有机电致发光材料、装置和调配物的制作方法
本申请是2015年5月5日提交的PCT申请第PCT/US15/29269号的部分接续申请,其要求2014年5月08日提交的美国临时申请第61/990,239号和2014年11月21日提交的美国临时申请第62/082,970号的优先权,所述申请的全部内容以引用的方式并入本文中。
技术领域
本发明大体上涉及新颖化合物;包含所述化合物的组合物;和所述化合物和组合物的应用,包括包含所述化合物和/或组合物的有机电致发光装置。
联合研究协议的各方
所要求的本发明是由达成联合大学公司研究协议的以下各方中的一或多者,以以下各方中的一或多者的名义和/或结合以下各方中的一或多者而作出:南加州大学和环宇显示器公司(Universal Display Corporation)。所述协议在作出所要求的本发明的日期当天和之前就生效,并且所要求的本发明是因在所述协议的范围内进行的活动而作出。
背景技术:
一般来说,OLED包含安置在阳极与阴极之间并且电连接到阳极和阴极的至少一个有机层。当施加电流时,阳极注入空穴并且阴极注入电子到有机层中。所注入的空穴和电子各自朝带相反电荷的电极迁移。当电子和空穴局限于同一分子上时,形成“激子”,其为具有激发能量状态的局部化电子-空穴对。当激子经由光电发射机制弛豫时,发射光。在一些情况下,激子可以局限于激元或激态复合物上。非辐射机制(例如热弛豫)也可能发生,但通常被视为不合需要的。
最初的OLED使用从单态发射光(“荧光”)的发射分子,如例如美国专利第4,769,292号中所公开,所述专利以全文引用的方式并入。荧光发射通常在小于10纳秒的时间范围中发生。
最近,已经论证了具有从三重态发射光(“磷光”)的发射材料的OLED。巴尔多(Baldo)等人的“从有机电致发光装置的高效磷光发射(Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices)”,自然(Nature),第395卷,第151-154页,1998;(“巴尔多-I”)和巴尔多等人的“基于电致磷光的非常高效绿色有机发光装置(Very high-efficiency green organic light-emitting devices based on electrophosphorescence)”,应用物理学报(Appl.Phys.Lett.),第75卷,第3期,第4-6页(1999)(“巴尔多-II”)以全文引用的方式并入。磷光可以称为“禁阻”跃迁,因为所述跃迁需要自旋态变化,并且量子力学指示这种跃迁是不利的。因此,磷光通常发生在超过至少10纳秒并且典型地大于100纳秒的时间帧中。如果磷光的自然辐射寿命太长,那么三重态可以通过非辐射机制而衰变,以便不发射光。往往还在非常低的温度下在含有杂原子、具有非共用电子对的分子中观察到有机磷光。2,2'-联吡啶是这种分子。非辐射衰变机制典型地是温度依赖性的,以使得在液氮温度下展现磷光的有机材料典型地在室温下不展现磷光。但,如巴尔多所证实,此问题可以通过选择在室温下发磷光的磷光化合物而得到解决。代表性发射层包括经掺杂或未经掺杂的磷光有机金属材料,例如美国专利第6,303,238号、第6,310,360号、第6,830,828号和第6,835,469号、美国专利申请公开第2002-0182441号和WO 2002/074015中所公开。
磷光前面可以是从三重态激发态跃迁为中间的非三重态状态,从所述非三重态状态发生发射衰变。举例来说,配位到镧系元素的有机分子往往从局域化于镧系金属上的激发态发磷光。然而,此类材料不直接从三重态激发态发磷光,而实际上从集中于镧系金属离子上的原子激发态发射。铕二酮络合物说明了一组这些类型的物质。
可以通过限制、优选地通过键结紧密接近于具有高原子数的原子的有机分子,优于荧光增强来自三重态的磷光。称为重原子效应的此现象通过称为自旋轨道耦合的机制而产生。可以从有机金属分子(例如三(2-苯基吡啶)铱(III))的激发的金属到配位体电荷转移(MLCT)状态观察到这种磷光跃迁。
出于若干原因,利用有机材料的光学电子装置变得越来越受欢迎。用以制造这样的装置的材料中的许多材料相对便宜,因此有机光学电子装置具有获得相对于无机装置的成本优势的潜力。另外,有机材料的固有性质(例如其柔性)可以使其非常适合具体应用,例如在柔性衬底上的制造。有机光学电子装置的实例包括有机发光装置(OLED)、有机光电晶体管、有机光伏打电池和有机光检测器。对于OLED,有机材料可以具有相对于常规材料的性能优点。举例来说,有机发射层发射光的波长通常可以容易地用适当的掺杂剂来调整。
OLED利用有机薄膜,其在电压施加于装置上时发射光。OLED正变为用于例如平板显示器、照明和背光应用中的越来越引人注目的技术。美国专利第5,844,363号、第6,303,238号和第5,707,745号中描述若干OLED材料和配置,所述专利以全文引用的方式并入本文中。
磷光性发射分子的一个应用是全色显示器。用于这种显示器的行业标准需要适于发射具体色彩(称为“饱和”色彩)的像素。具体地说,这些标准需要饱和的红色、绿色和蓝色像素。或者,OLED可经设计以发射白光。在常规液晶显示器中,使用吸收滤光器滤过来自白色背光的发射以产生红色、绿色和蓝色发射。相同技术也可以用于OLED。白色OLED可以是单EML装置或堆叠结构。可以使用本领域中所熟知的CIE坐标来测量色彩。
绿色发射分子的一个实例是三(2-苯基吡啶)铱、表示为Ir(ppy)3,其具有以下结构:
在此图和本文后面的图中,将从氮到金属(此处,Ir)的配价键描绘为直线。
如本文所用,术语“有机”包括聚合材料以及小分子有机材料,其可以用以制造有机光学电子装置。“小分子”是指不是聚合物的任何有机材料,并且“小分子”可能实际上相当大。在一些情况下,小分子可以包括重复单元。举例来说,使用长链烷基作为取代基不会将分子从“小分子”类别中去除。小分子还可以并入到聚合物中,例如作为聚合物主链上的侧基或作为主链的一部分。小分子还可以充当树枝状聚合物的核心部分,所述树枝状聚合物由建立在核心部分上的一系列化学壳层组成。树枝状聚合物的核心部分可以是荧光或磷光小分子发射体。树枝状聚合物可以是“小分子”,并且据信当前在OLED领域中使用的所有树枝状聚合物都是小分子。
如本文所用,“顶部”意指离衬底最远,而“底部”意指离衬底最近。在将第一层描述为“安置”在第二层“上”的情况下,第一层被安置为距衬底较远。除非规定第一层“与”第二层“接触”,否则第一与第二层之间可以存在其它层。举例来说,即使阴极和阳极之间存在各种有机层,仍可以将阴极描述为“安置在”阳极“上”。
如本文所用,“溶液可处理”意指能够以溶液或悬浮液的形式在液体介质中溶解、分散或输送和/或从液体介质沉积。
当据信配位体直接促成发射材料的光敏性质时,配位体可以称为“光敏性的”。当据信配位体并不促成发射材料的光敏性质时,配位体可以称为“辅助性的”,但辅助性的配位体可以改变光敏性的配位体的性质。
如本文所用,并且如本领域技术人员一般将理解,如果第一能级较接近真空能级,那么第一“最高占用分子轨道”(HOMO)或“最低未占用分子轨道”(LUMO)能级“大于”或“高于”第二HOMO或LUMO能级。由于将电离电位(IP)测量为相对于真空能级的负能量,因此较高HOMO能级对应于具有较小绝对值的IP(负得较少的IP)。类似地,较高LUMO能级对应于具有较小绝对值的电子亲和性(EA)(负得较少的EA)。在常规能级图上,真空能级在顶部,材料的LUMO能级高于同一材料的HOMO能级。“较高”HOMO或LUMO能级表现为比“较低”HOMO或LUMO能级靠近这个图的顶部。
如本文所用,并且如本领域技术人员一般将理解,如果第一功函数具有较高绝对值,那么第一功函数“大于”或“高于”第二功函数。因为通常将功函数测量为相对于真空能级的负数,因此这意指“较高”功函数负得较多。在常规能级图上,真空能级在顶部,将“较高”功函数说明为在向下方向上距真空能级较远。因此,HOMO和LUMO能级的定义遵循与功函数不同的惯例。
可以在以全文引用的方式并入本文中的美国专利第7,279,704号中找到关于OLED和上文所述的定义的更多细节。
技术实现要素:
根据本发明的一个方面,公开了一种化合物,其具有根据下式1的结构(LA)nMLm:
在式1中,M是具有大于40的原子量的金属,n具有至少1的值,并且m+n是可以与所述金属M连接的最大配位体数;
其中A是具有两到三个键联原子的键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;其中所述键联原子在两个键联原子之间形成至少一个单键;其中R1a到R1g各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中R1b到R1g连接的任一环原子可以被氮原子置换,
其中当所述环原子被氮原子置换时,所述相应R基团并不存在;并且其中L是被取代或未被取代的环金属化配位体。
根据本发明的另一方面,公开了一种有机发光装置。所述OLED包含阳极;阴极;和安置在所述阳极与所述阴极之间的有机层,其中所述有机层包含具有根据式1的结构的化合物。
根据本发明的另一方面,还公开了一种调配物,其包含具有根据式1的结构的化合物。
根据本发明的另一方面,公开了一种化合物,其具有根据以下展示的式(1a)的结构。
在式(1a)中,A是具有两到三个键联原子的键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;
其中Rab、Rga、R1b到R1f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;并且
其中Rab、Rga、R1b到R1f连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在。
根据本发明的另一方面,所述式(1a)化合物可以是具有由如下文所定义的系栓在一起的结构式式(2a)和式(2b)表示的结构的化合物:
其中A1和A2各自是具有两到三个键联原子的第一键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B和其组合,并且
其中Rac、Rgb和R2b到R2f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中所述化合物经由至少一个在Rab与Rac和/或Rga与Rgb之间形成的第二键联基团系栓在一起,其中至少一个第二键联基团具有一到三个键联原子,并且每个键联原子独立地选自由以下组成的群组:B、N、P、O、S、Se、C、Si、Ge和其组合;并且R1b到R1f和R2b到R2f连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在。
根据本发明的另一方面,所述式(1a)化合物可以是具有由以下展示的式(3a)表示的结构的化合物:
其中L2和L3各自独立地选自由以下组成的群组:单键、BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、SiR1R2和GeR1R2;
其中R3a到R3f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R1和R2独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中任两个相邻R1f、R3a、R3c、R3d、R1和R2任选地连接以形成环;其中L2与R1f、L2与R3a、或L2与R1f和R3a两者任选地连接以形成一或多个环;并且其中L3与R3c、L3与R3d、或L3与R3c和R3d两者任选地连接以形成一或多个环。
附图说明
当结合例示性实施例的附图阅读时,将更好地理解前述发明内容以及根据本发明的化合物、组合物和装置的例示性实施例的以下详细描述。然而,应理解,本发明不限于所示精确排列和手段。
在图中:
图1展示了例示性有机发光装置100;并且
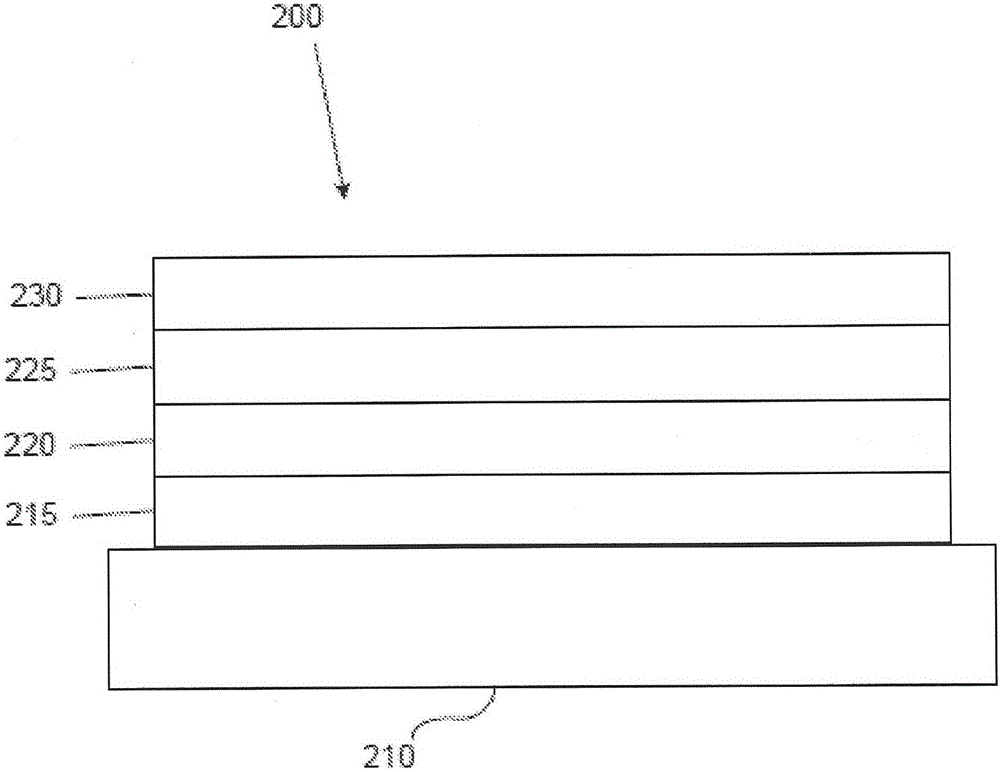
图2说明了根据本发明的例示性有机发光装置200。
图3a和3b说明了比较实例1的最小化断键几何构型(顶部)和最小化非断键几何构型(底部)的计算模型。
图4说明了比较化合物4的MALDI负模式质谱。最高强度峰对应于咪唑环的断裂。
图5说明了3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的x射线晶体结构。
图6说明了3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽的x射线晶体结构。
图7描绘了化合物49在77K和室温2-甲基THF溶剂和固态PMMA基质中的发射光谱。
具体实施方式
一般来说,OLED包含安置在阳极与阴极之间并且电连接到阳极和阴极的至少一个有机层。当施加电流时,阳极注入空穴并且阴极注入电子到有机层中。所注入的空穴和电子各自朝带相反电荷的电极迁移。当电子和空穴局限于同一分子上时,形成“激子”,其为具有激发能量状态的局部化电子-空穴对。当激子经由光电发射机制弛豫时,发射光。在一些情况下,激子可以局限于激元或激态复合物上。非辐射机制(例如热弛豫)也可能发生,但通常被视为不合需要的。
最初的OLED使用从单态发射光(“荧光”)的发射分子,如例如美国专利第4,769,292号中所公开,所述专利以全文引用的方式并入。荧光发射通常在小于10纳秒的时间范围中发生。
最近,已经论证了具有从三重态发射光(“磷光”)的发射材料的OLED。巴尔多(Baldo)等人的“从有机电致发光装置的高效磷光发射(Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices)”,自然,第395卷,第151-154页,1998;(“巴尔多-I”)和巴尔多等人的“基于电致磷光的非常高效绿色有机发光装置(Very high-efficiency green organic light-emitting devices based on electrophosphorescence)”,应用物理学报,第75卷,第3期,第4-6页(1999)(“巴尔多-II”)以全文引用的方式并入。以引用的方式并入的美国专利第7,279,704号第5-6列中更详细地描述磷光。
咪唑并菲啶是当连接到铂和铱金属两者时可以提供460nm发射的适用配位体。磷光咪唑并菲啶络合物可以按范围介于接近零到单一的可调光致发光量子产率提供深蓝色发射。不幸地,对于基于铱和铂两者的蓝色发射络合物,装置寿命都有限。我们在本文中提供了通过将一键定位在配位体上来提高咪唑并菲啶配位体的稳定性的策略,所述键通过计算理论、质谱片段分析和光氧化研究显示为弱键,这归因于多环环应力和电子结构。
图1展示了有机发光装置100。图不一定按比例绘制。装置100可以包括衬底110、阳极115、空穴注入层120、空穴输送层125、电子阻挡层130、发射层135、空穴阻挡层140、电子输送层145、电子注入层150、保护层155、阴极160和屏障层170。阴极160是具有第一导电层162和第二导电层164的复合阴极。装置100可以通过依序沉积所描述的层来制造。在以引用的方式并入的US 7,279,704的第6-10列中更详细地描述这些各种层以及实例材料的性质和功能。
这些层中的每一者有更多实例。举例来说,以全文引用的方式并入的美国专利第5,844,363号中公开柔性并且透明的衬底-阳极组合。经p掺杂的空穴输送层的实例是以50:1的摩尔比率掺杂有F4-TCNQ的m-MTDATA,如以全文引用的方式并入的美国专利申请公开案第2003/0230980号中所公开。以全文引用的方式并入的颁予汤普森(Thompson)等人的美国专利第6,303,238号中公开发射材料和主体材料的实例。经n掺杂的电子输送层的实例是以1:1的摩尔比率掺杂有Li的BPhen,如以全文引用的方式并入的美国专利申请公开案第2003/0230980号中所公开。以全文引用的方式并入的美国专利第5,703,436号和第5,707,745号公开了阴极的实例,其包括具有例如Mg:Ag等金属薄层与上覆的透明、导电、经溅镀沉积的ITO层的复合阴极。以全文引用的方式并入的美国专利第6,097,147号和美国专利申请公开案第2003/0230980号中更详细地描述阻挡层的原理和使用。以全文引用的方式并入的美国专利申请公开案第2004/0174116号中提供注入层的实例。可以在以全文引用的方式并入的美国专利申请公开案第2004/0174116号中找到保护层的描述。
图2展示了倒转的OLED 200。所述装置包括衬底210、阴极215、发射层220、空穴输送层225和阳极230。装置200可以通过依序沉积所描述的层来制造。因为最常见OLED配置具有安置在阳极上的阴极,并且装置200具有安置在阳极230下的阴极215,所以装置200可以称为“倒转”OLED。在装置200的对应层中,可以使用与关于装置100所描述的材料类似的材料。图2提供了可以如何从装置100的结构省略一些层的一个实例。
