DPP-4抑制剂化合物、聚合物及其制备方法和应用与流程
本发明涉及二肽基肽酶IV(DPP-4)抑制剂药物领域,具体涉及一类二肽基肽酶IV(DPP-4)抑制剂化合物和聚合物及其制备方法和应用。
背景技术:
肠降血糖素(incretin)是小肠内分泌细胞分泌的帮助机体在进食碳水化合物后产生餐后胰岛素反应的一种激素,其促胰岛素分泌的效应约占餐后胰岛素分泌总量的60%。至今已分离出2种肠降血糖素,即葡萄糖依赖性胰岛素释放肽(GIP)和胰高血糖素样肽-1(GLP-1),且有两类全新作用机制的降糖药上市,它们都通过提高2型糖尿病(T2DM)患者GLP-1水平从而发挥降糖作用。GLP-1受体激动剂能够代替GLP-1作用于其受体,并避免被二肽基肽酶-4(DPP-4)降解,如艾塞那肽、利拉鲁肽等;而DPP-4抑制剂则抑制DPP-4对GLP-1的降解作用,使内源性GLP-1在体内蓄积而达到降血糖作用,如西格列汀、维格列汀等。这两类药物具有保护胰岛细胞、降低体重等功能,同时很少引起低血糖反应,临床应用日益广泛。
GLP-1可被二肽基肽酶-4(dipeptidyl peptidase-4,DPP-4)快速降解(1~2min),之后由肾脏清除。DPP-4抑制剂(列汀类药物)通过竞争性结合DPP-4活化部位,降低酶的催化活性,抑制GLP-1降解,从而提升GLP-1在血液中的浓度,取得改善血糖控制、保护β细胞功能的效果。
迄今,世界范围内已上市7个DPP-4抑制剂:西格列汀(sitagliptin)、维格列汀(vildagliptin)、沙格列汀(saxagliptin)、阿格列汀(alogliptin)、利格列汀(linagliptin)、吉格列汀(gemigliptin)和替格列汀(teneligliptin)。其中,西格列汀、维格列汀和沙格列汀已在我国上市,阿格列汀和利格列汀已在我国提出临床申请,吉格列汀在我国则由LG Life Sciences授权双鹤制药开发。
DPP-4抑制剂可通过提高内源性GLP-1的生物学效应而纠正胰岛α细胞功能异常所致的胰升糖素不适当分泌。DPP-4抑制剂减少胰升糖素分泌的机制包括:升高的GLP-1直接作用于胰岛α细胞抑制胰升糖素分泌,通过刺激胰岛δ细胞分泌生长抑素和促进胰岛素分泌而发挥间接作用。
技术实现要素:
本发明所解决的现有技术的问题是:现有的DPP-4抑制剂种类有限,而且其在体内的半衰期短,药物的持续作用时间较短,作用疗效有待加强。
为了解决上述问题,本发明提供了一种DPP-4抑制剂化合物、聚合物以及药物组合物及其制备方法,同时本发明还提供了DPP-4抑制剂化合物或聚合物或由其作为活性组分形成的药物组合物在制备治疗和/或预防糖尿病、减肥或其他与DPP-4相关疾病药物方面的用途以及用于治疗和/或预防糖尿病、减肥或其他与DPP-4相关的疾病。
具体而言,本发明提供了如下技术方案:
技术方案1:
一种式(I)结构所示的化合物,X-R1-R2-Y(I)
其中,
X和Y分别为含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物形成的基团,或者为前述含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物药学上可以接受的盐或光学异构体形成的基团;其中X和Y相同或不同;
R1和R2分别为含有1-6个天然或非天然氨基酸的肽链片段、或者为巯基羧酸化合物基团、或者为经巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段,其中,R1和R2相同或不同。
技术方案2:
在所述式(I)结构式中,
X和Y分别为含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物形成的仲胺或叔胺基团,或者为前述含有氨基或亚氨基的DPP-4抑制剂或 者DPP-4抑制剂类似物药学上可以接受的盐或光学异构体形成的仲胺或叔胺基团;其中X和Y相同或不同;
