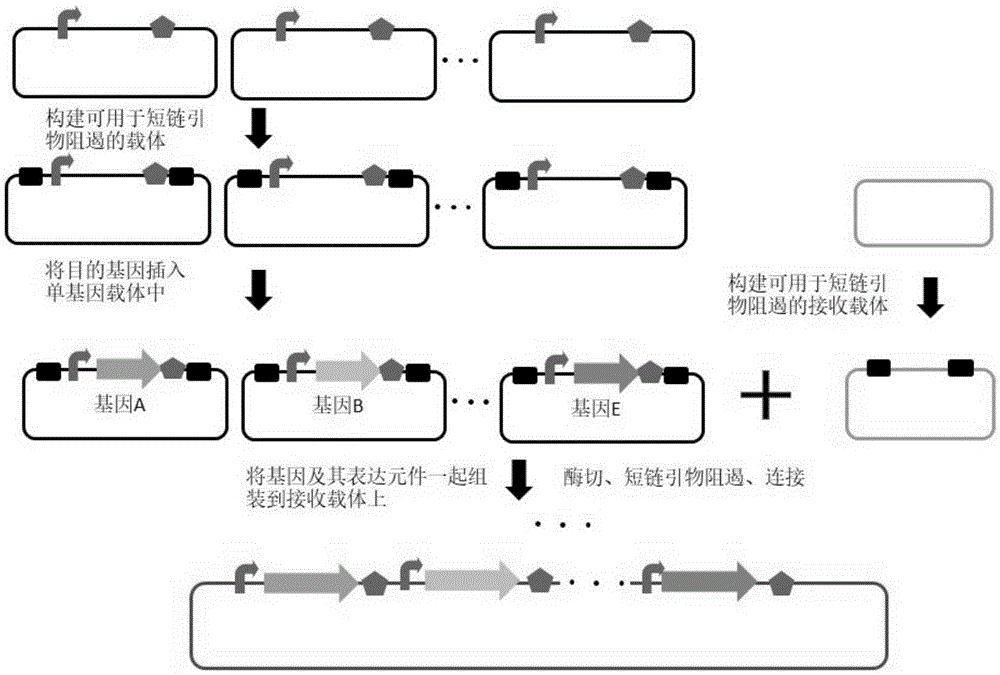
本发明从一个新的角度出发,提出了短链引物阻遏(Oligo blocking)的方法,用于提高DNA克隆以及组装的效率。以Golden gate DNA shuffling为例,每个待组装的DNA片段都被克隆于独立的载体模块上,然后将这些载体模块混合在一起,通过IIs型限制性内切酶酶切,将这些目标DNA片段释放下来,最后将这些被切下来的DNA片段按照设计好的顺序进行组装。在此过程中,单独的DNA片段会不断地连回原载体,然后再被切下来,影响组装效率。而本发明的短链引物阻遏(Oligo blocking)的方法则是一种解决已经被切下来的DNA片段重新连接回原载体的方法。如附图1所示,在原载体上设计两段DNA序列,将目标DNA片段与载体骨架序列隔开,两段DNA序列两侧分别设计有限制性内切酶位点,在进行DNA片段组装时,通过酶切,原载体会分为四部分,随后加入短链引物,设置温度梯度,在高温下,DNA双链解链,降温后,单链的短链引物会与原载体上添加的DNA序列中的一条链互补配对,经过该阻遏过程后,由于目的DNA片段的粘性末端与载体不匹配,被切下的目的DNA片段无法被连回原载体,有效的防止了酶切的DNA片段与原载体的自连的过程,因此提高了组装的效率。该方法还可扩展用于其他克隆方法,例如可以减少克隆步骤、提高克隆效率以及DNA片段的组装效率等。
附图说明
图1为短链引物阻遏原理。
图2为实施例1的流程图。其中,黑色圆角矩形代表DNA片段甲乙。
图3为使用短链引物阻遏前后DNA组装效率的比较。
图4为实施例2的流程图。其中,E代表限制性内切酶识别酶切位点,两个限制性内切酶之间的圆角矩形代表DNA片段甲乙。
图5为使用短链引物阻遏前后PCR产物连接效果的比较。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下述实施例中提到的DNA序列均为5’端到3’端方向。
实施例1、一种利用短链引物阻遏提高DNA克隆及组装效率的方法
本实施例中具体试验流程如附图2所示。
一、制备7个环状重组载体和接收载体x
环状重组载体1其从上游起包括DNA片段甲1、待拼接DNA片段1、DNA片段乙1和骨架载体(骨架载体的核苷酸序列为将序列1中4799到5663位核苷酸去除后的得到的序列);环状重组载体1中的DNA片段甲1为序列11中的第23-55位、DNA片段乙1为序列11中的第395-426位;;环状重组载体1中的DNA片段甲1与短链引物A相同或互补的核苷酸序列为序列11中的第30-51位、待拼接DNA片段1的核苷酸序列为序列11中的第52-398位、DNA片段乙1与短链引物B相同或互补的核苷酸序列为序列11中的第399-419位;
环状重组载体2其从上游起包括DNA片段甲2、待拼接DNA片段2、DNA片段乙2和骨架载体;环状重组载体2中的DNA片段甲2为序列13中的第23-55位、DNA片段乙2为序列13中的第1139-1170位;环状重组载体2中的DNA片段甲2与短链引物A相同或互补的核苷酸序列为序列13中的第30-51位、待拼接DNA片段2的核苷酸序列为序列13中的第52-1142位、DNA片段乙2与短链引物B相同或互补的核苷酸序列为序列13中的第1143-1163位;
环状重组载体3其从上游起包括DNA片段甲3、待拼接DNA片段3、DNA片段乙3和骨架载体;环状重组载体3中的DNA片段甲3为序列14中的第23-55位、DNA片段乙3为序列14中的第1472-1503位;环状重组载体3中的DNA片段甲3与短链引物A相同或互补的核苷酸序列为序列14中的第30-51位、待拼接DNA片段3的核苷酸序列为序列14中的第52-1475位、DNA片段乙3与短链引物B相同或互补的核苷酸序列为序列14中的第1476-1496位;
环状重组载体4其从上游起包括DNA片段甲4、待拼接DNA片段4、DNA片段乙4和骨架载体;环状重组载体4中的DNA片段甲4为序列15中的第23-55位、DNA片段乙4为序列15中的第1640-1671位;环状重组载体4中的DNA片段甲4与短链引物A相同或互补的核苷酸序列为序列15中的第30-51位、待拼接DNA片段4的核苷酸序列为序列15中的第52-1643位、DNA片段乙4与短链引物B相同或互补的核苷酸序列为序列15中的第1644-1664位;
环状重组载体5其从上游起包括DNA片段甲5、待拼接DNA片段5、DNA片段乙5和骨架载体;环状重组载体5中的DNA片段甲5为序列17中的第23-55位、DNA片段乙5为序列17中的第815-846位;环状重组载体5中的DNA片段甲5与短链引物A相同或互补的核苷酸序列为序列17中的第30-51位、待拼接DNA片段5的核苷酸序列为序列17中的第52-818位、DNA片段乙5与短链引物B相同或互补的核苷酸序列为序列17中的第819-839位;
环状重组载体6其从上游起包括DNA片段甲6、待拼接DNA片段6、DNA片段乙6和 骨架载体;环状重组载体6中的DNA片段甲6为序列19中的第23-55位、DNA片段乙6为序列19中的第1235-1266位;环状重组载体6中的DNA片段甲6与短链引物A相同或互补的核苷酸序列为序列19中的第30-51位、待拼接DNA片段6的核苷酸序列为序列19中的第52-1238位、DNA片段乙6与短链引物B相同或互补的核苷酸序列为序列19中的第1239-1259位;
环状重组载体7其从上游起包括DNA片段甲7、待拼接DNA片段7、DNA片段乙7和骨架载体,环状重组载体7中的DNA片段甲7为序列20中的第23-55位、DNA片段乙7为序列20中的第1211-1242位;环状重组载体7中的DNA片段甲7与短链引物A相同或互补的核苷酸序列为序列20中的第30-51位、待拼接DNA片段7的核苷酸序列为序列20中的第52-1214位、DNA片段乙7与短链引物B相同或互补的核苷酸序列为序列20中的第1215-1235位;
接收载体x其从上游起包括DNA片段甲x、待替换DNA片段x、DNA片段乙x和另一骨架载体(另一骨架载体的核苷酸序列为将序列9的第5045-5754位核苷酸去除后得到的序列);接收载体x中的DNA片段甲x为序列10中的第23-55位、待替换DNA片段x的核苷酸序列为序列10中的第56-743位、DNA片段乙x为序列10中的第744-775位;接收载体x中的DNA片段甲x与短链引物A相同或互补的核苷酸序列为序列10中的第27-48位、DNA片段乙x与短链引物B相同或互补的核苷酸序列为序列10中的第751-771位;
短链引物A的核苷酸序列为序列表中序列21;