图1和2中所说明的简单分层结构是作为非限制实例而提供,并且应理解,可以结合各种各样的其它结构使用本发明的实施例。所描述的具体材料和结构本质上是示范性的,并且可以使用其它材料和结构。可以基于设计、性能和成本因素,通过以不同方式组合所描述的各个层来实现功能性OLED,或可以完全省略若干层。还可以包括未具体描述的其它层。可以使用不同于具体描述的材料的材料。尽管本文所提供的实例中的许多实例将各种层描述为包含单一材料,但应理解,可以使用材料的组合(例如主体与掺杂剂的混合物)或更一般来说,混合物。并且,所述层可以具有各种子层。本文中给予各个层的名称不意欲具有严格限制性。举例来说,在装置200中,空穴输送层225输送空穴并且将空穴注入到发射层220中,并且可以被描述为空穴输送层或空穴注入层。在一个实施例中,可以将OLED描述为具有安置在阴极与阳极之间的“有机层”。此有机层可以包含单个层,或可以进一步包含如例如关于图1和2所描述的不同有机材料的多个层。
还可以使用未具体描述的结构和材料,例如包含聚合材料的OLED(PLED),例如以全文引用的方式并入的颁予弗兰德(Friend)等人的美国专利第5,247,190号中所公开。作为另一实例,可以使用具有单个有机层的OLED。OLED可以堆叠,例如如以全文引用的方式并入的颁予福利斯特(Forrest)等人的第5,707,745号中所描述。OLED结构可以脱离图1和2中所说明的简单分层结构。举例来说,衬底可以包括有角度的反射表面以改进出耦(out-coupling),例如如颁予福利斯特等人的美国专利第6,091,195号中所述的台式结构,和/或如颁予布利维克(Bulovic)等人的美国专利第5,834,893号中所述的凹点结构,所述专利以全文引用的方式并入。
除非另外规定,否则可以通过任何合适方法来沉积各种实施例的层中的任一者。对于有机层,优选方法包括热蒸发、喷墨(例如以全文引用的方式并入的美国专利第6,013,982号和第6,087,196号中所述)、有机气相沉积(OVPD)(例如以全文引用的方式并入的颁予福利斯特等人的美国专利第6,337,102号中所述)和通过有机蒸气喷射印刷(OVJP)的沉积(例如以全文引用的方式并入的美国专利第7,431,968号中所述)。其它合适沉积方法包括旋涂和其它基于溶液的工艺。基于溶液的工艺优选在氮或惰性气氛中进行。对于其它层,优选方法包括热蒸发。优选的图案化方法包括通过掩模的沉积、冷焊(例如以全文引用的方式并入的美国专利第6,294,398号和第6,468,819号中所述)和与例如喷墨和OVJD等沉积方法中的一些方法相关联的图案化。还可以使用其它方法。可以改进待沉积的材料,以使其与具体沉积方法相容。举例来说,可以在小分子中使用具支链或无支链并且优选含有至少3个碳的例如烷基和芳基等取代基,来增强其经受溶液处理的能力。可以使用具有20个或更多个碳的取代基,并且3-20个碳是优选范围。具有不对称结构的材料可以比具有对称结构的材料具有更好的溶液可处理性,因为不对称材料可以具有更低的再结晶倾向性。可以使用树枝状聚合物取代基来增强小分子经受溶液处理的能力。
根据本发明实施例制造的装置可以进一步任选地包含屏障层。屏障层的一个用途是保护电极和有机层免于因暴露于环境中的有害物质(包括水分、蒸气和/或气体等)而受损。屏障层可以沉积在衬底、电极上,沉积在衬底、电极下或沉积在衬底、电极旁,或沉积在装置的任何其它部分(包括边缘)上。屏障层可以包含单个层或多个层。屏障层可以通过各种已知的化学气相沉积技术形成,并且可以包括具有单一相的组合物以及具有多个相的组合物。任何合适材料或材料组合都可以用于屏障层。屏障层可以并入有无机化合物或有机化合物或两者。优选的屏障层包含聚合材料与非聚合材料的混合物,如以全文引用的方式并入本文中的美国专利第7,968,146号、PCT专利申请案第PCT/US2007/023098号和第PCT/US2009/042829号中所述。为了被视为“混合物”,构成屏障层的前述聚合材料和非聚合材料应在相同反应条件下和/或在同时沉积。聚合材料对非聚合材料的重量比率可以在95:5到5:95的范围内。聚合材料和非聚合材料可以由同一前体材料产生。在一个实例中,聚合材料与非聚合材料的混合物基本上由聚合硅和无机硅组成。
根据本发明的实施例而制造的装置可以并入到各种各样的电子组件模块(或单元)中,所述电子组件模块可以并入到多种电子产品或中间组件中。此类电子产品或中间组件的实例包括可以为终端用户产品制造商所利用的显示屏、照明装置(如离散光源装置或照明面板)等。此类电子组件模块可以任选地包括驱动电子装置和/或电源。根据本发明的实施例而制造的装置可以并入到各种各样的消费型产品中,所述消费型产品具有一或多种电子组件模块(或单元)并入于其中。此类消费型产品将包括含一或多个光源和/或某种类型的视觉显示器中的一或多者的任何种类的产品。此类消费型产品的一些实例包括平板显示器、计算机监视器、医疗监视器、电视机、告示牌、用于内部或外部照明和/或发信号的灯、平视显示器、全透明或部分透明的显示器、柔性显示器、激光印刷机、电话、手机、平板计算机、平板手机、个人数字助理(PDA)、可佩戴装置、膝上型计算机、数码相机、摄录像机、取景器、微型显示器、3-D显示器、交通工具、大面积墙壁、剧院或体育馆屏幕,或指示牌。可以使用各种控制机制来控制根据本发明而制造的装置,包括无源矩阵和有源矩阵。意欲将所述装置中的许多装置用于对人类来说舒适的温度范围中,例如18摄氏度到30摄氏度,并且更优选在室温下(20-25摄氏度),但可以在此温度范围外(例如-40摄氏度到+80摄氏度)使用。
本文所述的材料和结构可以应用于不同于OLED的装置中。举例来说,例如有机太阳能电池和有机光检测器等其它光电子装置可以使用所述材料和结构。更一般来说,例如有机晶体管等有机装置可以使用所述材料和结构。
如本文所用,术语“卤基”、“卤素”或“卤化物”包括氟、氯、溴和碘。
如本文所用,术语“烷基”意指直链或支链饱和非环烃基,其可以任选地被任何适合取代基取代。因此,根据本发明的烷基可以包含伯、仲、叔和季碳原子的任何组合。例示性烷基包括(但不限于)C1-C20烷基、C1-C18烷基、C1-C16烷基、C1-C14烷基、C1-C12烷基、C1-C10烷基、C1-C8烷基、C1-C6烷基、C1-C4烷基、C1-C3烷基和C2烷基。特定实例包括甲基、乙基、1-丙基、2-丙基、2-甲基-1-丙基、1-丁基、2-丁基、叔丁基、正辛基、正癸基和正十六烷基。
如本文所用,术语“杂烷基”是指其中一或多个碳原子被杂原子置换的如本文所述的烷基。适合杂原子包括氧、硫、氮、磷等。杂烷基的实例包括(但不限于)烷氧基、氨基、硫酯、聚(乙二醇)和被烷基取代的氨基。
如本文所用,术语“环烷基”涵盖环状烷基。优选的环烷基是含有3到7个碳原子的环烷基,并且包括环丙基、环戊基、环己基等。另外,环烷基可以是任选地被取代的。
如本文所用,术语“烯基”意指具有一或多个碳-碳双键的非环支链或非支链烃基。例示性烯基包括(但不限于)C1-C20烯基、C2-C18烯基、C2-C16烯基、C2-C14烯基、C2-C12烯基、C2-C10烯基、C2-C8烯基、C2-C6烯基、C2-C4烯基、C2-C3烯基和C2烯基。特定实例包括(但不限于)乙烯基、丙烯基、1-丁烯基、2-丁烯基、异丁烯基、1-戊烯基、2-戊烯基、3-甲基-1-丁烯基、2-甲基-2-丁烯基和2,3-二甲基-2-丁烯基。
如本文所用,术语“亚烷基”意指任选地被取代的饱和直链或支链烃基。例示性亚烷基包括(但不限于)C1-C20亚烷基、C2-C18亚烷基、C2-C16亚烷基、C2-C14亚烷基、C2-C12亚烷基、C2-C10亚烷基、C2-C8亚烷基、C2-C6亚烷基、C2-C4亚烷基、C2-C3亚烷基和C2亚烷基。亚烷基的特定实例包括(但不限于)亚甲基、二亚甲基和三亚甲基。
如本文所用,术语“炔基”意指具有至少一个碳-碳三键的非环支链或非支链烃。例示性炔基包括(但不限于)C1-C20炔基、C2-C18炔基、C2-C16炔基、C2-C14炔基、C2-C12炔基、C2-C10炔基、C2-C8炔基、C2-C6炔基、C2-C4炔基、C2-C3炔基和C2炔基。炔基的特定实例包括(但不限于)炔丙基和3-戊炔基、乙炔基、丙炔基、1-丁炔基、2-丁炔基、1-戊炔基、2-戊炔基和3-甲基-1-丁炔基。
如本文所用,术语“芳烷基”意指通过烷基桥连接的如本文所定义的一或多个芳基(例如,-烷基-(芳基)j,其中j是1、2或3)。芳烷基的特定实例包括(但不限于)苯甲基(—CH2-苯基,即,Bn)、二苯基甲基(—CH2—(苯基)2)和三苯甲基(—C-(苯基)3)。另外,芳烷基可以是任选地被取代的。
除非另外说明,否则如本文所用,术语“杂环”和所述术语的变化形式(包括“杂环基(heterocyclic group)”和“杂环基”)意指具有至少两种不同元素作为环成员原子的任选地被取代的单环或多环系统,并且其中单环或多环系统是饱和、不饱和或芳香族的。在一些实施例中,杂环包含碳原子和至少一个杂原子。在一些实施例中,杂环包含碳原子和至少一个选自氮、氧、硅、硒和硫的杂原子,并且其中氮、氧、硅、硒和硫杂原子可以任选地氧化,并且氮杂原子可以任选地季铵化。杂环的实例包括(但不限于)呋喃基、苯并呋喃基、噻吩基、苯并噻吩基、吡咯基、吲哚基、异吲哚基、氮杂吲哚基、吡啶基、喹啉基、异喹啉基、噁唑基、异噁唑基、苯并噁唑基、吡唑基、咪唑基、苯并咪唑基、噻唑基、苯并噻唑基、异噻唑基、哒嗪基、嘧啶基、吡嗪基、三嗪基、噌啉基、酞嗪基和喹唑啉基。因此,除了上文所列的芳香族杂芳基之外,杂环还包括(但不限于)吗啉基、吡咯烷酮基、吡咯烷基、哌嗪基、哌啶基、乙内酰脲基、戊内酰胺基、环氧乙烷基、氧杂环丁烷基、四氢呋喃基、四氢吡喃基、四氢吡啶基、四氢嘧啶基、四氢噻吩基、四氢硫吡喃基、四氢嘧啶基、四氢噻吩基和四氢硫吡喃基。
如本文所用,术语“芳基”意指任选地被取代的单环或多环芳香族烃。芳基的特定实例包括(但不限于)苯基、苯基、4-甲基苯基、2,6-二甲基苯基、萘基、蒽基和菲基。如本文所用,术语“芳基”或“芳香族基团”涵盖单环基团和多环系统。多环可以具有其中两个碳为两个邻接环(所述环是“稠合的”)共用的两个或更多个环,其中所述环中的至少一者是芳香族的,例如其它环可以是环烷基、环烯基、芳基、杂环和/或杂芳基。另外,芳基可以是任选地被取代的。
如本文所用,术语“杂芳基”意指具有至少一个杂原子和至少一个碳原子的任选地被取代的单环或多环芳香族烃。在一些实施例中,至少一个杂原子选自氮、氧、硅、硒和硫。杂芳基的特定实例包括(但不限于)呋喃基、苯并呋喃基、噻吩基、苯并噻吩基、吡咯基、吲哚基、异吲哚基、氮杂吲哚基、吡啶基、喹啉基、异喹啉基、噁唑基、异噁唑基、苯并噁唑基、吡唑基、咪唑基、苯并咪唑基、噻唑基、苯并噻唑基、异噻唑基、哒嗪基、嘧啶基、吡嗪基、三嗪基、噌啉基、酞嗪基和喹唑啉基。
烷基、环烷基、烯基、炔基、芳烷基、杂环基、芳基和杂芳基可以任选地被一或多个选自由以下组成的群组的取代基取代:氢、氘、卤素、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、环氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、醚基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合。
如本文所用,“被取代的”表示,不是H的取代基键结到相关位置,例如碳。因此,举例来说,在R1被单取代时,则一个R1必须不是H。类似地,在R1被二取代时,则两个R1必须不是H。类似地,在R1未被取代时,R1对于所有可用位置来说都是氢。
本文所述的片段(即氮杂-二苯并呋喃、氮杂-二苯并噻吩等)中的“氮杂”名称意指各别片段中的一或多个C-H基团可以被氮原子置换,例如并且无任何限制性地,氮杂三亚苯涵盖二苯并[f,h]喹喔啉和二苯并[f,h]喹啉。本领域的普通技术人员可以容易地预想上文所述的氮杂-衍生物的其它氮类似物,并且所有这些类似物都旨在由如本文中阐述的术语涵盖。
应理解,当将分子片段描述为取代基或以其它方式连接到另一部分时,其名称可以如同其是片段(例如苯基、亚苯基、萘基、二苯并呋喃基)一般或如同其是整个分子(例如苯、萘、二苯并呋喃)一般书写。如本文所用,这些不同的命名取代基或连接的片段的方式被视为等效的。
如本文所用,并且如本领域技术人员一般将理解,如果第一能级较接近真空能级,那么第一“最高占用分子轨道”(HOMO)或“最低未占用分子轨道”(LUMO)能级“大于”或“高于”第二HOMO或LUMO能级。由于将电离电位(IP)测量为相对于真空能级的负能量,因此较高HOMO能级对应于具有较小绝对值的IP(负得较少的IP)。类似地,较高LUMO能级对应于具有较小绝对值的电子亲和性(EA)(负得较少的EA)。在常规能级图上,真空能级在顶部,材料的LUMO能级高于同一材料的HOMO能级。“较高”HOMO或LUMO能级表现为比“较低”HOMO或LUMO能级更靠近这个图的顶部。
如本文所用,术语“三重态能量”是指对应于既定材料的磷光光谱中可辩别的最高能量特征的能量。最高能量特征未必是在磷光光谱中具有最大强度的峰,并且可以是例如这个峰的高能量侧上的明显肩部的局部最大值。
根据本发明的一个方面,公开了一种化合物,其具有根据以下展示的式1的结构(LA)nMLm。
在式I中,M是具有大于40的原子量的金属,n具有至少1的值,并且m+n是可以与所述金属连接的最大配位体数;
其中A是具有两到三个键联原子的键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;
其中所述键联原子在两个键联原子之间形成至少一个单键;
其中R1a到R1g各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中R1b到R1g连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在;并且
其中L是被取代或未被取代的环金属化配位体。
在式1化合物的一些实施例中,R1b到R1g连接的环原子之一是氮原子。在一些实施例中,R1e连接的环原子是氮原子。
在一个实施例中,所述化合物具有三重态激发态,并且其中当所述化合物处于所述三重态激发态时,所述键联基团A使N2与C1b之间的键结稳定免于裂解。
在一个实施例中,化合物的峰值发射波长小于500nm。在另一实施例中,化合物的峰值发射波长小于480nm。在又一实施例中,化合物的峰值发射波长范围介于400nm到500nm。
在式1化合物的一些实施例中,键联基团A是饱和基团。
在式1化合物的一个实施例中,键联基团A独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中取代基R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。任何相邻取代基是指可能形成环的任两个取代基。两个相邻取代基可以在相同原子或不同原子上。键联基团A可以选自由以下组成的群组:
在一些实施例中,其中键联基团A独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中取代基R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;至少一个相邻R1到R6连接以形成饱和五元环或饱和六元环。在一些实施例中,至少两个相邻R1到R6(如果存在的话)连接以形成饱和五元环或饱和六元环。在一些实施例中,每个R1到R6独立地选自由以下组成的群组:烷基、环烷基、芳基、杂芳基、其部分或完全氘化变体和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在一些实施例中,其中键联基团A独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中取代基R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;每个R1到R6独立地选自由以下组成的群组:甲基、乙基、丙基、1-甲基乙基、丁基、1-甲基丙基、2-甲基丙基、戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、环戊基、环己基、苯基、2,6-二甲基苯基、2,4,6-三甲基苯基、2,6-二异丙基苯基、其部分或完全氘化变体和其组合。在一些实施例中,每个R1到R6独立地选自由以下组成的群组:烷基、其部分或完全氘化变体和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在式1化合物的一些实施例中,R1a到R1g中的至少一者选自由以下组成的群组:烷基、环烷基、芳基、杂芳基、其部分或完全氘化变体和其组合。在其它实施例中,R1b、R1d和R1e中的至少一者选自由以下组成的群组:烷基、环烷基、芳基、杂芳基、其部分或完全氘化变体和其组合。在其它实施例中,R1d选自由以下组成的群组:烷基、环烷基、芳基、杂芳基、其部分或完全氘化变体和其组合。在其它实施例中,R1a选自由以下组成的群组:烷基、环烷基、芳基、杂芳基、其部分或完全氘化变体和其组合。