R1和R2分别为含有1-6个天然或非天然氨基酸的肽链片段、或者为巯基羧酸化合物基团、或者为经巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段,其中,R1和R2相同,R1和R2之间形成CO-NH、S-S或CO-S键,优选为S-S键。
技术方案3:
所述含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物选自西格列汀及其类似物、阿格列汀及其类似物、利格列汀及其类似物、沙格列汀及其类似物、维格列汀及其类似物、曲格列汀及其类似物、奥格列汀及其类似物或美格列汀及其类似物。
技术方案4:
在所述式(I)结构式中,
X和Y分别为选自西格列汀、阿格列汀、利格列汀、沙格列汀、维格列汀、曲格列汀、奥格列汀或美格列汀的仲胺或叔胺基团;R1和R2相同,均为巯基羧酸,R1和R2之间形成S-S、CO-NH或CO-S键。
技术方案5:
所述X和Y相同,均选自西格列汀、阿格列汀、利格列汀、沙格列汀、维格列汀、曲格列汀、奥格列汀或美格列汀的仲胺或叔胺基团;所述R1和R2相同,均为碳原子数为1-6的巯基羧酸基团,优选为巯基丙酸基团。
技术方案6:
西格列汀类似物结构通式如下所示:
式(1)
式(1)中,Ar为苯基或分别由卤素、碳原子数为1-6的烷基、碳原子数为1-6的烷氧基或氰基取代的苯基;X为N原子和-CR2;R1和R2各自独立地 选自以下基团中的一种:H,氰基,未取代或分别由1-5个卤素、氰基、羟基或羧基取代的碳原子数为1-10的烷基、苯基、五元或六元杂环;所述Ar优选为卤素取代苯基,还优选为氟或氯或溴取代苯基,更优选为三个卤素取代苯基,进一步优选为三个氟原子或氯取代苯基;所述R1优选为1-3个卤素取代烷基,更优选为三个卤素取代甲基;其中,所述西格列汀类似物不包括西格列汀自身;
其中阿格列汀类似物的结构通式如下所示:
式(2)
式(2)中,M为氮原子或-CR4,而R2,R3和R4各自独立的选自以下基团中的一种:非取代或者分别由氨基、氰基、羧酸取代的碳原子数为1-10的直链或支链烷基、碳原子数为3-12的环状烷基、碳原子数为3-12的含杂原子环状烷基或碳原子数为1-10的芳环烷基中的一种;其中,R3优选为氨基取代的碳原子数为3-12的含杂原子环烷基,所述杂原子选自氮或氧或硫,更优选为氨基取代的碳原子数为3-12的含氮原子环烷基,进一步更优选为氨基取代的氮杂环己基;其中L选自1-3个N原子、C原子、O原子或S原子的桥链,优选为C原子、S原子的桥链;X选自以下基团中的一种:非取代或者分别由氨基、氰基、羧酸取代的碳原子数为1-10的直链烷基,碳原子数为3-12的环状烷基,碳原子数为1-10的芳环烷基,优选为氨基、氰基取代的碳原子数为6-10的芳环烷基;其中,所述阿格列汀类似物不包括阿格列汀自身;
其中利格列汀的类似物结构通式如下所示:
式(3)
式(3)中,R1为萘基或分别由甲基、甲氧基、硝基或甲基氨基取代的萘基;2-苯乙烯基;连苯基;环己基羰基;苯并噻吩基或喹啉基;R1优选为萘基或分别由甲基、甲氧基取代的萘基;R2为甲基、异丙基、2-丙烯基、苯基、氰甲基或甲氧基羰基甲基,优选为甲基、苯基、氰甲基;R3为2-氰基苯基、2,6-二氰基苯基、2-甲基-2-丙烯基、2-氯-2-丙烯基、3-溴-2丙烯基、2-丁烯基、3-甲基-2-丁烯基、2,3-二甲基-2-丙烯基、2-丁炔基、1-环五亚乙基六胺甲基或2-呋喃甲基,优选为2-丁炔基、2-丁烯基、2-甲基-2-丙烯基,更优选为2-丁炔基;其中,所述利格列汀类似物不包括利格列汀自身;
其中沙格列汀的类似物结构通式如下所示:
式(4)