短链引物B的核苷酸序列为序列表中序列22;
环状重组载体1经IIs型限制性内切酶酶切产生待拼DNA片段1下游黏性末端与环状重组载体2经IIs型限制性内切酶酶切产生待拼DNA片段2上游黏性末端匹配;
环状重组载体2经IIs型限制性内切酶酶切产生待拼DNA片段2下游黏性末端与环状重组载体3经IIs型限制性内切酶酶切产生待拼DNA片段3上游黏性末端匹配;
依次类推;
环状重组载体m-1经IIs型限制性内切酶酶切产生的待拼DNA片段m-1下游黏性末端与环状重组载体m经IIs型限制性内切酶酶切产生的待拼DNA片段m上游黏性末端匹配;
接收载体x经IIs型限制性内切酶酶切产生的另一载体骨架上游黏性末端与环状重组载体1经IIs型限制性内切酶酶切产生的待拼接DNA片段1上游黏性末端匹配;
接收载体x经IIs型限制性内切酶酶切产生的另一载体骨架下游黏性末端与环状重组载体m经IIs型限制性内切酶酶切产生的待拼接DNA片段m下游黏性末端匹配;
上述环状重组载体和接收载体的具体制备方法如下:
(一)单基因载体的构建
1、1号单基因载体的构建
(1)PCR扩增
用EcoRI(EcoRI-HF NEB R3101L)和XhoI(NEB R0146L)双酶切载体A(载体A的核苷酸序列为序列1),胶回收小片段,以小片段为模板,采用引物YQO198和YQO199进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO198:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTTGGATAATACGACTCACT;
YQO199:CCGCTCGAGCTGCAGCGGCCGCTACTAGTACGTCTCGGTTGGCAGTGACTCCG TCTCTGCCTCAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃ 5min;16℃保存。
用EcoRI和XhoI对PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为1号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:1号单基因载体为将载体A的EcoRI和XhoI识别序列间的DNA片段替换为序列2所示的核苷酸分子(序列2所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列2所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
2、2号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板,采用引物YQO201和YQO202进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO201:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTAGGCTAATACGACTCACT;
YQO202:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTGGCACAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI(SpeI-HF NEB R3133L)对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为2号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:2号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列3所示的核苷酸分子(序列3所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列3所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
3、3号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板,采用引物YQO203和YQO204进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO203:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCT TGCCTAATACGACTCACT;
YQO204:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTAGTGCAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为3号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:3号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列4所示的核苷酸分子(序列4所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列4所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
4、4号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板,采用引物YQO205和YQO206进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO205:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTCACTTAATACGACTCACT;
YQO206:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTCGACCAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为4号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:4号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列5所示的核苷酸分子(序列5所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对,序列5所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