在式1化合物的一些实施例中,金属M选自由以下组成的群组:Re、Ru、Os、Rh、Ir、Pd、Pt和Au。在一些实施例中,金属M选自由以下组成的群组:Ir和Pt。
在式1化合物的一些实施例中,配位体LA选自由以下组成的群组:
在式1化合物的一些实施例中,配位体L选自由以下组成的群组:
其中每个X1到X13独立地选自由以下组成的群组:碳和氮;
其中X选自由以下组成的群组:BR'、NR'、PR'、O、S、Se、C=O、S=O、SO2、CR'R”、SiR'R”和GeR'R”;
其中R'和R”任选地稠合或连接以形成环;
其中每个Ra、Rb、Rc和Rd可以表示单取代基到可能最大数目的取代基或无取代基;
其中R'、R”、Ra、Rb、Rc和Rd各自独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合;并且
其中Ra、Rb、Rc和Rd的任两个相邻取代基任选地稠合或连接以形成环或形成多齿配位体。
在式1化合物的一些实施例中,配位体L选自由以下组成的群组:
其中Ra和Rb如上文所定义。
在式1化合物的一些实施例中,配位体L选自由以下组成的群组:
和其中Ra和Rb如上文所定义。
在式1化合物的一些实施例中,配位体L选自由以下组成的群组:
其中Ra、Rb和Rc如上文所定义。
在式1化合物的一些实施例中,配位体L选自由以下组成的群组:
在式1化合物的一些实施例中,化合物是(LA)3Ir,其中LA如上文所定义。
在式1化合物的一些实施例中,化合物是(LA)Ir(L)2或(LA)2Ir(L),其中LA和L如上文所定义。
在式1化合物的一些实施例中,其中LA如上文所定义,化合物是具有式Ir(LAi)3的化合物Ax;其中x=i,i是1到2758的整数。
在式1化合物的一些实施例中,其中LA如上文所定义,化合物是具有式Ir(LAi)(Lj)2的化合物By或具有式Ir(LAi)2(Lj)的化合物Cz;
其中y=39i+j-39,i是1到2758的整数,并且j是1到39的整数;
其中z=39i+j-39,i是1到2758的整数,并且j是1到39的整数;并且
其中L1到L39具有以下结构:
在式1化合物的一些实施例中,所述化合物选自由以下组成的群组:
在式1化合物的一些实施例中,化合物具有式2的结构:
其中M是Pt;
其中A1和A2各自独立地是具有两到三个键联原子的第一键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;
其中R1b到R1f和R2b到R2f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中R1b到R1f和R2b到R2f连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在;并且
其中Rab与Rac和/或Rga与Rgb可以键结以形成具有一到三个键联原子的第二键联基团,所述键联原子各自独立地选自由以下组成的群组:B、N、P、O、S、Se、C、Si、Ge或其组合。
在式2化合物的一些实施例中,所述第一键联基团A1和A2中的每一者独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中每个R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;并且其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在式2化合物的一些实施例中,所述化合物具有三重态激发态,并且其中当所述化合物处于所述三重态激发态时,所述键联基团使N2与C1b之间的键结稳定免于裂解。
在式2化合物的一些实施例中,化合物的峰值发射波长小于500nm。在一些实施例中,化合物的峰值发射波长小于480nm。在一些实施例中,化合物的峰值发射波长范围介于400nm到500nm。
在式2化合物的一些实施例中,第一键联基团A1和A2中的每一者独立地选自由以下组成的键联基团:
在式2化合物的一些实施例中,第二键联基团独立地选自由以下组成的群组:BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-、-C-SiR1R2-、SiR1R2和GeR1R2,其中每个R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、烯基、烯基、炔基、杂烷基、芳烷基、芳基、杂芳基和其组合;并且其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在式2化合物的一些实施例中,所述化合物选自由以下组成的群组:
在式1化合物的一些实施例中,化合物具有式3:
其中M是Pt;
其中L2和L3各自独立地选自由以下组成的群组:单键、BR、NR、PR、O、S、Se、C-O、S-O、SO2、CR1R2、SiR1R2和GeR1R2;
其中R3a到R3f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中任两个相邻R1f、R3a、R3c、R3d、R1和R2任选地连接以形成环;其中L2与R1f、L2与R3a、或L2与R1f和R3a两者任选地连接以形成一或多个环;并且
其中L3与R3c、L3与R3d、或L3与R3c和R3d两者任选地连接以形成一或多个环。
在式3化合物的一些实施例中,L2和L3各自独立地选自由以下组成的群组:BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、SiR1R2和GeR1R2。在式3化合物的一些实施例中,R1f或R3a与R1或R2连接以形成环。在式3化合物的一些实施例中,R3c或R3d与R1或R2连接以形成环。
在式3化合物的一些实施例中,所述化合物选自由以下组成的群组:
根据本发明的另一方面,公开了一种有机发光装置(OLED)。所述OLED包含阳极;阴极;和安置在所述阳极与所述阴极之间的有机层,其中所述有机层包含具有根据式1的结构(LA)nMLm的化合物:
其中M是具有大于40的原子量的金属,n具有至少1的值,并且m+n是可以与所述金属连接的最大配位体数;
其中A是具有两到三个键联原子的键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;
其中所述键联原子在两个键联原子之间形成至少一个单键;
其中R1a到R1g各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中R1b到R1g连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在;并且
其中L是被取代或未被取代的环金属化配位体。
在OLED的一些实施例中,所述OLED并入到选自由以下组成的群组的装置中:消费型产品、电子组件模块和照明面板。
在OLED的一些实施例中,所述有机层是发射层,并且所述化合物是发射掺杂剂或非发射掺杂剂。
在OLED的一些实施例中,所述有机层进一步包含主体,其中所述主体包含含有苯并稠合噻吩或苯并稠合呋喃的三亚苯;
其中所述主体中的任何取代基是独立地选自由以下组成的群组的非稠合取代基:CnH2n+1、OCnH2n+1、OAr1、N(CnH2n+1)2、N(Ar1)(Ar2)、CH=CH-CnH2n+1、C≡CCnH2n+1、Ar1、Ar1-Ar2、CnH2n-Ar1或无取代基;
其中n是1到10;并且
其中Ar1和Ar2独立地选自由以下组成的群组:苯、联苯、萘、三亚苯、咔唑和其杂芳香族类似物。
在OLED的一些实施例中,所述有机层进一步包含主体,其中所述主体包含至少一个选自由以下组成的群组的化学基团:三亚苯、咔唑、二苯并噻吩、二苯并呋喃、二苯并硒吩、氮杂三亚苯、氮杂咔唑、氮杂-二苯并噻吩、氮杂-二苯并呋喃和氮杂-二苯并硒吩。
在OLED的一些实施例中,所述有机层进一步包含主体,其中所述主体选自由以下组成的群组:
和其组合。
在OLED的一些实施例中,所述有机层进一步包含主体,其中所述主体包含金属络合物。
根据本发明的另一方面,还公开了一种调配物,其包含式1化合物。所述调配物可以包括一或多种本文中所公开的选自由以下组成的群组的组分:溶剂、主体、空穴注入材料、空穴输送材料和电子输送层材料。
根据本发明的另一方面,公开了一种化合物,其具有以下展示的式(1a)。
在式(1a)中,A是具有两到三个键联原子的键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B或其组合;
其中Rab、Rga、R1b到R1f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;并且
其中Rab、Rga、R1b到R1f连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在。
在式1a化合物的一些实施例中,键联基团A独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中每个R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在式(1a)化合物的一些实施例中,所述化合物具有三重态激发态,并且其中当所述化合物处于所述三重态激发态时,所述键联基团使N2与C1b之间的键结稳定免于裂解。
在式(1a)化合物的一些实施例中,所述键联基团A选自如上文所定义的键联基团。
在式(1a)化合物的一些实施例中,所述化合物选自由以下组成的群组:
在具有式(1a)的化合物的一些实施例中,所述化合物具有如下文所定义的系栓在一起的式(2a)和式(2b)的结构:
其中A1和A2各自是具有两到三个键联原子的第一键联基团,其中所述键联原子各自独立地选自由以下组成的群组:C、Si、O、S、N、B和其组合,并且
其中Rac、Rgb和R2b到R2f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中所述化合物经由至少一个在Rab与Rac和/或Rga与Rgb之间形成的第二键联基团系栓在一起,其中至少一个第二键联基团具有一到三个键联原子,并且每个键联原子独立地选自由以下组成的群组:B、N、P、O、S、Se、C、Si、Ge和其组合;并且其中R1b到R1f和R2b到R2f连接的任一环原子可以被氮原子置换,其中当所述环原子被氮原子置换时,所述相应R基团并不存在。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,在Rab与Rac之间形成所述至少一个第二键联基团。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,在Rga与Rgb之间形成所述至少一个第二键联基团。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,在Rga与Rgb和Rab与Rac之间形成所述至少一个第二键联基团。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,所述第一键联基团A1和A2中的每一者独立地选自由以下组成的群组:-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-和-C-SiR1R2-,其中每个R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,所述第一键联基团A1和A2中的每一者独立地选自如上文所定义的键联基团。
在具有如上文所定义的系栓在一起的式2a和式2b的结构的化合物的一些实施例中,所述第二键联基团独立地选自由以下组成的群组:BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、-CR1R2-CR3R4-、-CR1R2-CR3R4-CR5R6-、-CR1R2-NR3-、-CR1=CR2-CR3R4-、-O-SiR1R2-、-CR1R2-S-、-CR1R2-O-、-C-SiR1R2-、SiR1R2和GeR1R2,其中每个R1到R6可以相同或不同,并且独立地选自由以下组成的群组:氢、氘、烷基、环烷基、芳基、杂芳基和其组合;其中任何相邻R1到R6任选地连接以形成饱和五元环或饱和六元环。
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,所述化合物选自由以下组成的群组:
在具有如上文所定义的系栓在一起的式(2a)和式(2b)的结构的化合物的一些实施例中,所述化合物具有式(3a):
其中L2和L3各自独立地选自由以下组成的群组:单键、BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、SiR1R2和GeR1R2;
其中R3a到R3f各自独立地选自由以下组成的群组:氢、氘、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、CN、CF3、CO2R、C(O)R、C(O)NR2、NR2、NO2、OR、SR、SO2、SOR、SO3R、卤基、芳基、杂芳基、杂环基和其组合;
其中每个R1和R2独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳烷基、芳基、杂芳基和其组合;
其中任两个相邻R1f、R3a、R3c、R3d、R1和R2任选地连接以形成环;其中L2与R1f、L2与R3a、或L2与R1f和R3a两者任选地连接以形成一或多个环;并且
其中L3与R3c、L3与R3d、或L3与R3c和R3d两者任选地连接以形成一或多个环。
在具有式(3a)的化合物的一些实施例中,L2和L3独立地选自由以下组成的群组:BR1、NR1、PR1、O、S、Se、C=O、S=O、SO2、CR1R2、SiR1R2和GeR1R2。
在具有式(3a)的化合物的一些实施例中,R1f或R3a与R1或R2连接以形成环。
在具有式(3a)的化合物的一些实施例中,R3c或R3d与R1或R2连接以形成环。
在具有式(3a)的化合物的一些实施例中,所述化合物选自由以下组成的群组:
根据本发明的各种实施例的金属络合物可以展现多种所要特征。在一些实施例中,具有式1、式2或式3的结构的金属络合物可以展现高量子效率下、窄光谱宽度下和/或位于所要波长范围(例如可见范围或近红外范围)内的峰值发射波长下的光致发光。此外,这些光致发光特征可以在广泛范围的激发波长内相对恒定。在一些实施例中,式1、式2或式3的金属络合物可以具有其它所要特征,例如关于其带隙能量和电导率。此外,有利地,式1、式2或式3的金属络合物可以以低成本并且容易地由市售可得起始物质合成。在一些实施例中,式1、式2或式3的金属络合物可以展现相对低量子效率下的光致发光,但这对于某些应用仍可能是充足的。
在一些实施例中,具有式1、式2或式3的结构的金属络合物的峰值发射波长小于500nm。在一些实施例中,具有式1、式2或式3的结构的金属络合物的峰值发射波长小于480nm。在一些实施例中,具有式1、式2或式3的结构的金属络合物的峰值发射波长是400nm到500nm,包括400nm和500nm。
在一些实施例中,具有式1、式2或式3的结构的金属络合物具有三重态激发态以及当所述化合物处于所述三重态激发态时,以下展示的使N2与C1b之间的键结稳定免于裂解的所述键联基团A。
因此,在一些实施例中,具有式1、式2或式3的结构的金属络合物是磷光发光物质。在一些实施例中,具有式1、式2或式3的结构的金属络合物是荧光发光物质。在一些实施例中,具有式1、式2或式3的结构的金属络合物是荧光和磷光发光物质两者。
具有式1、式2或式3的结构的金属络合物适用于例如OLED,其利用材料当其通过电流激发时发射光的倾向。因此,在一些方面,本发明提供了一种有机发光材料,其包含至少一种具有式1、式2或式3的结构的金属络合物。在一些实施例中,本发明提供了一种有机发光材料,其包含至少两种选自具有式1、式2或式3的结构的化合物的金属络合物。
根据本发明的各种实施例的有机发光材料可以展现多种所要特征。在一些实施例中,所述有机发光材料可以展现高量子效率下、窄光谱宽度下和位于所要波长范围(例如可见范围或近红外范围)内的峰值发射波长下的光致发光。此外,这些光致发光特征可以在广泛范围的激发波长内相对恒定。所述有机发光材料可以具有其它所要特征,例如关于其带隙能量和电导率。有利地,所述有机发光材料可以以低成本并且容易地形成用于各种应用,包括消费型产品和照明面板。