式(4)中,x为1、y为0或者x为0、y为1,n为0或1,X为H或氰基,R1,R2,R3和R4各自独立地选自以下基团中的一种:H,非取代或者分别由卤素、直链或支链烷基、硝基、氰基、氨基或羟基取代的直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基,优选为直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基,更优选为直链或支链烷基、烯基、炔基、环烷基;其中,所述沙格列汀类似物不包括沙格列汀自身;
其中维格列汀的类似物结构通式如下所示:
式(5)
式(5)中,R1和R2分别独立地选自以下基团中的一种:H,氰基,非取代或者分别由卤素、硝基、氰基、氨基或羟基取代的直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基;其中,R1优选为氰基,非取代或者分别由氰基、羟基取代的直链或支链烷基、环烷基、芳烷基,R2优选为非取代或者分别由氰基、羟基取代的环烷基、芳烷基;其中,所述维格列汀类似物不包括维格列汀自身;
其中曲格列汀的类似物结构通式如下所示:
式(6)
式(6)中,R1和R2分别独立地选自以下基团中的一种:H,非取代或者分别由卤素、硝基、氰基、氨基或羟基取代的直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基;其中,R1优选为非取代或者卤素、氰基取代的环烷基、芳烷基,更优选为卤素或氰基取代的芳烷基;R2优选为直链或支链烷基,更优选为甲基、乙基、丙基;其中,所述曲格列汀类似物不包括曲格列汀自身;
其中奥格列汀的类似物结构通式如下所示:
式(7)
式(7)中,R1和R2分别独立地选自以下基团中的一种:H,非取代或者分别由卤素、硝基、氰基、氨基羟基取代的直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基;其中,R1优选为非取代或者卤素、氰基取代的环烷基、芳烷基,更优选为1-3个卤素取代的芳烷基;R2优选为非取代或者分别由氰基、羟基取代的环烷基、芳烷基;其中,所述奥格列汀类似物不包括奥格列汀自身;
其中美格列汀的类似物结构通式如下所示:
式(8)
式(8)中,R1、R2和R3分别独立地选自以下基团中的一种:H,氰基,非取代或者分别由卤素、硝基、氰基、氨基或羟基取代的直链或支链烷基、烯基、炔基、环烷基、二环烷基、三环烷基、羟烷基、芳烷基、杂环芳基;其中R1优选为氰基,非取代或者分别由氰基、卤素取代的环烷基、芳烷基,R2优选为卤素或氰基取代的芳烷基,R3优选为杂环烷基或卤素取代的杂环芳基,更优选为卤素取代的杂环芳基;其中,所述美格列汀类似物不包括美格列汀自身。
技术方案7:
所述含有巯基羧酸化合物为碳原子数为1-8的巯基羧酸、优选为巯基丙酸、巯基乙酸或者巯基丁酸,更优选为3-巯基丙酸;所述经巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段为经碳原子数为1-8的巯基羧酸修 饰的含有1-6个天然或非天然氨基酸的肽链片段,优选为巯代丙酰甘氨酸、巯代乙酰甘氨酸或者巯代丁酰甘氨酸,更优选为3-巯基丙酰甘氨酸。
技术方案8:
X和R1之间形成了选自C-N、C-O、C=N、C-C、C=C、C-S、S-S、CO-NH、CO-S中的一种化学键,优选为CO-NH、CO-S或S-S;
Y和R2之间形成了选自C-N、C-O、C=N、C-C、C=C、C-S、S-S、CO-NH、CO-S中的一种化学键,优选为CO-NH、CO-S或S-S;
R1和R2之间形成了选自C-N、C-O、C=N、C-C、C=C、C-S、S-S、CO-NH、CO-S中的一种化学键,优选为CO-NH、CO-S或S-S。
技术方案9:
X和R1之间形成C-N、C-O、C-S或S-S键;Y和R2之间形成C-N、C-O、C-S或S-S键;R1和R2之间形成CO-NH、CO-S或S-S键。