5、5号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板, 采用引物YQO207和YQO208进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO207:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTGTCGTAATACGACTCACT;
YQO208:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTGTGACAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为5号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:5号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列6所示的核苷酸分子(序列6所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列6所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
6、6号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板,采用引物YQO207和YQO209进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO207:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTGTCGTAATACGACTCACT;
YQO209:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTCTGCCAAAAAACCCCTCAAGACCC;
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为6号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:6号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列7所示的核苷酸分子(序列7所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列7所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
7、7号单基因载体的构建
(1)PCR扩增
用EcoRI和XhoI双酶切步骤1得到的1号单基因载体,胶回收小片段,以小片段为模板,采用引物YQO210和YQO208进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO210:CCGGAATTCGCGGCCGCTTCTAGAGCGTCTCGATCCGCAGTGTCTTGCGTCTCTGCAGTAATACGACTCACT;
YQO208:CTAGACTAGTACGTCTCGGTTGGCAGTGACTCCGTCTCTGTGACAAAAAACCCCTCAAGACCC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃5min;16℃保存。
用EcoRI和SpeI对上述PCR扩增产物和载体A进行双酶切,连接,得到重组载体,将其命名为7号单基因载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:7号单基因载体为将载体A的EcoRI和SpeI识别序列间的DNA片段替换为序列8所示的核苷酸分子(序列8所示的DNA分子的第24-45位核苷酸分子与短链引物A中第2-23位核苷酸分子互补配对;序列8所示的DNA分子的第897-917位核苷酸分子对应的互补链与短链引物B中第2-22位核苷酸分子互补配对),且保持载体A其他序列不变得到的载体。
(二)环状重组载体的构建
为实现多基因的共表达,需要将待表达的基因插入到对应编号的步骤一中制备的可用于短链引物阻遏的单基因载体中,分别得到环状重组载体。构建过程如下:
1、环状重组载体1的构建
以酿酒酵母BY4741的基因组DNA(BioVector NTCC Inc.货号:ATCC 201388D-5)为模板,采用YQO052和YQO053引物进行PCR扩增,得到PCR扩增产物,即为基因A。引物序列如下:
YQO052:5’-AGCGTGGGTCTCAGATGAATGTGAACCTTGCCGCGACGG-3’;
YQO053:5’-GTGCTGGGTCTCGGCTATTTACTAGCATTACTCTCTTTTTCTCC-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,40s;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)与1号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因A表达载体)。反应体系:5units BsaI(BsaI-HF NEB R3535L)、1U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,60min;50℃,15min;80℃,15min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因A表达载体为将1号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列11所示的DNA序列,且保持1号单基因载体其他序列不变后得到的载体。
2、环状重组载体2的构建
以合成DNA片段(核苷酸序列为序列12)为模板,采用引物YQO058和YQO059进行PCR扩增,得到PCR扩增产物,即为基因B。引物序列如下:
YQO058:5’-AgcgtgGGTCTCaGATGTTCCAGTTTGTTACTCCTGTG-3’;