在一些实施例中,相对于包含发光材料的发光层的总质量,根据本发明的发光材料中的光致发光物质(例如,一或多种具有式1、式2或式3的结构的金属络合物)的含量在0.1质量%到50质量%之间(包括0.1质量%和50质量%)。在一些实施例中,相对于包含发光材料的发光层的总质量,根据本发明的发光材料中的光致发光物质的含量在0.3质量%到40质量%之间(包括0.3质量%和40质量%)。在一些实施例中,相对于包含发光材料的发光层的总质量,根据本发明的发光材料中的光致发光物质的含量在0.5质量%到30质量%之间(包括0.5质量%和30质量%)。在一些实施例中,根据本发明的发光材料中的光致发光物质附接到聚合物链或并入在树枝状聚合物材料中。
IV.装置
在一些方面,本发明提供了一种有机电致发光装置,其包含至少一种具有式1、式2或式3的结构的金属络合物。在一些实施例中,根据本发明的有机电致发光装置包含第一有机发光装置,所述第一有机发光装置进一步包含阳极;阴极;有机层,其安置在所述阳极与所述阴极之间,并且包含至少一种具有式1、式2或式3的结构的金属络合物。在有机电致发光装置的一些优选实施例中,所述有机层进一步包含主体材料。在有机电致发光装置的一些优选实施例中,所述主体材料包含有机化合物。在有机电致发光装置的一些优选实施例中,所述主体材料包含具有含有至少一个选自由以下组成的群组的基团的分子的有机化合物:咔唑、二苯并噻吩、二苯并呋喃、氮杂咔唑、氮杂-二苯并噻吩和氮杂-二苯并呋喃。
一般来说,适用于本发明的有机电致发光装置的有机层可以取决于例如所述有机电致发光装置的应用和目的而具有任何适合层配置。因此,在有机电致发光装置的一些实施例中,所述有机层在透明电极或半透明电极上形成。在一些此类实施例中,所述有机层在所述透明电极或所述半透明电极的顶部表面或任何适合表面上形成。此外,所述有机层的适合形状、大小和/或厚度可以取决于例如所述有机电致发光装置的应用和目的而利用。具有衬底、阴极、阳极和有机层的本发明有机电致发光装置的配置的特定实例包括(但不限于)以下:
(A)阳极/空穴输送层/发光层/电子输送层/阴极;
(B)阳极/空穴输送层/发光层/阻挡层/电子输送层/阴极;
(C)阳极/空穴输送层/发光层/阻挡层/电子输送层/电子注入层/阴极;
(D)阳极/空穴注入层/空穴输送层/发光层/阻挡层/电子输送层/阴极;和
(E)阳极/空穴注入层/空穴输送层/发光层/阻挡层/电子输送层/电子注入层/阴极。
(F)阳极/空穴注入层/电子阻挡层/空穴输送层/发光层/阻挡层/电子输送层/电子注入层/阴极。
包括有机电致发光装置的衬底、阴极和阳极的其它装置配置描述于日本专利公开案第2008-270736号中。
<衬底>
可用于本发明的有机电致发光装置的适合衬底优选是当用于显示器应用时不散射或减少从有机层发射的光的衬底。当用于照明或某些显示器应用时,散射光的衬底是可接受的。在一些实施例中,所述衬底优选由展现优越耐热性、尺寸稳定性、耐溶剂性、电绝缘性质和/或可加工性的有机材料组成。
适用于本发明的衬底优选是不散射或减弱从有机化合物层发射的光的衬底。用于衬底的材料的特定实例包括(但不限于)无机材料,例如氧化锆稳定钇(YSZ)和玻璃;聚酯,例如聚对苯二甲酸乙二醇酯、聚邻苯二甲酸丁二醇酯和聚萘二甲酸乙二醇酯;和有机材料,例如聚苯乙烯、聚碳酸酯、聚醚砜、聚芳酯、聚酰亚胺、聚环烯烃、降冰片烯树脂、聚氯三氟乙烯等。
在一些实施例中,当玻璃用作衬底时,优选使用无碱玻璃。适合无碱玻璃的特定实例见于川口贵广(Takahiro Kawaguchi)的在2013年9月12日出版的美国专利申请公开案第2013/0237401号中。在一些实施例中,当碱石灰玻璃用作衬底时,优选的是使用在上面已经涂覆了二氧化硅屏障涂层等的玻璃。在一些实施例中,当有机材料用作衬底时,优选的是使用具有以下属性中的一或多者的材料:极佳的耐热性、尺寸稳定性、耐溶剂性、电绝缘性能和可加工性。
一般来说,关于衬底的形状、结构、大小等不存在特定限制,但这些属性中的任一者可以根据发光元件的应用、目的等适合地选择。一般来说,板样衬底作为衬底的形状是优选的。衬底的结构可以是单层结构或层合结构。此外,衬底可以由单一成员或两种或两种以上成员形成。
尽管衬底可以是透明并且无色的或透明并且有色的,但从衬底不散射或减弱从有机发光层发射的光的观点来看,衬底优选是透明并且无色的。在一些实施例中,湿气渗透预防层(气体阻挡层)可以提供于衬底的顶部表面或底部表面上。湿气渗透预防层(气体阻挡层)的材料的实例包括(但不限于)无机物质,例如氮化硅和氧化硅。湿气渗透预防层(气体阻挡层)可以根据例如高频溅镀法等形成。
在应用热塑性衬底的情况下,可以按需要进一步提供硬涂层层或涂层下的层。
<阳极>
任何阳极都可以用于本发明的有机电致发光装置,只要其充当向有机层中供应空穴的电极。在本发明的有机电致发光装置的一些实施例中,可以取决于例如有机电致发光装置的应用和目的而使用已知电极材料的任何适合形状、结构和/或大小。在一些实施例中,透明阳极是优选的。
阳极通常可以是任何材料,只要其具有作为用于向有机化合物层供应空穴的电极的功能,并且关于形状、结构、大小等不存在特定限制。然而,其可以根据发光元件的应用和目的适合地选自熟知电极材料之中。在一些实施例中,阳极以透明阳极形式提供。
阳极的材料优选包括例如金属、合金、金属氧化物、导电化合物和其混合物。功函数为4.0eV或4.0eV以上的材料是优选的。阳极材料的特定实例包括导电金属氧化物,例如掺杂有锑、氟等的锡氧化物(ATO和FTO)、氧化锡、氧化锌、氧化铟、氧化铟锡(ITO)和氧化铟锌(IZO);金属,例如金、银、铬、铝、铜和镍;这些金属和导电金属氧化物的混合物或层合物;无机导电材料,例如碘化铜和硫化铜;有机导电材料,例如聚苯胺、聚噻吩和聚吡咯;和这些无机或有机导电材料与ITO的层合物。其中,鉴于生产率、高导电性、透明度等,导电金属氧化物是优选的,并且尤其,ITO是优选的。
考虑材料构成阳极的适合性,阳极可以根据适当地选自以下的方法在衬底上形成:湿式方法,例如印刷法、涂布法等;物理方法,例如真空沉积法、溅镀法、离子电镀法等;和化学方法,例如CVD(化学气相沉积)和等离子体CVD法等。举例来说,当ITO被选择作为阳极的材料时,阳极可以根据DC或高频溅镀法、真空沉积法、离子电镀法等形成。
在本发明的有机电致发光元件中,待形成阳极的位置不受特别限制,但其可以根据发光元件的应用和目的适合地选择。阳极可以在衬底的任一侧上的整个表面或一部分表面上形成。
为了图案化以形成阳极,可以应用化学蚀刻法(例如光刻法)、物理蚀刻法(例如通过激光蚀刻)、通过叠加掩模的真空沉积或溅镀法、或剥离法或印刷法。
阳极的厚度可以根据构成阳极的材料适合地选择,并且因此无法明确地确定,但其通常在10nm到50μm并且优选50nm到20μm范围内。阳极层的厚度可以取决于用于其的材料而恰当地控制。阳极的电阻优选是103Ω/平方或103Ω/平方以下,并且更优选是102Ω/平方或102Ω/平方以下,更优选是30Ω/平方或30Ω/平方以下。在阳极透明的情况下,其可以是透明并且无色的,或透明并且有色的。为了从透明阳极侧提取发光,优选的是阳极的透光率是60%或60%以上,并且更优选是70%或70%以上。透明阳极的详细描述可以见于C.M.C.在1999年出版的由泽田丰(Yutaka Sawada)编辑的“TOUMEI DENNKYOKU-MAKU NO SHINTENKAI(透明电极薄膜的新颖开发(Novel Developments in Transparent Electrode Films))”中。
在具有低耐热性的塑料衬底用于本发明的情况下,优选的是ITO或IZO用以获得通过在150℃或150℃以下的低温下形成薄膜而制备的透明阳极。
<阴极>
任何阴极都可以用于本发明的有机电致发光装置,只要其充当向有机层中供应电子的电极。在本发明的有机电致发光装置的一些实施例中,可以取决于例如有机电致发光装置的应用和目的而使用已知电极材料的任何适合形状、结构和/或大小。在一些实施例中,透明阴极是优选的。
阴极通常可以是任何材料,只要其具有作为用于向有机化合物层注入电子的电极的功能,并且关于形状、结构、大小等不存在特定限制。然而,其可以根据发光元件的应用和目的适合地选自熟知电极材料之中。
构成阴极的材料包括例如金属、合金、金属氧化物、导电化合物和其混合物。功函数为4.0eV或4.0eV以上的材料是优选的。其特定实例包括碱金属(例如,Li、Na、K、Cs等)、碱土金属(例如,Mg、Ca等)、金、银、铅、铝、钠-钾合金、锂-铝合金、镁-银合金、稀土金属(例如铟和镱)等。其可以单独使用,但从满足稳定性和电子可注入性两者的观点来看,优选的是其中的两者或两者以上组合使用。
在一些实施例中,鉴于电子可注入性,作为构成阴极的材料,碱金属或碱土金属是优选的,并且鉴于极佳防腐稳定性,含有铝作为主要组分的材料是优选的。
术语“含有铝作为主要组分的材料”是指单独由铝构成的材料;包含铝和0.01重量%到10重量%碱金属或碱土金属的合金;或其混合物(例如,锂-铝合金、镁-铝合金等)。例示性阴极材料详细地描述于JP-A第2-15595号和第5-121172号中。
形成阴极的方法不受特别限制,但其可以根据熟知方法形成。举例来说,考虑材料构成阴极的适合性,阴极可以根据适当地选自以下的方法形成:湿式方法,例如印刷法、涂布法等;物理方法,例如真空沉积法、溅镀法、离子电镀法等;和化学方法,例如CVD和等离子体CVD法等。举例来说,当(一或多种)金属被选择作为(一或多种)阴极的材料时,其中的一或两者或两者以上可以根据溅镀法等同时或依序应用。
为了图案化以形成阴极,可以应用化学蚀刻法(例如光刻法)、物理蚀刻法(例如通过激光蚀刻)、通过叠加掩模的真空沉积或溅镀法、或剥离法或印刷法。
在本发明中,待形成阴极的位置不受特别限制,但其可以在整个或一部分有机化合物层上形成。
此外,由碱金属或碱土金属的氟化物、氧化物等制成的介电材料层可以按0.1nm到5nm的厚度插入阴极与有机化合物层之间。介电材料层可以被视为一类电子注入层。介电材料层可以根据例如真空沉积法、溅镀法、离子电镀法等形成。
阴极的厚度可以根据构成阴极的材料适合地选择,并且因此无法明确地确定,但其通常在10nm到5μm并且优选50nm到1μm范围内。
此外,阴极可以是透明或不透明的。透明阴极可以通过以下方式形成:制备具有1nm到10nm的小厚度的阴极材料,并且进一步使透明导电材料(例如ITO或IZO)层合到其上。
<保护层>
本发明的有机EL元件的全身可以通过保护层保护。任何材料都可以应用于保护层,只要材料具有防止会加速元件的磨损的成分(例如湿气、氧气等)渗透到元件中的功能。保护层的材料的特定实例包括金属,例如In、Sn、Pb、Au、Cu、Ag、Al、Ti、Ni等;金属氧化物,例如MgO、SiO、SiO2、Al2O3、GeO、NiO、CaO、BaO、Fe2O3、Y2O3、TiO2等;金属氮化物,例如SiNx、SiNxOy等;金属氟化物,例如MgF2、LiF、AlF3、CaF2等;聚乙烯;聚丙烯;聚甲基丙烯酸甲酯;聚酰亚胺;聚脲;聚四氟乙烯;聚氯三氟乙烯;聚二氯二氟乙烯;氯三氟乙烯与二氯二氟乙烯的共聚物;通过使含有四氟乙烯与至少一种共聚单体的单体混合物共聚获得的共聚物;各自在共聚主链中具有环状结构的含氟共聚物;各自具有1%或1%以上的吸水系数的吸水材料;各自具有0.1%或0.1%以下的吸水系数的湿气渗透预防物质;等。
关于形成保护层的方法不存在特定限制。举例来说,可以应用真空沉积法、溅镀法、反应性溅镀法、MBE(分子束外延)法、团簇离子束法、离子电镀法、等离子体聚合法(高频激发离子电镀法)、等离子体CVD法、激光CVD法、热CVD法、气体源CVD法、涂布法、印刷法或转移法。
<密封>
本发明的整个有机电致发光元件可以用密封盖密封。此外,吸湿剂或惰性液体可以用以密封密封盖与发光元件之间所界定的空间。尽管吸湿剂不受特别限制,但其特定实例包括氧化钡、氧化钠、氧化钾、氧化钙、硫酸钠、硫酸钙、硫酸镁、五氧化二磷、氯化钙、氯化镁、氯化铜、氟化铯、氟化铌、溴化钙、溴化钒、分子筛、沸石、氧化镁等。尽管惰性液体不受特别限制,但其特定实例包括石蜡;液体石蜡;基于氟的溶剂,例如全氟烷烃、全氟胺、全氟醚等;基于氯的溶剂;硅酮油;等。
<驱动>
在本发明的有机电致发光元件中,当DC(按需要可以含有AC组分)电压(通常2伏特到15伏特)或DC施加到阳极和阴极上时,可以获得发光。对于本发明的有机电致发光元件的驱动方法,JP-A第2-148687号、第6-301355号、第5-29080号、第7-134558号、第8-234685号和第8-241047号;日本专利第2784615号、美国专利第5,828,429号和第6,023,308号中描述的驱动方法是可适用的。
<应用>
根据本文所述的本发明的实施例制造的装置可以并入到各种各样的消费型产品中,所述消费型产品包括(但不限于)平板显示器、计算机监视器、电视机、告示牌、用于内部或外部照明和/或发信号的灯、平视显示器、全透明显示器、柔性显示器、激光印刷机、电话、手机、个人数字助理(PDA)、膝上型计算机、数码相机、摄录像机、取景器、微型显示器、交通工具、大面积墙壁、剧院或体育馆屏幕,或指示牌。
<有机层>
适用于本发明的有机电致发光装置的有机层可以包含多个层,包括例如发光层、主体材料、电荷输送层、空穴注入层和空穴输送层。阻挡层还可以包括例如空穴(和或激子)阻挡层(HBL)或电子(和或激子)阻挡层(EBL)。在本发明的有机电致发光装置的一些实施例中,每个有机层可以通过以下方法形成:干式成膜法,例如沉积法或溅镀法;或溶液涂布工艺,例如转移法、印刷法、旋涂法或棒涂法。在本发明的有机电致发光装置的一些实施例中,有机层的至少一个层优选通过溶液涂布工艺形成。
A.发光层
发光材料:
根据本发明的发光材料优选包括至少一种具有式1、式2或式3的结构的金属络合物。相对于构成发光层的化合物的总质量,本发明的有机电致发光装置的一些实施例包含约0.1质量%到约50质量%的量的发光材料。在一些实施例中,相对于构成发光层的化合物的总质量,本发明的有机电致发光装置包含约1质量%到约50质量%的量的发光材料。在一些实施例中,相对于构成发光层的化合物的总质量,本发明的有机电致发光装置包含约2质量%到约40质量%的量的发光材料。在一些实施例中,相对于发光层中所含有的化合物的全部量,发光层中的发光材料的总量优选是约0.1重量%到约30重量%。在一些实施例中,鉴于耐久性和外部量子效率,发光层中的发光材料的总量优选是约1重量%到约20重量%。在一些实施例中,发光层中的主体材料的总量优选是约70重量%到约99.9重量%。在一些实施例中,鉴于耐久性和外部量子效率,发光层中的主体材料的总量优选是约80重量%到99重量%。在一些实施例中,可以在发光层内使用渐变的发光层或渐变的界面。渐变可以例如通过以不形成一种层向另一层的突然变化的方式混合两种或两种以上独特材料而形成。渐变发光层和或界面已经显示可增加装置寿命并且此装置架构对于增加PHOLED寿命和一般性能可以是有益的。在此情况下,发光材料可以按约0质量%到约100质量%的量存在于发光层内的任何既定位置。
在一些实施例中,本发明中的发光层可以包括发光材料和发光层中所含有的主体材料,其呈通过单重态激子发光(荧光)的荧光发射材料与主体材料的组合,或通过三重态激子发光(磷光)的磷光发光材料与主体材料的组合。在一些实施例中,本发明中的发光层可以包括发光材料和发光层中所含有的主体材料,其呈磷光发光材料与主体材料的组合。
在一些实施例中,所述第一化合物可以是发射掺杂剂。在一些实施例中,所述化合物可以经由磷光、荧光、热激活延迟荧光(即TADF,也称为E型延迟荧光)、三重态-三重态消灭或这些工艺的组合产生发射。
B.主体材料
适用于本发明的主体材料可以是空穴输送主体材料(有时称为空穴输送主体)和/或电子输送主体材料(有时称为电子输送主体)。
所述有机层还可以包括主体。在一些实施例中,两种或两种以上主体是优选的。在一些实施例中,所使用的主体可以是在电荷输送中起极小作用的a)双极,b)电子输送,c)空穴输送,或d)宽带隙材料。在一些实施例中,所述主体可以包括金属络合物。所述主体可以是含有苯并稠合噻吩或苯并稠合呋喃的三亚苯。所述主体中的任何取代基可以是独立地选自由以下组成的群组的非稠合取代基:CnH2n+1、OCnH2n+1、OAr1、N(CnH2n+1)2、N(Ar1)(Ar2)、CH=CH-CnH2n+1、C≡C-CnH2n+1、Ar1、Ar1-Ar2和CnH2n-Ar1,或无取代基。在前述取代基中,n可以在1到10范围内变化;并且Ar1和Ar2可以独立地选自由以下组成的群组:苯、联苯、萘、三亚苯、咔唑和其杂芳香族类似物。在一些实施例中,所述主体还可以是无机化合物。举例来说,含Zn的无机材料,例如ZnS。