技术方案10:
X和R1之间形成C=N、C=C或CO-NH键;Y和R2之间形成C=N、C=C或CO-NH键;R1和R2之间形成CO-NH、CO-S或S-S键。
技术方案11:
X和R1之间形成CO-NH键;Y和R2之间形成CO-NH键;R1和R2之间形成S-S、CO-S或CO-NH键。
技术方案12:
本发明同时提供了一种聚合物,采用技术方案1-11任一所述的化合物作为重复结构单元,其通式表示为[X-R1-R2-Y]n,其中n是2~10的整数。
技术方案13:
所述重复结构单元之间采用选自C-N、C-O、C=N、C-C、C=C、C-S、S-S、CO-NH、CO-S中的一种化学键,优选为CO-NH、CO-S或S-S键。
技术方案14:
本发明同时提供了以上任一所述化合物的制备方法,包括如下步骤:
将含有氨基或亚氨基的DPP-4抑制剂、或者含有氨基或亚氨基的DPP-4抑制剂类似物、或者含有氨基或亚氨基的DPP-4抑制剂药学上可接受的盐或光学异构体分别与含有1-6个天然或非天然氨基酸的肽链片段、或者巯基羧酸 化合物、或者经巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段混合,在偶联剂的作用下进行反应。
技术方案15:
所述偶联剂选自1,3-二环己基碳二亚胺、N,N’-二异丙基碳二亚胺、1-(3-二甲氨基丙基)-3-乙基羰基二胺甲碘、N,N-二异丙基乙胺、N-甲基吗啡啉、1-羟基苯并三唑、2-(7-偶氮苯并三氮唑)-N,N,N’,N’-四甲基脲六氟磷酸酯、苯并三氮唑-N,N,N’,N’-四甲基脲六氟磷酸酯、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯、2-(1H-苯并三偶氮L-1-基)-1,1,3,3-四甲基脲四氟硼酸酯、5-降冰片烯-2,3-二羰基-N,N,N’,N’-四甲基脲四氟硼酸酯、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷中的一种或几种以上。
技术方案16:
本发明还提供了一种药物组合物,其特征在于,该药物组合物包含治疗有效量的游离形式或可药用盐形式的技术方案1-11所述的化合物或者技术方案12-13所述的聚合物作为活性成分;和一种或多种药用载体物质和/或稀释剂。
技术方案17:
技术方案1-11任一所述的化合物或者技术方案12-13任一所述的聚合物或者技术方案16所述的药物组合物在制备治疗和/或预防糖尿病、减肥或其他与DPP-4相关疾病的药物方面的用途。
技术方案18:
技术方案1-11任一所述的化合物或者技术方案12-13任一所述的聚合物或者技术方案16所述的药物组合物用于治疗和/或预防糖尿病、减肥或其他与DPP-4相关的疾病。
需要说明的是,本发明提到的环烷基,是指具有一个环的环烷基。
将八种不同的DPP-4抑制剂及其类似物与含有1-6个氨基酸的肽链片段进行化学键合,或者与经巯基羧酸化合物修饰的含有1-6个氨基酸的肽链片段进行化学键合,或者直接与巯基羧酸化合物反应,制备得到DPP-4抑制剂化合物,同时还可以以制备得到的化合物作为重复结构单元,然后通过其他化学键,尤其是二硫键、酰硫键或酰胺键键合的方式形成DPP-4抑制剂二聚或多聚物。
发明人对目前已上市的DPP-4抑制剂进行了大量的筛选,选用不同的DPP-4抑制剂及其类似物、DPP-4抑制剂有效官能团和DPP-4抑制剂药学上可接受的盐、异构体、前药、衍生物及其有效官能团的衍生物,将化合物中的氨基或亚氨基活性官能团分别和1-6个氨基酸或含有巯基羧酸化合物的短肽进行酰胺化反应,DPP-4抑制剂修饰后的肽链通过二硫键或简单的其他化学键进行二聚或多聚,制备得到结构新型的DPP-4抑制剂多聚物,从而完成了本发明。同时通过动物实验进一步说明本发明的化合物可应用于制备治疗和/或预防糖尿病药物或其他与DPP-4相关疾病药物方面。