YQO059:5’-GtgctgGGTCTCgGCTAAACCCATCTCCATAAACAGC-3’;
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,2min;30个循环;68℃7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)与2号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因B表达载体)。反应体系:2units BsaI(BsaI-HF NEB R3535L)、0.5U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,5min;1个循环;37℃,5min,25℃5min;5个循环;50℃,5min;80℃,5min;降温后加入0.5U连接酶;25℃40min;10℃保存。将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因B表达载体为将2号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列13所示的DNA片段,且保持2号单基因载体其他序列不变后得到的载体。
3、环状重组载体3的构建
以酿酒酵母BY4741的基因组DNA为模板,采用YQO056和YQO057引物进行PCR扩增,得到PCR扩增产物,即为基因C。引物序列如下:
YQO056:5’-agcgtgGGTCTCaGATGAACATCCTTTTACAGGATCC-3’;
YQO057:5’-GtgctgGGTCTCgGCTATGATGACTGCATCTTAATTATC-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,2min;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)和3号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因C表达载体)。反应体系:2units BsaI(BsaI-HF NEB R3535L)、0.5U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1x T4连接酶缓冲液(Thermo Scientific B69)。反应程序:37℃,5min;1个循环;37℃,5min,25℃ 5min;5个循环;50℃,5min;80℃,5min;降温后加入0.5U连接酶;25℃ 40min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因C表达载体为将3号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列14所示的DNA片段,且保持3号单基因载体其他序列不变后得到的载体。
4、环状重组载体4的构建
以酿酒酵母BY4741的基因组DNA为模板,采用YQO065和YQO055引物进行PCR扩增,得到PCR扩增产物,即为基因D。引物序列如下:YQO065:
5’-agcgtgGGTCTCaGATGGAAGGGAAAAGACCTAATTTACCACAG-3’;
YQO055:5’-GTGCTGGGTCTCGGCTATTGCTGTAGGGCAATTTGCTCCAAC-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,2min;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)和4号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因D表达载体)。反应体系:5units BsaI(BsaI-HF NEB R3535L)、1U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,60min;50℃,15min;80℃,15min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因D表达载体为将4号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列15所示的DNA片段,且保持4号单基因载体其他序列不变后得到的载体。
5、环状重组载体5的构建
以合成片段(核苷酸序列为序列16)为模板,采用YQO322和YQO051引物进行PCR扩增,得到PCR扩增产物,即为基因E。引物序列如下:
YQO322:5’-AGCGTGGGTCTCAGATGCATCATCATCACCACCACGATTACGATATCC-3’;
YQO051:5’-GTGCTGGGTCTCGGCTAGTTCAAGAAACCTTTACAATTAGG-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,1min;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)和5号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因E表达载体)。反应体系:5units BsaI(BsaI-HF NEB R3535L)、1U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,60min;50℃,15min;80℃,15min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因E表达载体为将5号单基因载体的SphI和XhoI识别序列间的DNA 片段替换为序列表中序列17所示的DNA片段,且保持5号单基因载体其他序列不变后得到的载体。
6、环状重组载体6的构建
以合成片段(核苷酸序列为序列18)为模板,采用YQO322和YQO051引物进行PCR扩增,得到PCR扩增产物,即为基因F。引物序列如下:
YQO322:5’-AGCGTGGGTCTCAGATGCATCATCATCACCACCACGATTACGATATCC-3’