所述主体可以是包含至少一个选自由以下组成的群组的化学基团的化合物:三亚苯、咔唑、二苯并噻吩、二苯并呋喃、二苯并硒吩、氮杂三亚苯、氮杂咔唑、氮杂-二苯并噻吩、氮杂-二苯并呋喃和氮杂-二苯并硒吩。所述主体可以包括金属络合物。所述主体可以是选自由以下组成的群组的特定化合物:
和其组合。
空穴输送主体材料
空穴输送主体材料的特定实例包括(但不限于)吡咯、咔唑、氮杂咔唑、吡唑、吲哚、氮杂吲哚、咪唑、聚芳基烷、吡唑啉、吡唑啉酮、苯二胺、芳胺、氨基取代的查耳酮、苯乙烯基蒽、芴酮、腙、芪、硅氮烷、芳香族叔胺化合物、苯乙烯胺化合物、芳香族二亚甲基化合物、卟啉化合物、聚硅烷化合物、聚(N-乙烯咔唑)、苯胺共聚物、导电高分子寡聚物(例如噻吩寡聚物、聚噻吩等)、有机硅烷、碳薄膜、其衍生物等。一些优选的主体材料包括咔唑衍生物、吲哚衍生物、咪唑衍生物、芳香族叔胺化合物和噻吩衍生物。
电子输送主体材料的特定实例包括(但不限于)吡啶、嘧啶、三嗪、咪唑、吡唑、三唑、噁唑、噁二唑、芴酮、蒽醌二甲烷、蒽酮、二苯醌、噻喃二氧化物、碳化二亚胺、亚芴基甲烷、二苯乙烯吡嗪、氟取代的芳香族化合物、萘、苝等的芳香环四羧酸酐、酞菁、其衍生物,包括多种由8-喹啉醇衍生物的金属络合物、金属酞菁和具有苯并噁唑或苯并噻唑作为配位体的金属络合物表示的金属络合物。
优选的电子输送主体是金属络合物、唑衍生物(苯并咪唑衍生物、咪唑并吡啶衍生物等)和吖嗪衍生物(吡啶衍生物、嘧啶衍生物、三嗪衍生物等)。
C.薄膜厚度
在一些实施例中,发光层的薄膜厚度优选是约10nm到约500nm。在一些实施例中,取决于例如所要亮度均匀性、驱动电压和亮度,发光层的薄膜厚度优选是约20nm到约100nm。在一些实施例中,发光层被配置成具有最佳化电荷从发光层通过到相邻层而不降低发光效率的厚度。在一些实施例中,发光层被配置成具有维持最小驱动电压最大发光效率的厚度。
D.层配置
发光层可以由单一层或两个或两个以上层组成,并且相应层可以产生不同发光色彩的发光。此外,在发光层具有层合结构的情况下,尽管配置层合结构的每个层的薄膜厚度不受特别限制,但优选每个发光层的总薄膜厚度属于前述范围内。在一些实施例中,可以在层内使用渐变层或渐变界面。
E.空穴注入层和空穴输送层
空穴注入层和空穴输送层是作用以接收来自阳极或阳极侧的空穴和将空穴输送到发射层的层。待引入空穴注入层或空穴输送层中的材料不受特别限制,但可以使用低分子化合物或高分子化合物中的任一者。
空穴注入层和空穴输送层中所含有的材料的特定实例包括(但不限于)吡咯衍生物、咔唑衍生物、氮杂咔唑衍生物、吲哚衍生物、氮杂吲哚衍生物、咪唑衍生物、聚芳基烷衍生物、吡唑啉衍生物、吡唑啉酮衍生物、苯二胺衍生物、芳胺衍生物、氨基取代的查耳酮衍生物、苯乙烯基蒽衍生物、芴酮衍生物、腙衍生物、芪衍生物、硅氮烷衍生物、芳香族叔胺化合物、苯乙烯胺化合物、芳香族二亚甲基化合物、酞菁化合物、卟啉化合物、有机硅烷衍生物、碳等。
受电子掺杂剂可以引入本发明的有机EL元件中的空穴注入层或空穴输送层中。作为待引入空穴注入层或空穴输送层中的受电子掺杂剂,可以使用无机化合物或有机化合物中的任一者,只要化合物具有受电子性质和氧化有机化合物的功能。
具体来说,无机化合物包括金属卤化物,例如氯化铁、氯化铝、氯化镓、氯化铟、五氯化锑等,和金属氧化物,例如五氧化二钒、三氧化钼等。
在采用有机化合物的情况下,优选可以应用具有取代基的化合物,所述取代基例如硝基、卤素、氰基、三氟甲基等;醌化合物;酸酐化合物;富勒烯;等。
空穴注入和空穴输送材料的特定实例包括例如以下的专利文件中描述的化合物:JP-A第6-212153号、第11-111463号、第11-251067号、第2000-196140号、第2000-286054号、第2000-315580号、第2001-102175号、第2001-160493号、第2002-252085号、第2002-56985号、第2003-157981号、第2003-217862号、第2003-229278号、第2004-342614号、第2005-72012号、第2005-166637号、第2005-209643号等。
空穴注入和空穴输送材料的特定实例包括有机化合物:六氰基丁二烯、六氰基苯、四氰基乙烯、四氰基对醌二甲烷、四氟四氰基对醌二甲烷、对四氟醌、对四氯醌、对四溴醌、对苯醌、2,6-二氯苯醌、2,5-二氯苯醌、1,2,4,5-四氰基苯、1,4-二氰基四氟苯、2,3-二氯-5,6-二氰基苯醌、对二硝基苯、间二硝基苯、邻二硝基苯、1,4-萘醌、2,3-二氯萘醌、1,3-二硝基萘、1,5-二硝基萘、9,10-蒽醌、1,3,6,8-四硝基咔唑、2,4,7-三硝基-9-芴酮、2,3,5,6-四氰基吡啶和富勒烯C60。其中,六氰基丁二烯、六氰基苯、四氰基乙烯、四氰基对醌二甲烷、四氟四氰基对醌二甲烷、对四氟醌、对四氯醌、对四溴醌、2,6-二氯苯醌、2,5-二氯苯醌、2,3-二氯萘醌、1,2,4,5-四氰基苯、2,3-二氯-5,6-二氰基苯醌和2,3,5,6-四氰基吡啶是更优选的,并且四氟四氰基对醌二甲烷。
由于一或多种受电子掺杂剂可以引入本发明的有机EL元件中的空穴注入层或空穴输送层中,所以这些受电子掺杂剂可以单独或以两者或两者以上的组合使用。尽管所用的这些受电子掺杂剂的确切量将取决于材料的类型,但空穴输送层或空穴注入层的总重量的约0.01重量%到约50重量%是优选的。在一些实施例中,这些受电子掺杂剂的量在空穴输送层或空穴注入层的总重量的约0.05重量%到约20重量%范围内。在一些实施例中,这些受电子掺杂剂的量在空穴输送层或空穴注入层的总重量的约0.1重量%到约10重量%范围内。
在一些实施例中,鉴于降低驱动电压或针对光学出耦最佳化,空穴注入层的厚度和空穴输送层的厚度各自优选是约500nm或500nm以下。在一些实施例中,空穴输送层的厚度优选是约1nm到约500nm。在一些实施例中,空穴输送层的厚度优选是约5nm到约50nm。在一些实施例中,空穴输送层的厚度优选是约10nm到约40nm。在一些实施例中,空穴注入层的厚度优选是约0.1nm到约500nm。在一些实施例中,空穴注入层的厚度优选是约0.5nm到约300nm。在一些实施例中,空穴注入层的厚度优选是约1nm到约200nm。
空穴注入层和空穴输送层可以由包含一或两种或两种以上上文所提及的材料的单层结构;或由多个具有均匀组成或非均匀组成的层组成的多层结构组成。
F.电子注入层和电子输送层
电子注入层和电子输送层是具有从阴极或阴极侧接收电子和将电子输送到发光层的功能的层。用于这些层的电子注入材料或电子输送材料可以是低分子化合物或高分子化合物。适用于电子注入和电子输送层的材料的特定实例包括(但不限于)吡啶衍生物、喹啉衍生物、嘧啶衍生物、吡嗪衍生物、酞嗪衍生物、菲咯啉衍生物、三嗪衍生物、三唑衍生物、噁唑衍生物、噁二唑衍生物、咪唑衍生物、芴酮衍生物、蒽醌二甲烷衍生物、蒽酮衍生物、二苯醌衍生物、噻喃二氧化物衍生物、碳化二亚胺衍生物、亚芴基甲烷衍生物、二苯乙烯吡嗪衍生物、苝、萘等的芳香环四羧酸酐、酞菁衍生物、由8-喹啉醇衍生物的金属络合物、金属酞菁和含有苯并噁唑或苯并噻唑作为配位体的金属络合物表示的金属络合物、由硅咯(Silole)例示的有机硅烷衍生物等。
电子注入层或电子输送层可以含有供电子掺杂剂。适用于电子注入层或电子输送层的供电子掺杂剂包括可以使用的任何适合材料,只要其具有供电子性质以及还原有机化合物的性质。供电子掺杂剂的特定实例包括碱金属,例如Li;碱土金属,例如Mg;过渡金属,包括稀土金属;和还原性有机化合物。供金属掺杂剂的其它实例包括功函数为4.2V或4.2V以下的金属,例如Li、Na、K、Be、Mg、Ca、Sr、Ba、Y、Cs、La、Sm、Gd、Yb等。还原性有机化合物的特定实例包括含氮化合物、含硫化合物、含磷化合物等。
供电子掺杂剂可以单独或以两者或两者以上的组合使用。在一些实施例中,供电子掺杂剂以范围介于电子输送层材料或电子注入层材料的总重量的约0.1重量%到约99重量%的量含于电子注入层或电子输送层中。在一些实施例中,供电子掺杂剂以范围介于电子输送层材料或电子注入层材料的总重量的约1.0重量%到约80重量%的量含于电子注入层或电子输送层中。在一些实施例中,供电子掺杂剂以范围介于电子输送层材料或电子注入层材料的总重量的约2.0重量%到约70重量%的量含于电子注入层或电子输送层中。
鉴于驱动电压的降低,电子注入层的厚度和电子输送层的厚度各自优选是500nm或500nm以下。电子输送层的厚度优选是1nm到500nm,更优选是5nm到200nm,并且甚至更优选是10nm到100nm。电子注入层的厚度优选是0.1nm到200nm,更优选是0.2nm到100nm,并且甚至更优选是0.5nm到50nm。
电子注入层和电子输送层可以由包含一或两种或两种以上上文所提及的材料的单层结构;或由多个具有均匀组成或非均匀组成的层组成的多层结构组成。
与其它材料的组合
本文描述为可用于有机发光装置中的具体层的材料可以与存在于所述装置中的多种其它材料组合使用。举例来说,本文所公开的发射掺杂剂可以与多种主体、输送层、阻挡层、注入层、电极和其它可能存在的层结合使用。下文描述或提及的材料是可以与本文所公开的化合物组合使用的材料的非限制性实例,并且本领域技术人员可以容易地查阅文献以鉴别可以组合使用的其它材料。
HIL/HTL:
本发明中所用的空穴注入/输送材料不受特别限制,并且可以使用任何化合物,只要化合物典型地用作空穴注入/输送材料即可。所述材料的实例包括(但不限于):酞菁或卟啉衍生物;芳香族胺衍生物;吲哚并咔唑衍生物;含有氟烃的聚合物;具有导电性掺杂剂的聚合物;导电聚合物,例如PEDOT/PSS;衍生自例如膦酸和硅烷衍生物的化合物的自组装单体;金属氧化物衍生物,例如MoOx;p型半导体有机化合物,例如1,4,5,8,9,12-六氮杂三亚苯六甲腈;金属络合物,和可交联化合物。
HIL或HTL中所用的芳香族胺衍生物的实例包括(但不限于)以下通式结构:
Ar1到Ar9中的每一者选自由芳香族烃环化合物组成的群组,所述化合物例如为苯、联苯、联三苯、三亚苯、萘、蒽、萉、菲、芴、芘、苣、苝和薁;由芳香族杂环化合物组成的群组,所述化合物例如为二苯并噻吩、二苯并呋喃、二苯并硒吩、呋喃、噻吩、苯并呋喃、苯并噻吩、苯并硒吩、咔唑、吲哚并咔唑、吡啶基吲哚、吡咯并二吡啶、吡唑、咪唑、三唑、噁唑、噻唑、噁二唑、噁三唑、二噁唑、噻二唑、吡啶、哒嗪、嘧啶、吡嗪、三嗪、噁嗪、噁噻嗪、噁二嗪、吲哚、苯并咪唑、吲唑、吲哚并噁嗪、苯并噁唑、苯并异噁唑、苯并噻唑、喹啉、异喹啉、噌啉、喹唑啉、喹喔啉、萘啶、酞嗪、喋啶、二苯并哌喃、吖啶、吩嗪、吩噻嗪、吩噁嗪、苯并呋喃并吡啶、呋喃并二吡啶、苯并噻吩并吡啶、噻吩并二吡啶、苯并硒吩并吡啶和硒吩并二吡啶;和由2到10个环状结构单元组成的群组,所述结构单元为选自芳香族烃环基和芳香族杂环基的相同类型或不同类型的基团,并且直接或经由氧原子、氮原子、硫原子、硅原子、磷原子、硼原子、链结构单元和脂族环基中的至少一者彼此键结。其中每个Ar进一步被选自由以下组成的群组的取代基取代:氢、氘、卤基、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合。
在一个方面,Ar1到Ar9独立地选自由以下组成的群组:
其中k是1到20的整数;X101到X108是C(包括CH)或N;Z101是NAr1、O或S;Ar1具有以上定义的相同基团。
HIL或HTL中所用的金属络合物的实例包括(但不限于)以下通式:
其中Met是金属,其可以具有大于40的原子量;(Y101-Y102)是双齿配位体,Y101和Y102独立地选自C、N、O、P和S;L101是辅助性配位体;k'是1到可以与金属连接的最大配位体数的整数值;并且k'+k"是可以与金属连接的最大配位体数。
在一个方面,(Y101-Y102)是2-苯基吡啶衍生物。在另一方面,(Y101-Y102)是碳烯配位体。在另一方面,Met选自Ir、Pt、Os和Zn。在另一方面,金属络合物具有小于约0.6V的相对于Fc+/Fc对的溶液态最小氧化电位。
EBL:
电子阻挡层(EBL)可以用以减少离开发射层的电子和/或激子的数目。与缺乏阻挡层的类似装置相比,这种阻挡层在装置中的存在可以产生实质上较高的效率和或较长的寿命。此外,阻挡层可以用以将发射限制于OLED的所要区域。在一些实施例中,与最接近EBL界面的发射体相比,EBL材料具有较高LUMO(较接近真空能级)和/或较高三重态能量。在一些实施例中,与最接近EBL界面的主体中的一或多者相比,EBL材料具有较高LUMO(较接近真空能级)和或较高三重态能量。在一个方面,EBL中所使用的化合物含有与下文描述的主体之一所使用相同的分子或相同的官能团。
主体:
本发明的有机EL装置的发光层优选地至少含有金属络合物作为发光材料,并且可以含有使用金属络合物作为掺杂剂材料的主体材料。在一些实施例中,两种或两种以上主体是优选的。在一些实施例中,所使用的主体可以是在电荷输送中起极小作用的a)双极,b)电子输送,c)空穴输送,或d)宽带隙材料。主体材料的实例不受特别限制,并且可以使用任何金属络合物或有机化合物,只要主体的三重态能量大于掺杂剂的三重态能量即可。虽然下表将优选用于发射各种颜色的装置的主体材料加以分类,但可以与任何掺杂剂一起使用任何主体材料,只要三重态准则满足即可。
用作主体的金属络合物的实例优选具有以下通式:
其中Met是金属;(Y103-Y104)是双齿配位体,Y103和Y104独立地选自C、N、O、P和S;L101是另一配位体;k'是1到可以与金属连接的最大配位体数的整数值;并且k'+k"是可以与金属连接的最大配位体数。
在一个方面,金属络合物是:
其中(O-N)是具有与O和N原子配位的金属的双齿配位体。
在另一方面,Met选自Ir和Pt。在另一方面,(Y103-Y104)是碳烯配位体。
用作主体的有机化合物的实例选自由芳香族烃环化合物组成的群组,所述化合物例如为苯、联苯、联三苯、三亚苯、萘、蒽、萉、菲、芴、芘、苣、苝和薁;由芳香族杂环化合物组成的群组,所述化合物例如为二苯并噻吩、二苯并呋喃、二苯并硒吩、呋喃、噻吩、苯并呋喃、苯并噻吩、苯并硒吩、咔唑、吲哚并咔唑、吡啶基吲哚、吡咯并二吡啶、吡唑、咪唑、三唑、噁唑、噻唑、噁二唑、噁三唑、二噁唑、噻二唑、吡啶、哒嗪、嘧啶、吡嗪、三嗪、噁嗪、噁噻嗪、噁二嗪、吲哚、苯并咪唑、吲唑、吲哚并噁嗪、苯并噁唑、苯并异噁唑、苯并噻唑、喹啉、异喹啉、噌啉、喹唑啉、喹喔啉、萘啶、酞嗪、喋啶、二苯并哌喃、吖啶、吩嗪、吩噻嗪、吩噁嗪、苯并呋喃并吡啶、呋喃并二吡啶、苯并噻吩并吡啶、噻吩并二吡啶、苯并硒吩并吡啶和硒吩并二吡啶;和由2到10个环状结构单元组成的群组,所述结构单元为选自芳香族烃环基和芳香族杂环基的相同类型或不同类型的基团,并且直接或经由氧原子、氮原子、硫原子、硅原子、磷原子、硼原子、链结构单元和脂族环基中的至少一者彼此键结。其中每个基团进一步被选自由以下组成的群组的取代基取代:氢、氘、卤基、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合。
在一个方面,主体化合物在分子中含有以下基团中的至少一者:
其中R101到R107独立地选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合,当其是芳基或杂芳基时,其具有与上述Ar类似的定义。k是0到20或1到20的整数;k'"是0到20的整数。X101到X108选自C(包括CH)或N。
Z101和Z102选自NR101、O或S。
HBL:
空穴阻挡层(HBL)可以用以减少离开发射层的空穴和/或激子的数目。与缺乏阻挡层的类似装置相比,这种阻挡层在装置中的存在可以产生实质上较高的效率和/或较长的寿命。此外,阻挡层可以用以将发射限于OLED的所要区域。在一些实施例中,与最接近HBL界面的发射体相比,HBL材料具有较低HOMO和或较高三重态能量。
在一个方面,HBL中所用的化合物含有用作上述主体的相同分子或相同官能团。
在另一方面,HBL中所用的化合物在分子中含有以下基团中的至少一者:
其中k是1到20的整数;L101是另一配位体,k'是1到3的整数。
ETL:
电子输送层(ETL)可以包括能够输送电子的材料。电子输送层可以是本质的(未掺杂)或经掺杂的。掺杂可以用以增强导电性。ETL材料的实例不受特别限制,并且可以使用任何金属络合物或有机化合物,只要其典型地用以输送电子即可。
在一个方面,ETL中所用的化合物在分子中含有以下基团中的至少一者:
其中R101选自由以下组成的群组:氢、氘、卤基、烷基、环烷基、杂烷基、芳烷基、烷氧基、芳氧基、氨基、硅烷基、烯基、环烯基、杂烯基、炔基、芳基、杂芳基、酰基、羰基、羧酸基、酯基、腈基、异腈基、硫基、亚磺酰基、磺酰基、膦基和其组合,当其是芳基或杂芳基时,其具有与上述Ar类似的定义。Ar1到Ar3具有与上述Ar类似的定义。k是1到20的整数。X101到X108选自C(包括CH)或N。