R1和R2之间若通过二硫键进行桥联时,可在氧化剂的作用下进行,其中氧化剂选自碘、溴、过氧化氢、过氧乙酸、过氧化尿素中的一种。当然,也可以直接通过购买得到已经制备好的R1和R2之间桥联的化合物。
本发明的有益效果在于:将DPP-4抑制剂或其类似物直接修饰至一段1至6个天然、非天然氨基酸或巯基羧酸化合物的肽链片段上而后进行二聚或多聚,得到DPP-4抑制剂化合物或聚合物,此类聚合体提供了一种DPP-4抑制剂的同源或异源二聚体或多聚体,具有比已上市的DPP-4抑制剂类药物更长时间的作用曲线和更低的清除率,此类化合物以通过抑制DPP-4对于GLP-1的降解活性,具体表现为阻断DPP-4对于GLP-1或者GLP-1类似物的氨基末端二位氨基酸的快速降解,从而延长了GLP-1在体内的半衰期。
附图说明
图1为本发明实施例一制备得到的目标化合物的质谱图。
图2为本发明实施例二制备得到的目标化合物的质谱图。
具体实施方式
如上所述,本发明的目的在于:提供一种DPP-4抑制剂化合物、聚合物以及药物组合物、它们的制备方法和用途。
具体而言,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I)
其中,
X和Y分别为含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物形成的基团,或者为前述含有氨基或亚氨基的DPP-4抑制剂或者DPP-4抑制剂类似物药学上可以接受的盐或光学异构体形成的基团;其中X和Y相同或不同;
R1和R2分别为含有1-6个天然或非天然氨基酸的肽链片段、或者为巯基羧酸化合物基团、或者为经巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段,其中,R1和R2相同或不同。
其中,DPP-4抑制剂选自西格列汀、阿格列汀、利拉列汀、沙格列汀、维格列汀、曲格列汀、奥格列汀或美格列汀。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为西格列汀或前述式(1)表示的西格列汀类似物形成的仲胺或叔胺基团,最优选西格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为阿格列汀或前述式(1)表示的阿格列汀类似物形成的仲胺或叔胺基团,最优选阿格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为利格列汀或前述式(1)表示的利格列汀类似物形成的仲胺或叔胺基团,最优选利格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为沙格列汀或前述 式(1)表示的沙格列汀类似物形成的仲胺或叔胺基团,最优选沙格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为维格列汀或前述式(1)表示的维格列汀类似物形成的仲胺或叔胺基团,最优选维格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为曲格列汀或前述式(1)表示的曲格列汀类似物形成的仲胺或叔胺基团,最优选曲格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为奥格列汀或前述式(1)表示的奥格列汀类似物形成的仲胺或叔胺基团,最优选奥格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为美格列汀或前述式(1)表示的美格列汀类似物形成的仲胺或叔胺基团,最优选美格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸基团,更优选为碳原子数为1-6的巯基羧酸基团、最优选为巯基丙酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