YQO051:5’-GTGCTGGGTCTCGGCTAGTTCAAGAAACCTTTACAATTAGG-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,2min;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)和6号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因F表达载体)。反应体系:5units BsaI(BsaI-HF NEB R3535L)、1U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,60min;50℃,15min;80℃,15min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因F表达载体为将6号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列19所示的DNA片段,且保持6号单基因载体其他序列不变后得到的载体。
7、环状重组载体7的构建
以酿酒酵母BY4741的基因组DNA为模板,采用YQO240和YQO241引物进行PCR扩增,得到PCR扩增产物,即为基因G。引物序列如下:
YQO240:5’-AGCGTGGGTCTCAGATGAATCCAACTACAGGAGAGGACGTAT-3’;
YQO241:5’-GTGCTGGGTCTCGGCTACAAACCTCTTCTTAAAATTTCCTCATAGTATTG-3’。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xbuffer for KOD-Plus-(TOYOBO)、MgSO4(TOYOBO)2mM、KOD-Plus-(TOYOBO)0.02units/μl、引物(终浓度均为0.5mM)、基因组模板150ng。
PCR反应条件如下:94℃,5min;1个循环;94℃,30s,55℃,30s,68℃,2min;30个循环;68℃ 7min;10℃保存。
将PCR扩增产物用琼脂糖凝胶电泳分析,回收目的条带,得到回收产物。
将回收产物(100ng)与7号单基因载体(20ng)加入到反应体系中反应,得到反应产物(单基因G表达载体)。反应体系:5units BsaI(BsaI-HF NEB R3535L)、1U T4 DNA连接酶(Thermo Scientific EL0011)、0.1mg/ml BSA(NEB B9001S)、1xT4连接酶缓冲液(Thermo Scientific B69);反应程序:37℃,60min;50℃,15min;80℃,15min;10℃保存。
将反应产物直接转化大肠杆菌DH5α,经酶切鉴定后送测序。
经测序表明:单基因G表达载体为将7号单基因载体的SphI和XhoI识别序列间的DNA片段替换为序列表中序列20所示的DNA片段,且保持7号单基因载体其他序列不变后得到的载体。
(三)接收载体的构建
以载体B(载体B的核苷酸序列为序列9)为模板,采用YQO213和YQO214引物进行PCR扩增,得到PCR扩增产物。引物序列如下:
YQO213:CATGCATGCGAATTCGCGGCCGCTTCTAGAGTGGAAGAGACGCAAGACACTGCGGATCGAGACGCTCTAGAGGATCCG;
YQO214:CTAGACTAGTAGTGAAGAGACGGAGTCACTGCCAACCGAGACGGTCGACCTGCAGAC。
PCR反应体系(终浓度):dNTP(GenStar A114-01)0.2mM、1xQ5 reaction buffer(NEB B9027S)、Q5(High-Fidelity DNA Polymerase NEB M0491S)0.02units/μl、引物(终浓度均为0.5mM)、胶回收产物模板30ng。
PCR反应程序如下:98℃,2min;98℃,10s,52℃,30s,72℃,30s;5个循环;98℃,10s,55℃,30s,72℃,30s;5个循环;98℃,10s,58℃,30s,72℃,30s;20个循环;72℃ 5min;16℃保存。
用SphI(SphI-HF NEB R3182L)和SpeI对上述PCR扩增产物和载体B进行双酶切,连接,得到重组载体,将其命名为接收载体,并将其转化大肠杆菌OneccdB SurvivalTM2 T1R,经酶切鉴定后送测序。
测序结果表明:接收载体为将载体B的SphI和SpeI识别序列间的DNA片段替换为序列10所示的核苷酸分子(序列10所示的DNA分子的第27-48位核苷酸分子对应的互补链与短链引物A中第2-23位核苷酸分子互补配对;序列10所示的DNA分子的第751-771位核苷酸分子与短链引物B中第2-22位核苷酸分子互补配对),且保持载体B其他序列不变得到的载体。
二、用IIs型限制性内切酶酶切m个环状重组载体和接收载体x,得到酶切产物;将酶切产物和短链引物组(短链引物A和短链引物B,短链引物A与DNA片段甲全长或部分片段相同或互补,短链引物B与DNA片段乙全长或部分片段相同或互补)退火,得到退火产物;连接酶连接退火产物,实现m个待拼接DNA片段的拼接,且使拼接后的长DNA片段替换接收载体上待替换DNA片段x。
本实施例中的m为5或者6。具体步骤如下
1、五个环状重组载体和一个接收载体的组装
(1)无短链引物阻遏的组装
将环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4和环状重组载体5与接收载体(150ng,环状重组载体与接收载体为等摩尔量)加入到酶切体系中,55℃酶切1小时,得到酶切产物。酶切体系:BsmBI(NEB R0580S,5units)和1xNEB buffer 3.1。
运行程序:83℃ 6min;80℃ 3min;75℃ 1min30sec;70℃ 1min30sec;65℃ 1min30sec;60℃ 1min30sec;55℃ 1min30sec;50℃ 1min30sec;16℃保存。
然后向上述酶切产物中加入连接所需试剂:PEG4000(Thermo Scientific 00135879)5%(质量-体积浓度),T4连接酶(Thermo Scientific EL0011)1U、ATP 1mM、DTT 5mM。16℃连接过夜,得到连接产物。
将多基因共表达载体(连接产物)直接转化大肠杆菌DH5α,并经菌落PCR鉴定组装效率(即正确的PCR条带占总数的比例,PCR扩增的是接收载体与目的基因两侧的接头处)。
(2)有短链引物阻遏的组装