在另一方面,ETL中所用的金属络合物包括(但不限于)以下通式:
其中(O-N)或(N-N)是具有与原子O、N或N、N配位的金属的双齿配位体;L101是另一配位体;k'是1到可以与金属连接的最大配位体数的整数值。
电荷产生层(CGL):
在串联或堆叠OLED中,CGL对性能起基本作用,其由分别用于注入电子和空穴的经n掺杂的层和经p掺杂的层组成。电子和空穴由CGL和电极供应。CGL中消耗的电子和空穴由分别从阴极和阳极注入的电子和空穴再填充;随后,双极电流逐渐达到稳定状态。典型CGL材料包括输送层中使用的n和p导电性掺杂剂。
在OLED装置的每个层中所用的任何上述化合物中,氢原子可以部分或完全氘化。因此,任何具体列出的取代基(例如(但不限于)甲基、苯基、吡啶基等)涵盖其非氘化、部分氘化和完全氘化形式。类似地,取代基类别(例如(但不限于)烷基、芳基、环烷基、杂芳基等)也涵盖其非氘化、部分氘化和完全氘化形式。
除本文所公开的材料外和/或与本文所公开的材料组合,OLED中还可以使用许多空穴注入材料、空穴输送材料、主体材料、掺杂剂材料、激子/空穴阻挡层材料、电子输送材料和电子注入材料。可以与本文所公开的材料组合用于OLED中的材料的非限制性实例在下表A中列出。表A列出材料的非限制性类别、每种类别的化合物的非限制性实例和公开所述材料的参考文献。
表A
本发明通过以下实例更详细地阐述,但不希望因此限制其。本领域的技术人员将能够基于描述在无发明性步骤的情况下制备其它电子装置,并且将因此能够在整个所要求的范围中实施本发明。
实例
除非另外指示,否则以下合成在干燥溶剂中在保护性气体大气下进行。金属络合物另外在排除光的情况下进行处置。溶剂和试剂可以购自例如西格玛-阿尔德里奇(Sigma-ALDRICH)或ABCR。
实例1:根据流程1制备3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的合成。
流程1
A.合成4-氯丁醛:
将草酰氯(22.54ml,263mmol)于DCM(400ml)中的溶液在iPrOH/CO2浴中冷却。借助注射器缓慢添加DMSO(37.3ml,525mmol),并且冷搅拌1小时。逐滴添加4-氯丁-1-醇(19g,175mmol)于50mL DCM中的溶液。将冷混合物搅拌一小时,然后缓慢添加三乙胺(110ml,788mmol)。将悬浮液冷搅拌30分钟,然后使其升温到室温。将反应物用水淬灭,酸化,并且分离有机物。去除溶剂接着蒸馏得到呈无色油状的产物,8g。
B.合成2-溴-4-氯丁醛:
将4-氯丁醛(7.939g,74.5mmol)溶解于DCM(300ml)中,并且在冰浴中冷却。经约1小时添加二溴(4.00ml,78mmol)于DCM(50ml)中的溶液。在添加之后,将红色溶液冷搅拌30分钟,然后缓慢升温到室温,并且再搅拌一小时。添加水,分离有机物,并且干燥和去除溶剂得到呈浅黄色油状的粗产物,1.57g(80%)。
C.合成4-溴菲啶-6-胺:
2,6-二溴苯胺(15.33g,61.1mmol)、2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲腈(7.0g,30.6mmol)和单水合磷酸钾(21.11g,92mmol)组合于二噁烷(120ml)和水(7.49ml)中。将混合物脱气,然后添加(dppf)PdCl2络合物,添加DCM(0.749g,0.917mmol),并且使混合物回流4小时。将黑色混合物分配在EtOAc与水/盐水之间。将有机层用盐水洗涤,干燥,并且去除溶剂。溶解于500mL EtOAc中,接着通过硅胶塞使用EtOAc洗脱和去除溶剂得到橙色残余物,通过柱色谱法纯化所述残余物,得到呈黄/橙色固体状的产物,5.86g,70%。
D.合成5-溴-3-(2-氯乙基)咪唑并[1,2-f]菲啶:
将4-溴菲啶-6-胺(5.86g,21.46mmol)、2-溴-4-氯丁醛(5.36g,28.9mmol)和碳酸氢钠(3.60g,42.9mmol)组合于2-丙醇(102ml)和水(5.11ml)中。将悬浮液在室温下搅拌4小时,然后在回流下搅拌16小时。在真空下去除溶剂,并且将残余物涂布于硅藻土上。柱色谱法得到产物和起始脒的混合物,将所述混合物用于DCM中的过量乙酰氯和三乙胺处理。在处理之后,通过重复萃取到庚烷中从乙酰胺萃取所要产物,得到3.93g黄色粘性残余物(51%)。
E.合成3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将5-溴-3-((2-氯乙基)咪唑并[1,2-f]菲啶(3.93g,10.93mmol)溶解于THF(200ml)中,在冰浴中冷却,并且缓慢添加异丙基氯化镁于THF中的溶液(2.0M,6.01ml,12.02mmol)。将溶液冷搅拌30分钟,然后升温到室温,并且再搅拌2小时。将反应物淬灭,萃取到DCM中,并且使用柱色谱法纯化反应产物,得到1.90g浅米色结晶固体(71%)。
3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的X射线结构展示于图5中。3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的晶体结构可以由下表中列出的一或多种特征界定。
aR1=Σ||Fo|-|Fc||/Σ|Fo|;wR2=[Σ[w(Fo2-Fc2)2]/Σ[w(Fo2)2]]1/2;GOF=[Σw(|Fo|-|Fc|)2/(n-m)]1/2
实例2:合成4,4-二甲基-3,4-二氢-1,2a1-二氮杂-4-硅苯并[fg]乙烯合蒽和3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽:
根据以下流程2制备以上配位体。
流程2
A.合成5-溴咪唑并[1,2-f]菲啶:
将4-溴菲啶-6-胺(4.0g,14.7mmol)溶解于100mL iPrOH中。添加氯乙醛(50%水溶液,3.6g,22mmol,1.5当量),接着添加NaHCO3(2.5g,2当量),并且使混合物回流2小时,然后在冰浴中冷却。将茶色固体过滤出,用MeOH洗涤。改变接收烧瓶,并且将固体用水洗涤,得到清洁灰白色产物,3.2g。将水性洗液用EtOAc萃取,并且将这些萃取物与来自初始过滤的醇洗液合并。去除溶剂,得到1.3g橙色固体,使所述固体由EtOAc再结晶,得到呈茶色针状的更清洁产物,0.46g。总产率:3.5g(80%)。
B.合成3,5-二溴咪唑并[1,2-f]菲啶:
将5-溴咪唑并[1,2-f]菲啶(2.0g,6.73mmol)溶解于DMF(125ml)中,然后在氮气下缓慢添加NBS(1.318g,7.40mmol)于10mL DMF中的溶液。在室温下搅拌3小时、然后平缓加热16小时之后,将反应混合物分配于300mL水与EtOAc之间。进一步用EtOAc萃取水层,用水洗涤有机物,并且通过柱色谱法分离呈浅黄色固体状的产物,1.99g(79%)。
C.合成5-溴-3-((氯甲基)二甲基硅烷基)咪唑并[1,2-f]菲啶:
将3,5-二溴咪唑并[1,2-f]菲啶(0.48g,1.28mmol)和氯(氯甲基)二甲基硅烷(0.17ml,1.28mmol)溶解于THF(25ml)中,并且在iPrOH/CO2浴中冷却。缓慢添加丁基锂于己烷中的溶液(2.5M,0.51ml,1.28mmol),将混合物冷搅拌30分钟,然后使其升温到室温。添加盐水以淬灭反应物,将有机物萃取到EtOAc中并且通过柱色谱法纯化,得到呈无色粘性残余物形式的产物,0.16g(31%)。
D.合成4,4-二甲基-3,4-二氢-1,2a1-二氮杂-4-硅苯并[fg]乙烯合蒽和3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽:
将5-溴-3-((氯甲基)二甲基硅烷基)咪唑并[1,2-f]菲啶(0.13g,0.322mmol)溶解于THF(25ml)中,并且在冰浴中冷却。缓慢添加异丙基氯化镁于THF(2.0M,0.18ml,0.36mmol)中的溶液,然后升温到室温。用盐水淬灭反应物,用DCM萃取有机物,并且色谱分离混合物,得到16mg呈粘性残余物形式的4,4-二甲基-3,4-二氢-1,2a1-二氮杂-4-硅苯并[fg]乙烯合蒽(17%)和33mg呈结晶固体状的3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽(36%)。
此外,通过高效液相色谱法(Tosoh TSKgel ODS-100Z)升华纯化并且分析此实例中所用的所有有机材料,并且使用在254nm下具有99.9%或99.9%以上的吸收强度面积比的材料。
3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽的X射线结构展示于图6中。3,3-二甲基-3,4-二氢-1,2a1-二氮杂-3-硅苯并[fg]乙烯合蒽的晶体结构可以由下表中列出的一或多种特征界定。
aR1=Σ||Fo|-|Fc||/Σ|Fo|;wR2=[Σ[w(Fo2-Fc2)2]/Σ[w(Fo2)2]]1/2;GOF=[Σw(|Fo|-|Fc|)2/(n-m)]1/2
实例3:合成6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的铂(II)络合物:
流程3
A.合成2-溴-5-甲氧基苯甲腈:
使2-溴-5-甲氧基苯甲醛(100g,0.47mol,1当量)、羟胺盐酸盐(64.8g,0.93mol,2当量)、乙酸钠(76.42g,0.93mol,2当量)和冰乙酸(500mL)的混合物回流16小时。在减压下去除乙酸,并且将残余物用二氯甲烷(约400mL)萃取。将有机层用饱和盐水(3×200mL)洗涤,经硫酸钠干燥,并且在减压下浓缩。将所得残余物用庚烷(50mL)湿磨,并且将固体用额外庚烷(2×50mL)洗涤,得到呈白色粉末状的所要产物(82.6g,86%产率)。
B.合成5-甲氧基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲腈:
向2-溴-5-甲氧基苯甲腈(82.6g,0.39mol,1当量)、双(频哪醇根基)二硼(109.1g,0.43mol,1.1当量)和乙酸钾(115.3g,1.17mol,3当量)于1,4-二噁烷(400mL)和DMSO(40mL)的混合物中的混合物充氮气1小时。添加Pd(dppf)Cl2(7.13g,5mol%),并且将反应混合物在60℃下温和地加热2小时,然后回流16小时。通过硅藻土过滤混合物,并且用异丙醇和庚烷洗涤从滤液分离的固体,得到呈灰白色固体状的所要产物(57.41g,57%产率)。从滤液分离额外产物(约10g)。
C.合成4-溴-2-异丙基-8-甲氧基菲啶-6-胺:
向5-甲氧基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲腈(57.41g,0.22mol,1当量)、2,6-二溴-4-异丙基苯胺(64.92g,0.22mol,1当量)和磷酸钾(153.1g,0.66mol,3当量)于甲苯与水的4:1混合物(1250mL)中的混合物充氮气1小时。添加反式Pd(PPh3)2Cl2(7.8g,11mmol,0.05当量),并且使反应混合物回流20小时。添加额外磷酸钾(77g,0.33mol,1.5当量)和反式Pd(PPh3)2Cl2(1g,1.43mmol,0.0065当量),并且使反应混合物再回流3小时。分离各层,并且用热水(2×400mL)洗涤有机层。将有机层经硫酸钠干燥,并且在减压下浓缩。将所得固体依序用二氯甲烷和庚烷湿磨。柱色谱法得到所要产物(30g)。
D.合成6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4二氢-二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将4-溴-2-异丙基-8-甲氧基菲啶-6-胺(8.9g,25.8mmol,1当量)、单水合对甲苯磺酸(348mg)、新鲜制备之2-溴-4-氯丁醛(24g,129mmol,5当量)和异丙醇(500mL)的悬浮液在室温下搅拌2.5小时。添加碳酸钠(6.5g,77.4mmol,3当量)和去离子水(32ml),并且使反应混合物回流16小时。在冷却到室温之后,将反应混合物体积在减压下减少到约100mL。将混合物用乙酸乙酯(350mL)稀释,并且用饱和盐水(200mL)洗涤。将有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到8.44g产物(76%产率)。
E.合成6-异丙基-10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
向6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4二氢-二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(8.44g,19.6mmol,1.0当量)于无水THF(250mL)中的溶液充氮气30分钟。在冷却到0℃之后,逐滴添加于THF中的2M异丙基氯化镁(14.7mL,29.4mmol,1.5当量)。使反应混合物升温达到室温,并且搅拌16小时。用水(10mL)淬灭反应物,并且在减压下去除THF。将残余物用乙酸乙酯(400mL)稀释,并且用饱和盐水(2×200mL)洗涤。经硫酸钠干燥有机层,并且通过柱色谱法纯化残余物,得到3.6g产物(58%产率)。
F.合成6-异丙基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-10-醇:
在-78℃下逐滴添加三溴化硼(5.4mL,56.78mmol,5当量)到6-异丙基-10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(3.6g,11.36mmol,1当量)于二氯甲烷(200mL)中的溶液中。使反应物升温到室温,并且搅拌16小时。将反应混合物小心地倾入300ml冰水中,并且将所得固体过滤并且依序用水(70mL)、乙酸乙酯(40mL)和庚烷(40mL)洗涤,得到3.6g产物(定量产率)。
G.合成4'-溴-2-硝基-1,1'-联苯:
添加碳酸钾(84g,608mmol,3.0当量)于水(450mL)中的溶液到2-碘-硝基苯(50g,200mmol,1.0当量)和4-溴苯硼酸(40.7g,202mol,1.0当量)于1,2-二甲氧基乙烷(660mL)中的混合物中。向反应物充氮气5.0分钟。添加四(三苯基膦)钯(0)(2.32g,2mmol,1mol%),并且再向混合物充氮气10分钟。在回流16小时之后,将反应物冷却到室温,并且分离各层。用乙酸乙酯(500mL)萃取水层。将经合并的有机萃取物用饱和盐水(500mL)洗涤,经硫酸钠干燥,过滤,并且在减压下浓缩。将残余物溶解于含25%乙酸乙酯的庚烷(300mL)中,并且通过硅胶垫真空过滤(135g)。用含25%乙酸乙酯的庚烷(3×350mL)冲洗垫。在减压下浓缩经合并的滤液,得到橙色固体。将此残余物悬浮于庚烷(150mL)中,并且加热到40℃后持续20分钟。使悬浮液冷却到室温持续1.0小时。将固体通过真空过滤收集,用庚烷(50mL)洗涤,并且干燥,得到呈黄色固体状的4'-溴-2-硝基-1,1'-联苯(49.16g,88.4%产率)。
H.合成2-溴-9H-咔唑:
经5分钟添加三苯基膦(156.3g,596mmol,2.5当量)到4'-溴-2-硝基-1,1'-联苯(66.25g,238mmol,1.0当量)于1,2-二氯苯(460mL)中的溶液中。向反应物充氮气5分钟,然后使其回流16小时。将反应物冷却到室温,并且真空蒸馏以去除大部分1,2-二氯苯(450mL)。将此暗色残余物溶解于乙酸乙酯(1.5L)中,并且在50℃下用脱色碳(50g)处理30分钟。在冷却之后,将混合物通过硅藻土(200g)过滤,然后用乙酸乙酯洗液(2×650mL)洗涤。将经合并的滤液在减压下浓缩到约500mL体积。将溶液冷却到室温,并且在1.5小时之后,将所得浅茶色固体(三苯基氧化膦)通过过滤移出并且舍弃。在减压下浓缩滤液。将残余物溶解于甲醇(600mL)中,并且在室温下储存16小时。将所得茶色固体过滤,用甲醇(2×100mL)洗涤,并且在真空下在40℃下干燥,得到呈浅茶色固体状的2-溴-9H-咔唑(33.5g,57.2%产率)。
I.合成2-溴-9-(4-异丙基吡啶-2-基)-9H-咔唑:
向2-溴-9H-咔唑(13.9g,56.5mmol,1当量)、4-异丙基-2-氯吡啶(15.86g,101.7mmol,1.8当量)、L-脯氨酸(1.3g,11.3mmol,0.2当量)、碘化铜(1)(0.95g,5.65mmol,0.1当量)、碳酸钾(19.48g,141.25mmol,2.5当量)和DMSO(80mL)的悬浮液充氮气5分钟。将混合物在95℃下加热16小时。添加额外4-异丙基-2-氯吡啶(1.58g,10.12mmol,0.18当量),将反应混合物在155℃下再加热24小时。将反应混合物冷却到室温,用乙酸乙酯(750mL)稀释,并且通过硅藻土(70g)真空过滤。用乙酸乙酯洗液(2×100mL)洗涤硅藻土垫。将经合并的滤液用饱和盐水(3×500mL)洗涤,经硫酸钠干燥,过滤,并且在减压下浓缩。通过柱色谱法纯化此残余物,得到1.8g呈褐色油状的产物(8.6%产率)。
J.合成6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将6-异丙基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-10-醇(1.5g,4.93mmol,1当量)、2-溴-9-(4-异丙基吡啶-2-基)-9H-咔唑(1.8g,4.93mmol,1当量)、磷酸钾(5.68g,24.65mmol,5当量)、碘化铜(I)(0.47g,2.47mmol,0.5当量)、吡啶甲酸(1.52g,12.33mmol,2.5当量)和DMSO(150mL)的混合物在150℃下加热4.5小时。在冷却到室温之后,将反应混合物倾入水(700mL)中,并且用乙酸乙酯(4×150mL)萃取。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到呈茶色固体状的产物,1.25g(43%产率)。
K.合成6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪的铂(II)络合物:
将6-异丙基-10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(400mg,0.68mmol,1当量)溶解于60ml冰乙酸中,并且充氮气30分钟。然后添加K2PtCl4(283mg,0.68mmol,1当量),并且使反应混合物回流40小时。在冷却到室温之后,将橙色沉淀物过滤,并且依序用水(3×15mL)和庚烷(10ml×2次)洗涤。将粗产物(340mg)溶解于10ml二氯甲烷中,并且通过硅胶塞过滤以去除残余K2PtCl4,用额外二氯甲烷(10mL)洗脱。将滤液减少到其一半体积并且用庚烷(10mL)稀释。将产物过滤,并且用10%二氯甲烷于庚烷中的溶液(10mL)湿磨,得到呈淡黄色固体状的产物(140mg,26%产率)。从乙酸和二氯甲烷/庚烷滤液分离额外产物。
实例4:合成(3-苯基-1H-吡唑)2Ir(MeOH)2(OTf)
流程4
A.合成(3-苯基-1H-吡唑)2IrCl2二聚体
将水合氯化铱(6.00g,17.02mmol)和1-苯基-1H-吡唑(5.89g,40.9mmol)组合于2-乙氧基乙醇(120ml)和水(40ml)中。在氮气下将反应混合物加热到回流后持续16小时。将所得固体过滤出,并且用甲醇洗涤,并且干燥,得到8.3g铱二聚体。
将实例4A的铱二聚体(8.3g,8.07mmol)溶解于100mL DCM中,并且添加三氟甲磺酸银(4.36g,16.96mmol)于20mL甲醇中的溶液。将反应混合物在室温下在氮气下搅拌1小时。通过硅藻土过滤混合物,并且用DCM洗涤滤饼。蒸发滤液,得到10.85g(3-苯基-1H-吡唑)2Ir(MeOH)2(OTf)(97%)。
实例5:根据流程5制备例示性化合物35。
流程5
A.合成咪唑并[1,2-f]菲啶
向2-苯基-1H-咪唑(10.0g,69.3mmol,1当量)、1,2-二溴苯(19.63g,83.2mmol,1.2当量)、碳酸铯(67.79g,208.0mmol,3当量)、Xantphos(4.01g,6.9mmol,0.1当量)和四(三苯基膦)钯(8.01g,6.9mmol,0.1当量)于DMF(550mL)中的混合物充氮气流15分钟。将混合物在140℃下加热24小时,然后在减压下浓缩。将残余物通过柱色谱法纯化为呈浅黄色固体状的咪唑并[1,2-f]菲啶(10g,67%产率)。
B.合成3-溴咪唑并[1,2-f]菲啶
在0℃下添加N-溴琥珀酰亚胺(1.62g,9.1mmol,1当量)到15(1.99g,9.1mmol,1当量)于DMF(32mL)中的溶液中。在室温下搅拌18小时之后,将反应物用水(300mL)稀释,并且依序用含10%二氯甲烷的甲基叔丁基醚(3×500mL)、乙酸乙酯(2×300mL)和二氯甲烷(400mL)萃取。将经合并的有机层经硫酸钠干燥,过滤,并且在减压下浓缩。通过柱色谱法纯化残余物,得到呈灰白色固体状的3-溴咪唑并[1,2-f]菲啶(1.66g,65%产率)。
C.合成2-(咪唑并[1,2-f]菲啶-3-基)乙酸叔丁酯
添加二-μ-溴双(三-叔丁基膦)二钯(I)(2.01g,2.5mmol,0.05当量)到16(15.4g,51.8mmol,1当量)于无水四氢呋喃(220mL)中的溶液中,并且向溶液充氮气流15分钟。在氮气下添加含0.5M于乙醚中的2-叔丁氧基-2-氧代乙基溴化锌(155mL,77.7mmol,1.5当量)。将反应物在60℃下搅拌16小时。添加额外0.5M 2-叔丁氧基-2-氧代乙基氯化锌溶液(155mL,77.7mmol,1.5当量)和二-μ-溴双(三-叔丁基膦)-二钯(I)(2.01g,2.5mmol,0.05当量),并且将反应物在60℃下搅拌,直到LC/MS分析指示其完成。在减压下浓缩反应混合物。将残余物溶解于二氯甲烷(1L)中,并且通过硅藻土垫过滤。在减压下浓缩滤液。通过柱色谱法纯化残余物,得到呈橙色固体状的2-(咪唑并[1,2-f]菲啶-3-基)乙酸叔丁酯(5g,30%产率)。
D.合成2-(咪唑并[1,2-f]菲啶-3-基)乙酸甲酯盐酸盐
将17(2.8g,8.4mmol,1当量)于1.25M HCl(55mL,68.7mmol,6.5当量)中于甲醇中的溶液在60℃下搅拌16小时。在减压下浓缩反应混合物。将残余物用乙醚洗涤,并且在40℃下在真空下干燥16小时,得到呈灰白色固体状的2-(咪唑并[1,2-f]菲啶-3-基)乙酸甲酯盐酸盐(2.5g,100%产率)。
E.合成2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸甲酯
在5℃下依序添加氢化钠于矿物油中的60%分散液(2.45g,61.2mmol,5当量)和碘甲烷(2mL,32.1mmol,2.6当量)到2-(咪唑并[1,2-f]菲啶-3-基)乙酸甲酯盐酸盐(4.0g,12.24mmol,1当量)于无水DMF(45mL)中的溶液中。将混合物在冷却浴中搅拌30分钟,升温到室温,并且搅拌6小时。添加额外碘甲烷(1.2mL,19.2mmol,1.6当量)。将反应物在室温下搅拌过周末,用甲醇(32mL)淬灭,并且在减压下浓缩。将残余油用二氯甲烷(350mL)稀释,并且用水(100mL)洗涤。用二氯甲烷(2×100mL)萃取水层。将经合并的有机层用饱和氯化铵(100mL)洗涤,经硫酸钠干燥,过滤,并且在减压下浓缩。通过柱色谱法纯化残余物,得到呈灰白色固体状的2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸甲酯(1.6g,41%产率)。
F.合成2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸
将2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸甲酯(1.6g,5.0mmol,1当量)于甲醇(100mL)中的溶液用1N氢氧化钠水溶液(30mL,30mmol,6当量)处理,并且进一步用水(100mL)稀释。在回流5天之后,将反应物在减压下浓缩。将残余物溶解于水(100mL)中,并且用浓HCl酸化到pH 5-6。将所得白色悬浮液用异丙醇与二氯甲烷的1比2混合物(4×200mL)萃取。将经合并的有机层经硫酸钠干燥,过滤,在减压下浓缩。将残余物在高真空下在40℃下干燥16小时,得到呈白色固体状的2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸(1.3g,82%产率)。
G.合成2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酰氯
添加亚硫酰氯(1mL,13.7mmol,2当量)和无水DMF(0.05mL,0.6mmol,0.11当量)到2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酸(1.3g,4.2mmol,1当量)于无水二氯甲烷(100mL)中的悬浮液中。在室温下搅拌16小时之后,将混合物在减压下浓缩,得到呈灰白色固体状的2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酰氯(1.37g,100%产率)。
H.合成3,3-二甲基二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4(3H)-酮
将2-(咪唑并[1,2-f]菲啶-3-基)-2-甲基丙酰氯(1.37g,4.2mmol,1当量)和无水氯化铝(6.0g,44.9mmol,10当量)于无水二氯甲烷(60mL)中的混合物在室温下搅拌6小时。将反应物用冰水浴冷却,用冰淬灭,用饱和碳酸氢钠(300mL)稀释,并且用二氯甲烷(4×400mL)萃取。将经合并的有机层经硫酸钠干燥,过滤,并且在减压下浓缩。使用柱色谱法纯化残余物,得到呈白色固体状的3,3-二甲基二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4(3H)-酮(1g,81%产率)。
I.合成3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-醇
在5℃下以一份方式添加硼氢化钠(0.24g,6.3mmol,2当量)到3,3-二甲基二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4(3H)-酮(0.9g,3.1mmol,1当量)于乙醇(70mL)中的溶液中。将反应物在室温下搅拌1.5小时,并且然后用丙酮(2mL)淬灭。在减压下浓缩反应混合物。将残余物溶解于甲基叔丁基醚(300mL)中,用饱和碳酸氢钠(2×60mL)和饱和盐水(60mL)洗涤。将有机层经硫酸钠干燥,过滤,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到呈白色固体状的3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-醇(0.9g,100%产率)。
J.二硫代碳酸邻(3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-酯)S-甲酯
在0℃下添加氢化钠(0.48g,20.2mmol,5当量)于矿物油中的60%分散液到3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-醇(0.71g,2.46mmol,1当量)于无水THF(70mL)中的溶液中。在5℃下搅拌30分钟之后,添加咪唑(0.0168g,0.24mmol,0.1当量)于无水四氢呋喃(3.2mL)中的溶液,接着逐滴添加二硫化碳(0.89mL,14.8mmol,6当量)。经30分钟使反应物缓慢升温到12℃。逐滴添加(放热)碘甲烷(0.92mL,14.7mmol,6当量),并且将反应物在室温下搅拌1小时。将反应混合物冷却到5℃,用饱和盐水(140mL)稀释,并且用二氯甲烷(5×100mL)萃取。将经合并的有机层经硫酸钠干燥,过滤,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到呈白色固体状的二硫代碳酸邻(3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-酯)S-甲酯(0.86g,93%产率)。
K.合成3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪
将二硫代碳酸邻(3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-4-酯)S-甲酯(0.98g,2.6mmol,1当量)、2,2'-氮杂双(2-甲基丙腈)(0.098g,0.6mmol,0.2当量)和三丁基氢化锡(1.81mL,6.7mmol,2.6当量)于无水甲苯(70mL)中的溶液在80℃下搅拌3.5小时。在冷却到室温之后,将反应混合物在减压下在35℃下浓缩,并且吸收到硅胶(10g)上。通过柱色谱法纯化粗物质,得到呈白色固体状的3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.53g,72%产率)。
L.合成(3-氯丙基)(甲基)硫烷
将甲硫醇钠(6.14g,88mmol)溶解于150mL EtOH中,在冰浴中冷却,然后添加1-溴-3-氯丙烷(8.6ml,87mmol)。使溶液升温到室温,并且搅拌2小时。过滤沉淀的固体,并且使滤液在真空下冷凝。在真空下蒸馏残余物,得到呈无色油状的产物,36%。
M.合成三-[(3-甲硫基)丙基]铱(III)
通过将由(3-氯丙基)(甲基)硫烷和镁屑制成的格林纳(Grignard)与IrCl3(THT)3一起于THF中搅拌,接着进行柱色谱法,合成三-[(3-甲硫基)丙基]铱(III),得到白色固体,32%。
N.合成化合物35
将来自实例5M的三-[(3-甲硫基)丙基]铱(III)(0.020g,0.044mmol)和来自实例5K的3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.036g,0.131mmol)组合于乙二醇(0.5ml)中,通过抽真空/回填循环脱气,并且在回流下搅拌,变黄然后变黑。将冷却的残余物分配于水与DCM之间,将有机物干燥并且涂布于硅藻土上。通过柱色谱法纯化,得到4mg呈米色固体状的化合物35(9%)。
实例6:如流程6中那样进行化合物48的合成。
流程6
将来自实例4的(3-苯基-1H-吡唑)2Ir(MeOH)2(OTf)(0.031g,0.045mmol)和来自实例5K的3,3-二甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.024g,0.090mmol)组合于2-乙氧基乙醇(0.5ml)中,进行快速抽真空/回填三次,然后在回流下在氮气下加热2小时。将反应混合物溶解于DCM中,涂布于硅藻土上,并且通过柱色谱法纯化,得到呈近似无色残余物形式的化合物35,6mg(18%)。
实例7:根据以下流程7进行化合物49的合成。
流程7
A.合成1-甲基菲啶-6-胺:
向2-溴-3-甲基苯胺(38.8g,208mmol,1当量)、(氯(2-二环己基膦-2',6′-二甲氧基-1,1′-联苯)[2-(2'-氨基-1,1′-联苯)]钯(II)(2.99g,4.16mmol,0.02当量)、2-二环己基-膦-2',6′-二甲氧基联苯(1.71g,4.16mmol,0.02当量)于THF(832mL)中的混合物充氮气15分钟。添加(2-氰基苯基)溴化锌溶液(500mL,0.5M于THF中,250mmol,1.2当量)到混合物中,并且使反应物回流16小时。在冷却到室温之后,将反应物用饱和盐水(10mL)稀释,并且在减压下浓缩。将固体溶解于含10%甲醇的二氯甲烷(500mL)和24重量%氢氧化钠水溶液(500mL)中。分离各层,并且用二氯甲烷(3×500mL)萃取水溶液。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。将褐色固体依序用含25%MTBE的庚烷(1.5L)和二氯甲烷(5×25mL)湿磨,得到呈浅黄色固体状的26(10.7g,25%产率,>95%纯度)。
B.合成8-甲基咪唑并[1,2-f]菲啶:
使1-甲基菲啶-6-胺(10.7g,51mmol,1当量)、50重量%氯乙醛水溶液(16mL,102mmol,2当量)、碳酸钠(13.5g,128mmol,2.5当量)于异丙醇(340mL)中的混合物回流2小时。将反应物冷却到4℃,并且用二氯甲烷(250mL)和饱和碳酸氢钠(500mL)稀释。分离各层,并且用二氯甲烷(3×250mL)萃取水层。将经合并的有机物层经硫酸钠干燥,并且在减压下浓缩,得到呈褐色固体状的粗8-甲基咪唑并[1,2-f]菲啶(23.8g),其随后使用。