其中,在本发明的一种优选实施方式中,本发明提供了一种式(I)结构所示的化合物,X-R1-R2-Y(I),其中,X和Y相同均为西格列汀或前述式(1)表示的西格列汀类似物形成的仲胺或叔胺基团,最优选西格列汀形成的仲胺或叔胺基团;其中,R1和R2相同,均为巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段,更优选为碳原子数为1-6的巯基羧酸化合物修饰的含有1-6个天然或非天然氨基酸的肽链片段、最优选为巯基丙酰甘氨酸基团;其中R1和R2之间形成S-S、CO-NH或CO-S键。
下面结合附图和具体的实施例对本发明作进一步的详细的说明。但是,这些实施例仅是用于说明本发明,而不是对本发明范围的限制。
其中,实施例中所用的试剂和仪器的厂商型号如下:
西格列汀,阿格列汀,利格列汀,维格列汀,沙格列汀,美格列汀,曲格列汀,奥格列汀,均购自安润医药科技(苏州)有限公司;
3,3’-二硫代二丙酸,购自Aladdin;
二甲基甲酰胺,购自浙江江山化工股份有限公司;
六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(PyBop),购自苏州昊帆生物科技有限公司;
N,N’-二异丙基乙胺(DIEA),购自苏州吴帆生物科技有限公司;
质谱仪,型号MALDI-TOF 4700,厂商AB SCIEX;
BP211D Sarorius电子天平,厂商:德国赛多利斯股份有限公司;
BA-90生化分析仪,型号HL-YQ-4,厂商:深圳迈瑞;
高效液相色谱仪,型号Ultimate-U3000,厂商DIONEX;
其中实施例中所用到的HPLC的检测条件为:
用十八烷基硅烷键合硅胶(5μm,250×4.6mm)为填充剂;以0.1%TFA溶液为流动相A,以乙腈为流动相B,进行梯度洗脱;流速为每分钟1.0ml;检测波长为220nm;柱温30℃。取供试品溶液20μl,注入液相色谱仪,记录色谱图。
(一)化学合成实验
实施例一
化学反应式
将西格列汀1.01mmol加入到50ml圆底烧瓶中,采用10ml二甲基甲酰胺(DMF)将其溶解,再将3,3’-二硫代二丙酸0.50mmol,六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(PyBop)0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,通过HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.37g。通过质谱仪检测质荷比为,MSm/z:991.225(M+H),其质谱谱图如图1所示。
实施例二
化学反应式
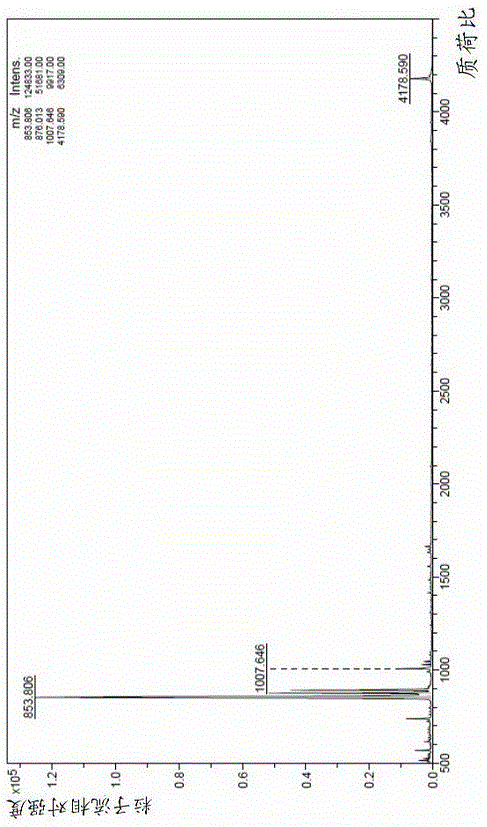
将阿格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.33g。通过质谱仪检测质荷比为,MSm/z:853.806(M+H),其质谱谱图如图2所示。
实施例三
化学反应式
将利格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.47g。通过质谱仪检测质荷比为,MSm/z:1119.476(M+H)。
实施例四
化学反应式
将维格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌 20min,过滤,得到类白色固体,干燥,得到目标化合物0.29g。通过质谱仪检测质荷比为,MSm/z:781.375(M+H)。
实施例五
化学反应式
将沙格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.31g。通过质谱仪检测质荷比为,MSm/z:835.485(M+H)。