将环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4和环状重组载体 5与接收载体(150ng,环状重组载体与接收载体为等摩尔量)加入酶切体系中,55℃酶切1小时,得到酶切产物。酶切体系:BsmBI(NEB R0580S,5units)和1xNEB buffer 3.1。
向上述酶切产物中加入短链引物A和短链引物B(引物的终浓度均为1μM)进行反应,得到阻遏反应产物。短链引物A:AAGAGACGCAAGACACTGCGGATA(序列21);短链引物B:AAGAGACGGAGTCACTGCCAACA(序列22);运行程序:83℃ 6min;80℃ 3min;75℃ 1min30sec;70℃ 1min30sec;65℃ 1min30sec;60℃ 1min30sec;55℃ 1min30sec;50℃ 1min30sec;16℃保存。
最后向上述阻遏反应产物中加入连接所需试剂:PEG4000(Thermo Scientific 00135879)5%(质量-体积浓度),T4连接酶(Thermo Scientific EL0011)1U、ATP 1mM、DTT 5mM。16℃连接过夜,得到连接产物。
将多基因共表达载体(连接产物)直接转化大肠杆菌DH5α,经菌落PCR鉴定组装效率(即正确的PCR条带占总数的比例,PCR扩增的是接收载体与目的基因两侧的接头处)。PCR鉴定的第一对引物如下:YQO031:CATTCACCACCCTGAATTGACTC,YQO060:AgcgtgGGTCTCGTTTCTGCATCAAAGATCCGTATGAG;PCR鉴定的第二对引物如下:YQO119:GCAGCCGGATCCCATAGTTATATC,YQO326:GAACAGATAGCTGAAGACATAGTATGG。
2、六个环状重组载体与接收载体的组装
(1)无短链引物阻遏的组装
将环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4、环状重组载体6和环状重组载体7与接收载体(150ng,环状重组载体与接收载体为等摩尔量)加入到酶切体系中,55℃酶切1小时,得到酶切产物。酶切体系:BsmBI(NEB R0580S,5units)和1xNEB buffer 3.1。
运行程序:83℃ 6min;80℃ 3min;75℃ 1min30sec;70℃ 1min30sec;65℃ 1min30sec;60℃ 1min30sec;55℃ 1min30sec;50℃ 1min30sec;16℃保存。
然后向酶切产物中加入连接所需试剂:PEG4000(Thermo Scientific 00135879)5%(质量-体积浓度),T4连接酶(Thermo Scientific EL0011)1U、ATP 1mM、DTT 5mM。16℃连接过夜,得到连接产物。
将多基因共表达载体(连接产物)直接转化大肠杆菌DH5α,经菌落PCR鉴定组装效率(即正确的PCR条带占总数的比例,PCR扩增的是接收载体与目的基因两侧的接头处)。
(2)有短链引物阻遏的组装
将环状重组载体1、环状重组载体2、环状重组载体3、环状重组载体4、环状重组载体6和环状重组载体7与接收载体(150ng,环状重组载体与接收载体为等摩尔量)加入到酶切体系中,55℃酶切1小时,得到酶切产物。酶切体系:BsmBI(NEB R0580S,5units)和1xNEB buffer 3.1。
向上述酶切产物中加入短链引物A和短链引物B(引物的终浓度均为1μM)进行反应,得到阻遏反应产物。短链引物A:AAGAGACGCAAGACACTGCGGATA(序列21);短链引物B:AAGAGACGGAGTCACTGCCAACA(序列22);运行程序:83℃ 6min;80℃ 3min;75℃ 1min30sec;70℃ 1min30sec;65℃ 1min30sec;60℃ 1min30sec;55℃ 1min30sec;50℃ 1min30sec;16℃保存。
最后向阻遏反应产物中加入连接所需试剂:PEG4000(Thermo Scientific 00135879)5% (质量-体积浓度),T4连接酶(Thermo Scientific EL0011)1U、ATP 1mM、DTT 5mM。16℃连接过夜,得到连接产物。
将多基因共表达载体(连接产物)直接转化大肠杆菌DH5α,经菌落PCR鉴定组装效率(即正确的PCR条带占总数的比例,PCR扩增的是接收载体与目的基因两侧的接头处)。PCR鉴定的第一对引物如下:YQO031:CATTCACCACCCTGAATTGACTC,YQO060:AgcgtgGGTCTCGTTTCTGCATCAAAGATCCGTATGAG;PCR鉴定的第二对引物如下:YQO119:GCAGCCGGATCCCATAGTTATATC,YQO325:GATGACGAGGAAAGACTTCCTTG。
三、短链引物阻遏对DNA片段组装效率的影响
多基因共表达载体转化后的菌落生长情况及菌落PCR鉴定的结果如图3所示:从图中可以看出,五个环状重组载体和一个接收载体的在有短链引物阻遏的条件下的组装效率为91.7%,五个环状重组载体和一个接收载体的在没有短链引物阻遏的条件下的组装效率为58.3%。六个环状重组载体和一个接收载体的在有短链引物阻遏的条件下的组装效率为95.8%,六个环状重组载体和一个接收载体的在没有短链引物阻遏的条件下的组装效率为33.3%。说明在加入了短链引物阻遏后再进行组装,可以明显提升组装效率,从而证明我们的短链引物阻遏方法确实可以提高DNA组装效率。
实施例2、短链引物阻遏的方法在多片段的拼接中的应用
本实施例的原理如附图4所示。
一、制备7个DNA片段
DNA片段1的核苷酸序列如序列表中序列25所示,DNA片段1依次包括DNA片段甲1、待拼接DNA片段1和DNA片段乙1;DNA片段1中的DNA片段甲1与短链引物C相同或互补的核苷酸序列为序列25中第27-53位核苷酸分子;待拼接DNA片段1的核苷酸序列为序列25中的第54-256位、DNA片段乙1与短链引物D相同或互补的核苷酸序列为序列25中的第257-281位;
DNA片段2的核苷酸序列如序列表中序列26所示,DNA片段2依次包括DNA片段甲2、待拼接DNA片段2和DNA片段乙2;DNA片段2中的DNA片段甲2与短链引物C相同或互补的核苷酸序列为序列26中第27-53位核苷酸分子;待拼接DNA片段2的核苷酸序列为序列26中的第54-254位、DNA片段乙2与短链引物D相同或互补的核苷酸序列为序列26中的第255-279位;
DNA片段3的核苷酸序列如序列表中序列27所示,DNA片段3依次包括DNA片段甲3、待拼接DNA片段3和DNA片段乙3;DNA片段3中的DNA片段甲3与短链引物C相同或互补的核苷酸序列为序列27中第27-53位核苷酸分子;待拼接DNA片段3的核苷酸序列为序列27中的第54-298位、DNA片段乙3与短链引物D相同或互补的核苷酸序列为序列27中的第299-323位;
DNA片段4的核苷酸序列如序列表中序列28所示,DNA片段4依次包括DNA片段甲4、待拼接DNA片段4和DNA片段乙4;DNA片段4中的DNA片段甲4与短链引物C相同或互补的核苷酸序列为序列28中第27-53位核苷酸分子;待拼接DNA片段4的核苷酸序列为序列28中的第54-298位、DNA片段乙4与短链引物D相同或互补的核苷酸序列为序列28中的第299-323位;
DNA片段5的核苷酸序列如序列表中序列29所示,DNA片段5依次包括DNA片段甲5、待拼接DNA片段5和DNA片段乙5;DNA片段5中的DNA片段甲5与短链引物C相同或互补的核苷酸序列为序列29中第27-53位核苷酸分子;待拼接DNA片段5的核苷酸序列为序列29中的第54-298位、DNA片段乙5与短链引物D相同或互补的核苷酸序列为序列29中的第299-323位;
DNA片段6的核苷酸序列如序列表中序列30所示,DNA片段6依次包括DNA片段甲6、待拼接DNA片段6和DNA片段乙6;DNA片段6中的DNA片段甲6与短链引物C相同或互补的核苷酸序列为序列30中第27-53位核苷酸分子;待拼接DNA片段6的核苷酸序列为序列30中的第54-304位、DNA片段乙6与短链引物D相同或互补的核苷酸序列为序列30中的第305-329位;
DNA片段7的核苷酸序列如序列表中序列31所示,DNA片段7依次包括DNA片段甲7、待拼接DNA片段7和DNA片段乙7;DNA片段7中的DNA片段甲7与短链引物C相同或互补的核苷酸序列为序列31中第27-53位核苷酸分子;待拼接DNA片段7的核苷酸序列为序列31中的第54-253位、DNA片段乙7与短链引物D相同或互补的核苷酸序列为序列31中的第254-278位;
每个DNA片段甲的两端均为IIs型限制性内切酶序列,且IIs型限制性内切酶酶切DNA片段产生的DNA片段甲上游黏性末端与DNA片段甲下游黏性末端不同且不配对;
每个DNA片段乙的两端均为IIs型限制性内切酶序列,且IIs型限制性内切酶酶切DNA片段产生的DNA片段乙上游黏性末端与DNA片段乙下游黏性末端不同且不配对;
DNA片段1经IIs型限制性内切酶酶切产生待拼DNA片段1下游黏性末端与DNA片段2经IIs型限制性内切酶酶切产生待拼DNA片段2上游黏性末端匹配;
DNA片段2经IIs型限制性内切酶酶切产生待拼DNA片段2下游黏性末端与DNA片段3经IIs型限制性内切酶酶切产生待拼DNA片段3上游黏性末端匹配;
依次类推;
DNA片段m-1经IIs型限制性内切酶酶切产生的待拼DNA片段m-1下游黏性末端与DNA片段m经IIs型限制性内切酶酶切产生的待拼DNA片段m上游黏性末端匹配;
短链引物C的核苷酸序列为序列23;
短链引物D的核苷酸序列为序列24;
上述DNA片段1、DNA片段2、DNA片段3、DNA片段4、DNA片段5、DNA片段6、DNA片段7的制备方法具体如下:
1、DNA片段1
以载体1为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段1,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段1的核苷酸序列如序列表中序列25所示,DNA片段1中的DNA片段甲1为序列25中第20‐57位核苷酸分子、DNA片段乙1为序列25中的第253‐288位。其中,序列25所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列25所示的DNA分子的第257-281位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
2、DNA片段2
以载体2为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段2,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段2的核苷酸序列如序列表中序列26所示,DNA片段2中的DNA片段甲2为序列26中第20‐57位核苷酸分子、DNA片段乙2为序列26中的第251‐286位。其中,序列26所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列26所示的DNA分子的第255-279位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
3、DNA片段3
以载体3为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段3,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段3的核苷酸序列如序列表中序列27所示,DNA片段3中的DNA片段甲3为序列27中第20‐57位核苷酸分子、DNA片段乙3为序列27中的第295‐330位。其中,序列27所示的DNA分子的27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列27所示的DNA分子的第299-323位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
4、DNA片段4
以载体4为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段4,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段4的核苷酸序列如序列表中序列28所示,DNA片段4中的DNA片段甲4为序列28中第20‐57位核苷酸分子、DNA片段乙4为序列28中的第295‐330位。其中,序列28所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列28所示的DNA分子的第299-323位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
5、DNA片段5