实施例六
化学反应式
将美格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌 20min,过滤,得到类白色固体,干燥,得到目标化合物0.34g。通过质谱仪检测质荷比为,MSm/z:815.297(M+H)。
实施例七
化学反应式
将曲格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.28g。通过质谱仪检测质荷比为,MSm/z:889.352(M+H)。
实施例八
化学反应式
将奥格列汀1.01mmol加入到50ml圆底烧瓶中,采用10mlDMF将其溶解,再将3,3’-二硫代二丙酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.35g。通过质谱仪检测质荷比为,MSm/z:971.258(M+H)。
实施例九
化学反应式
将西格列汀1.01mmol加入到50ml圆底烧瓶中,采用10ml二甲基甲酰胺(DMF)将其溶解,再将3,3’-二硫代二丙氨酰二甘氨酸0.50mmol,PyBop 0.79g(1.52mmol),DIEA 0.50ml(3.04mmol)加入到反应液中,室温搅拌反应2h,待HPLC检测原料反应完,将反应液慢慢倾倒入10倍反应液体积的水中,有大量白色固体析出,搅拌20min,过滤,得到类白色固体,干燥,得到目标化合物0.32g。通过质谱仪检测质荷比为,MSm/z:1103.260(M+H)。
(二)动物实验
下面的方法是采用腹腔注射链脲霉素(STZ)建立大鼠糖尿病模型,通过皮下注射实施例一到九制备得到的DPP-4抑制剂化合物来评价它们的降糖作用,从而作进一步的筛选。
1.材料
1.1供试品及配制方法
配制前紫外灯将配制区照射30min进行消毒灭菌,然后分别称取1.04mg实施例一到实施例九制备得到的DPP-4抑制剂化合物,分别加入10.4ml溶媒,配制成浓度为100ug/ml的供试品溶液。
其中,溶媒的配制方法如下:取Na2HPO4·2H2O 0.71g加入450ml蒸馏水溶解,加入7.0g丙二醇和2.75g苯酚,用1mol/LNaOH溶液调整PH为7.7,蒸馏水定容至500ml混匀即可。
1.2造模用药及配制方法
名称:链脲霉素(STZ)
纯度:≥98%(HPLC)
规格:1.0g/瓶
批号:WXBB7151V
性状:白色或类白色或淡黄色结晶性粉末
保存条件:-20℃
生产单位:Sigma-Aldrich
配制方法:配制前用紫外灯将配制区照射30min,称取STZ加入0.1mol/L柠檬酸钠-柠檬酸缓冲液(PH=4.2),配制成10mg/ml的溶液(冰浴中避光配制)。
1.3实验动物及饲养管理
Wistar大鼠20只,雄性,体重180~220g,购于山东鲁抗医药有限公司,许可证号:SCXK(鲁)20130001。
动物入室检疫3天,温度20~26℃,湿度40~70%,换气次数:10~15次/h,每天照明12h。
2.实验方法
2.1模型建立
将实验大鼠禁食8h后测定体重,并随机分为空白对照组和模型组,其中模型组大鼠腹腔注射STZ(60mg/kg),空白对照组腹腔注射等体积柠檬酸钠-柠檬酸缓冲液;期间,观察大鼠进食情况、尿量和体重变化,并于4d后采血测空腹血糖。若出现多饮、多食、多尿、消瘦,血糖大于16.7mmol/L为造模成功。
2.2实验分组及给药
分别取正常大鼠和糖尿病大鼠,,分别定义为正常对照组和给药组,其中正常对照组大鼠按照1ml/kg的量,皮下注射溶媒;给药组按照100ug/kg的量皮下注射供试品溶液。
3.观察指标及血糖测定
测定时间:给药后的第3-7d
例数:全例
检查方法:眼眶静脉采血,常规制备血清,采用BA-90血生化分析仪进行测定血糖值。
4.统计学处理
采用EXCEL2013和SPSS13.0软件对数据进行录入和统计分析。结果如表1所示。
表1DPP-4抑制剂化合物对糖尿病大鼠降糖作用的筛选评价
±S;*p<0.05,**p<0.01与各给药组与给药前比较;★★p<0.01给药后给药组与正常对照组比较。
5.实验结果
动物实验结果数据见表1,从表1的结果可以看出:
正常对照组药后血糖与药前相比无明显差异(P>0.05),说明本发明所用溶媒本身不会给大鼠带来任何血糖值的变化;而给药之后血糖值测定结果表明,给药组与正常对照组相比均有明显差异(P<0.01)。
其中,实施例一作用7天后给药组血糖值与给药前相比下降,而且表现出显著性差异(P<0.01),给药后给药组血糖值与正常对照组相比无明显差异(P>0.05)。测定结果表明,实验例一制备得到的药物表现出良好的降糖效果,其在测定的时间内,能够使大鼠血糖降低到正常值,表现出良好的降糖效果,同时由于药物作用7天后其血糖值与正常值几乎没有差别,可以看出制备合成的化合物有着更长时间的作用曲线,可以将药物进一步研究并应用于制备糖尿病或与血糖相关的疾病。
实施例四作用7天后和实施例六作用5天后给药组血糖值与给药前相比下降,有明显差异(P<0.05,P<0.01),给药后给药组血糖值与正常对照组相比有明显差异(P<0.01),表明虽然在测定的时间内,血糖值虽然没有降低到正常值,但是实施例四和实施例六制备的药物仍然表现出良好的降糖效果,使我们有理由相信同样可以将药物应用于制备糖尿病或者与血糖相关的疾病。
实施例二作用3天后、实施例三作用3天后、实施例五作用3天后、实施例七作用7天后、实施例八作用3天后和实施例九作用5天后给药组血糖值与给药前相比并无明显差异(P>0.05),给药后给药组血糖值与正常对照组相比有明显差异(P<0.01)。测定结果表明,虽然以上实施例制备得到的药物在测 定的时间内,并没有表现出明显的降糖效果,但是实施例二、实施例七以及实施例九的血糖值相比较给药前还是表现为下降趋势。这提示我们制备得到的药物很可能用于治疗糖尿病或者与血糖相关的疾病。由于实验数据检测的误差,实施例三、实施例五、实施例八给药后与给药前相比血糖值略有波动上升,而从理论上分析实施例三、五、八制备得到的化合物及其类似物的结构,不难得出这些化合物存在能阻断DPP-4酶酶切GLP-1,从而提升GLP-1在血液中的浓度,取得改善血糖控制、保护β细胞功能的效果的潜在可能性。因此即便是实施例三、实施例五、实施例八的实验结果不太理想,但是由于利格列汀、沙格列汀、奥格列汀本身已经成药,同时结合其他实施例的实验结果,同样使我们有理由相信实施例三、五、八制备得到的化合物同样具备成药性。
最后说明的是,以上所述实施例仅为本发明的较佳实施例,仅用以说明本发明的技术方案,并不用于局限本发明,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,凡在本发明的精神和原则之内所做的修改、等同替换和改进等,均包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 可聚合化合物和其在液晶显示器中的用途的制作方法与工艺
- 可聚合化合物及其在液晶显示器中的用途的制作方法与工艺
- 能聚合的液晶化合物,用于光学膜的组合物,以及光学膜、补偿膜、抗反射膜和显示装置的制作方法
- 对非酚类聚合物使用酚类脂肪酸化合物的方法与流程
- 化合物、聚合物、着色剂、感光性树脂组成物、感光性树脂层以及彩色滤光片的制造方法与工艺
- 包含改性的二有机硅氧烷聚合物的化合物的制造方法与工艺
- 用于氯乙烯聚合物树脂的新型偶联尿嘧啶化合物的制造方法与工艺
- 聚合性化合物、组合物、聚合物、光学各向异性体、液晶显示元件和有机EL元件的制造方法与工艺
- 可聚合的化合物和其在液晶显示器中的用途的制造方法与工艺
- 免疫调节剂‑聚合物化合物的制造方法与工艺