以载体5为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段5,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段5的核苷酸序列如序列表中序列29所示,DNA片段5中的DNA片段甲5为序列29中第20‐57位核苷酸分子、DNA片段乙5为序列29中的第294‐330位。其中,序列29所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列29所示的DNA分子的第299-323位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
6、DNA片段6
以载体6为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段6,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段6的核苷酸序列如序列表中序列30所示,DNA片段6中的DNA片段甲6为序列30 中第20‐57位核苷酸分子、DNA片段乙6为序列30中的第301‐336位。其中,序列30所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列30所示的DNA分子的第305-329位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对;
7、DNA片段7
以载体7为模板,采用引物1和引物2(引物1:TGTAAAACGACGGCCAGT;引物2:CAGGAAACAGCTATGAC)进行PCR扩增,得到PCR扩增产物,记作DNA片段7,该DNA片段包括待拼接的目的片段,且目的片段的两侧为用于与短链引物互补配对的DNA序列;DNA片段7的核苷酸序列如序列表中序列31所示,DNA片段7中的DNA片段甲7为序列31中第20‐57位核苷酸分子、DNA片段乙7为序列31中的第250‐285位。其中,序列31所示的DNA分子的第27-53位核苷酸分子与短链引物C的第2-28位核苷酸分子互补配对;序列31所示的DNA分子的第254-278位核苷酸分子的互补链与短链引物D的第2-26位核苷酸分子互补配对。
二、多片段的拼接
用BsaI酶切m个DNA片段,得到酶切产物;再用短链引物C和短链引物D对酶切产物进行退火(短链引物C与DNA片段甲全长或部分片段相同或互补),短链引物D与DNA片段乙全长或部分片段相同或互补),得到退火产物;连接酶连接退火产物,得到全长DNA片段,实现m个目的DNA片段的拼接。
本实施例中的m为3或5或7。具体步骤如下:
1、拼接模板的获得
将DNA片段1、DNA片段2和DNA片段3等质量比混合,记为模板1;
将DNA片段1、DNA片段2、DNA片段3、DNA片段4和DNA片段5等质量比混合,记为模板2;
将DNA片段1、DNA片段2、DNA片段3、DNA片段4、DNA片段5、DNA片段6和DNA片段7等质量比混合,记为模板3。
2、拼接体系的配制
按照是否添加用于阻遏的短链引物分成如下6组拼接体系:
拼接体系1:1*NEB buffer4、50-100ng(100ng)模板1、100ug/ml BSA、5U BsaI-HF(NEB);
拼接体系2:1*NEB buffer4、50-100ng(100ng)模板2、100ug/ml BSA、5U BsaI-HF(NEB);
拼接体系3:1*NEB buffer4、50-100ng(100ng)模板3、100ug/ml BSA、5U BsaI-HF(NEB);
拼接体系4:1*NEB buffer4、0.5uM oligo1、0.5uM oligo2、50-100ng(100ng)待拼接的DNA片段1、100ug/ml BSA、5U BsaI-HF(NEB);
拼接体系5:1*NEB buffer4、0.5uM oligo1、0.5uM oligo2、50-100ng(100ng)待拼接的DNA片段2、100ug/ml BSA、5U BsaI-HF(NEB);
拼接体系6:1*NEB buffer4、0.5uM oligo1、0.5uM oligo2、50-100ng(100ng)待拼接的DNA片段3、100ug/ml BSA、5U BsaI-HF(NEB)。
其中Oligo短链引物序列如下:
短链引物C:AAGAGACCTGTGCCAAGAGACTGCGGATA(序列23);
短链引物D:AAGAGACCGTGCGAGTTACTGCCAACA(序列24)。
将上述6组拼接体系先置于37℃条件下酶切1小时;在将体系进行引物阻遏反应,得到 反应产物。引物阻遏的反应条件为65℃ 20分钟;50℃ 5分钟;室温5分钟。
3、多个DNA片段的拼接
将步骤2中的反应产物加入连接反应体系中进行连接反应,得到连接产物。上述连接反应体系:PEG4000(Thermo Scientific 00135879)5%(质量-体积浓度),T4连接酶(Thermo Scientific EL0011)1U、ATP 1mM、DTT 5mM;连接反应条件:室温连接1小时,65℃ 10分钟失活连接酶。
三、多片段拼接的效率
将上述得到的连接产物进行电泳,连接产物的电泳检测结果图5所示。其中,泳道1:拼接体系1(3片段拼接,不添加短链引物);泳道2:拼接体系1(5片段拼接,不添加短链引物);泳道3:拼接体系3(7片段拼接,不添加短链引物);泳道4:拼接体系4(3片段拼接,添加短链引物);泳道5:拼接体系5(5片段拼接,添加短链引物);泳道6:拼接体系6(7片段拼接,添加短链引物);泳道7:2-log marker(NEB)。从电泳结果可以看出,添加短链引物后,正确拼接的产物量明显增加,从而本发明的短链引物阻遏方法确实可以提高小片段DNA的拼接成长片段的效率。
- 还没有人留言评论。精彩留言会获得点赞!
- 一组小熊猫基因的微卫星引物的制作方法与工艺
- 羊肚菌交配型决定基因鉴定专用引物及出菇能力预测方法与流程
- 一种KRAS基因的检测引物组、其构成的反应体系及应用的制作方法与工艺
- MTHFR‑A1298C基因多态性快速检测引物、分子信标、试剂盒及检测方法与流程
- 用于检测人类VKORC1与CYP2C9基因多态性的引物、探针、试剂盒及方法与流程
- 一种牛卷毛基因快速筛选的引物、方法及试剂盒与流程
- KCNQ1OT1基因、aHIF基因及其引物的应用以及用于诊断冠心病的试剂盒的制作方法与工艺
- 用于检测人类ALDH2基因多态性的引物、探针、试剂盒及方法与流程
- 一种检测耳聋基因的引物及其应用的制造方法与工艺
- 海岛棉跨膜蛋白基因、引物及其应用的制造方法与工艺