C.合成3-溴-8-甲基咪唑并[1,2-f]菲啶:
将粗8-甲基咪唑并[1,2-f]菲啶(23.8g)、N-溴琥珀酰亚胺(9.1g,51mmol,1当量)于二氯甲烷(306mL)中的混合物在室温下搅拌2小时。添加水(500mL),并且分离各层。用二氯甲烷(3×500mL)萃取水溶液。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。将固体预吸收到硅胶上,并且通过柱色谱法纯化,得到呈淡褐色固体状的3-溴-8-甲基咪唑并[1,2-f]菲啶(12g,98%纯度)。
D.合成8-甲基-3-(2-甲基丙-1-烯-1-基)咪唑并[1,2-f]菲啶:
向3-溴-8-甲基咪唑并[1,2-f]菲啶(12g,38.5mmol,1当量)、4,4,5,5-四甲基-2-(2-甲基丙-1-烯-1-基)-1,3,2-二氧杂硼杂环戊烷(10.5g,58mmol,1.5当量)和碳酸钾(16g,115.5mmol,3当量)于1,4-二噁烷与水的5比1混合物(185mL)中的混合物充氮气15分钟。添加(氯(2-二环己基膦-2',6′-二甲氧基-1,1′-联苯)[2-(2'-氨基-1,1′-联苯)]钯(II)(4.16g,5.78mmol,0.15当量)和2-二环己基膦-2′,6′-二甲氧基联苯(2.38g,5.78mmol,0.15当量),并且使反应物回流36小时。在冷却到室温之后,将反应物用水(200mL)稀释。分离各层,并且用乙酸乙酯(3×200mL)萃取水溶液。将经合并的有机物层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗固体,得到呈淡褐色固体状的8-甲基-3-(2-甲基丙-1-烯-1-基)咪唑并[1,2-f]菲啶(8.5g,70%产率,90%纯度)。
E.合成4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将8-甲基-3-(2-甲基丙-1-烯-1-基)咪唑并[1,2-f]菲啶(1.6g,5.69mmol,1当量)和无水氯化铝(3.8g,28.4mmol,5当量)于二氯甲烷(57mL)中的混合物在室温下搅拌16小时。将反应物在冰浴中冷却,并且逐滴添加水(10mL)。分离各层,并且用二氯甲烷(3×50mL)萃取水层。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗固体,得到呈淡黄色固体状的4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(1.43g,88%产率,98%纯度)。
F.合成2-溴-4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(500mg,1.75mmol,1当量)和N-溴琥珀酰亚胺(311mg,1.75mmol,1当量)于二氯甲烷(11mL)中的混合物在室温下搅拌2小时。将反应物用水(20mL)和二氯甲烷(10mL)稀释。分离各层,并且用二氯甲烷(3×20mL)萃取水溶液。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化残余物,得到呈淡褐色固体状的2-溴-4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(575mg,90%产率,97%纯度)。
G.合成2,4,4,7-四甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
向2-溴-4,4,7-三甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(265mg,0.73mmol,1当量)、三甲基硼氧杂环己烷(0.6mL,4.4mmol,6当量)和碳酸钾(608mg,4.4mmol,6当量)于1,4-二噁烷与水的10比1混合物(7mL)中的混合物充氮气15分钟。添加(氯(2二环己基膦-2′,6′-二甲氧基-1,1′-联苯)[2-(2′-氨基-1,1′-联苯)]钯(II)(108mg,0.15mmol,0.2当量)和2-二环己基膦-2′,6′-二甲氧基联苯(62mg,0.15mmol,0.2当量),并且使反应物回流16小时。在冷却到室温之后,将反应物用水(10mL)和乙酸乙酯(10mL)稀释。分离各层,并且用乙酸乙酯(3×20mL)萃取水溶液。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化残余物,得到呈浅黄色固体状的2,4,4,7-四甲基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(100mg,46%产率,95%纯度)。
H.合成化合物49:
以与化合物35类似的方法合成化合物49,得到13mg黄色粉末(15%)。
实例8:根据以下流程8进行化合物50的合成。
流程8
A.合成1-氯菲啶-6-胺:
将3-氯-2-碘苯胺(8.77g,34.6mmol)、CyJohnPhos(0.462g,1.319mmol)和Pd(CH3CN)2Cl2(0.171g,0.659mmol)的混合物溶解于二噁烷(80ml)中。借助注射器依序添加三乙胺(13.78ml,99mmol)和4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(10.04ml,69.2mmol)到溶液中。使反应物回流4小时。将反应物冷却到室温,并且添加2-溴苯甲腈(6g,33.0mmol)、S-Phos Pd G2(0.475g,0.659mmol)、S-Phos(0.271g,0.659mmol)和碳酸钾(9.11g,65.9mmol)的固体混合物到反应混合物中,接着添加二噁烷(20ml)和水(20ml),并且将反应物加热到85℃后持续16小时。将粗产物用DCM萃取,并且降低真空度,得到橙色油。将其溶解于THF(80mL)中,并且在0℃下添加氢化钠(1.978g,49.4mmol),并且搅拌20分钟。将反应物用盐水淬灭,并且用DCM萃取。蒸发反应混合物,接着用乙醚湿磨,得到呈灰白色固体状的1-氯菲啶-6-胺(52%产率)。
B.合成8-氯咪唑并[1,2-f]菲啶:
将1-氯菲啶-6-胺(864mg,3.78mmol)、2-氯乙醛(50重量%水溶液,1.02mL,7.56mmol)和碳酸氢钠(635mg,7.56mmol)组合于iPrOH中,并且回流1小时。将混合物冷却到室温,并且倾入水中,并且过滤(99%产率)。
C.合成8-苯基咪唑并[1,2-f]菲啶:
将8-氯咪唑并[1,2-f]菲啶(955mg,3.78mmol)、苯基硼酸(829mg,6.80mmol)、S-Phos Pd G2(109mg,0.151mmol)、S-Phos(62.1mg,0.151mmol)和碳酸钾(522mg,3.78mmol)的混合物抽真空并且用氮气回填数次。添加二噁烷(20ml)和水(4ml),并且使其回流1小时。将粗产物用DCM和盐水萃取,并且通过柱色谱法纯化,得到产物(99%产率)。
D.合成3-溴-8-苯基咪唑并[1,2-f]菲啶:
将8-苯基咪唑并[1,2-f]菲啶(1.15mg,3.91mmol)和NBS(0.765g,4.30mmol)组合于DMF中,并且在室温下搅拌30分钟,接着用水淬灭。将所得固体过滤并且在真空中干燥,得到75%产率的3-溴-8-苯基咪唑并[1,2-f]菲啶。
E.合成3-(2-甲基丙-1-烯-1-基)-8-苯基咪唑并[1,2-f]菲啶:
将3-溴-8-苯基咪唑并[1,2-f]菲啶(980mg,2.63mmol)、SPhos Pd G2(76mg,0.105mmol)、SPhos(43.1mg,0.105mmol)和碳酸钾(363mg,2.63mmol)的混合物抽真空并且用氮气回填数次。添加甲苯(15ml)、水(3ml)和4,4,5,5-四甲基-2-(2-甲基丙-1-烯-1-基)-1,3,2-二氧杂硼杂环戊烷(1.077ml,5.25mmol),并且在回流下加热16小时。将产物用DCM和盐水萃取,并且通过柱色谱法纯化,得到20%产率的3-(2-甲基丙-1-烯-1-基)-8-苯基咪唑并[1,2-f]菲啶。
F.合成4,4-二甲基-7-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将3-(2-甲基丙-1-烯-1-基)-8-苯基咪唑并[1,2-f]菲啶(160mg,0.459mmol)溶解于DCM(10ml)中,并且添加三氯化铝(184mg,1.378mmol)。将反应物在室温下搅拌40分钟。将混合物用KOH(aq)/盐水淬灭,并且用DCM萃取数次。通过柱色谱法纯化产物,得到63%产率的4,4-二甲基-7-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪。
G.合成化合物50:
将来自实例4的(3-苯基-1H-吡唑)2Ir(MeOH)2(OTf)(0.03g,0.043mmol)和4,4-二甲基-7-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.030g,0.087mmol)组合于2-乙氧基乙醇(0.5ml)中,快速抽真空/用氮气回填三次,然后在回流下在氮气下加热2小时。通过柱色谱法纯化产物,得到56%产率的化合物50。
实例9:根据以下流程9进行化合物108的合成。
流程9
A.合成(4-((三异丙基硅烷基)氧基)苯基)氨基甲酸叔丁酯:
依序添加三异丙基氯硅烷(32mL,0.15mol,1.2当量)和三乙胺(21mL,0.15mol,1.2当量)到(4-羟苯基)氨基甲酸叔丁酯(26.1g,0.125mol,1当量)于THF(200mL)中的溶液中。将反应混合物在室温下搅拌16小时。过滤反应物,并且用THF(2×30mL)洗涤固体。在减压下浓缩经合并的滤液。通过柱色谱法纯化粗产物,得到呈黄色油状的(4-((三异丙基硅烷基)氧基)苯基)氨基甲酸叔丁酯(39.66g,87%产率)。
B.合成4-((三异丙基硅烷基)氧基)苯胺:
在室温下添加三氟乙酸(41.51mL,0.54mol,5当量)到(4-((三异丙基硅烷基)氧基)苯基)氨基甲酸叔丁酯(39.66g,0.1085mol,1当量)于二氯甲烷(400mL)中的溶液中。在搅拌16小时之后,在减压下去除溶剂。使残余物与甲苯(3×50mL)共沸。经硅胶纯化粗产物,得到4-((三异丙基硅烷基)氧基)苯胺(25g,87%产率)。
C.合成2,6-二溴-4-((三异丙基硅烷基)氧基)苯胺:
在0℃下逐滴添加溴(8.2mL,0.16mol,2.5当量)到4-((三异丙基硅烷基)氧基)苯胺(17g,64.4mmol,1当量)于二氯甲烷和甲醇的1:1混合物(60mL)中的溶液中。使反应混合物升温达到室温,并且搅拌16小时。将反应混合物用二氯甲烷(200mL)稀释,并且依序用1M NaOH(2×100mL)和饱和盐水(2×100mL)洗涤。将有机层经硫酸钠干燥并且在减压下浓缩,得到呈褐色油状的2,6-二溴-4-((三异丙基硅烷基)氧基)苯胺(26.37g,97%产率),其随后使用。
D.合成4-溴-8-甲氧基-2-((三异丙基硅烷基)氧基)菲啶-6-胺:
向5-甲氧基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲腈(16.14g,62.3mmol,1当量)、51(26.37g,62.3mmol,1当量)和磷酸钾(43.04g,0.187mol,3当量)于甲苯和水的4比1混合物(500mL)中的混合物充氮气1小时。添加反式Pd(PPh3)2Cl2(2.8g,3.11mmol,0.05当量),并且使反应混合物回流20小时。添加额外5-甲氧基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲腈(2.2g,8.5mmol,0.14当量)和反式Pd(PPh3)2Cl2(0.3g,0.43mmol,0.0069当量),并且使反应混合物再回流4小时。分离各层,并且用热水(2×200mL)洗涤有机层。将有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化残余物,得到20%产率的4-溴-8-甲氧基-2-((三异丙基硅烷基)氧基)菲啶-6-胺。
E.合成5-溴-3-(2-氯乙基)-11-甲氧基-7-((三异丙基硅烷基)氧基)咪唑并[1,2-f]菲啶:
将4-溴-8-甲氧基-2-((三异丙基硅烷基)氧基)菲啶-6-胺(5.95g,12.53mmol,1当量)、单水合对甲苯磺酸(175mg)和新鲜制备的2(6.67g,62.63mmol,5当量)于异丙醇(500mL)中的悬浮液在室温下搅拌2小时。添加碳酸钠(3.25g,37.6mmol,3当量)和去离子水(12ml),并且使反应混合物回流16小时。在冷却到室温之后,将反应混合物体积在减压下减少到约60ml。将混合物用乙酸乙酯(300mL)稀释,并且用饱和盐水(200mL)洗涤。将有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到5-溴-3-(2-氯乙基)-11-甲氧基-7-((三异丙基硅烷基)氧基)咪唑并[1,2-f]菲啶(5.53g,79%产率)。
F.合成10-甲氧基-6-((三异丙基硅烷基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
向5-溴-3-(2-氯乙基)-11-甲氧基-7-((三异丙基硅烷基)氧基)咪唑并[1,2-f]菲啶(5.53g,9.84mmol,1.0当量)于无水THF(300mL)中的溶液充氮气30分钟。在冷却到0℃之后,借助注射器逐滴添加2M于THF中的异丙基氯化镁(7.4mL,14.76mmol,1.5当量)。使反应混合物升温到室温,并且搅拌16小时。用水(10mL)淬灭反应物,并且在减压下去除THF。用二氯甲烷(500mL)萃取残余物。将有机层用水(2×200mL)洗涤,经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到10-甲氧基-6-((三异丙基硅烷基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(3g,68%产率)。
G.合成10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-醇:
逐滴添加含三水合四丁基氟化铵的THF(30mL)到10-甲氧基-6-((三异丙基硅烷基)氧基)-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(3g,6.72mmol,1当量)于THF(100mL)中的溶液中。在室温下搅拌16小时之后,在减压下去除溶剂,并且用二氯甲烷(80mL)萃取残余物。用饱和盐水(2×100mL)洗涤有机层。在用饱和盐水洗涤后,大量沉淀物开始在有机层中形成。将沉淀物过滤并且用庚烷(2×10mL)洗涤,得到纯10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-醇(1.83g,94%产率)。
H.合成三氟甲磺酸10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-酯:
在0℃下依序添加三氟乙酸酐(1.14mL,6.77mmol,1.1当量)和吡啶(0.744mL,9.24mmol,1.5当量)到10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-醇(1.79g,6.16mmol,1当量)于二氯甲烷(100mL)中的混合物中。在搅拌15分钟之后,使反应物升温到室温,并且搅拌6小时。将反应混合物用二氯甲烷(200mL)稀释并且用水(3×100mL)洗涤。经硫酸钠干燥有机层,并且在减压下去除溶剂。将残余物用庚烷和二氯甲烷的10比1混合物(10mL)湿磨,得到三氟甲磺酸10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-酯(2.17g,83%产率)。
I.合成10-甲氧基-6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
向三氟甲磺酸10-甲氧基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-6-酯(0.65g,1.54mmol,1当量)、苯基硼酸(0.188g,1.54mmol,1当量)和磷酸钾(1.06g,4.62mmol,3当量)于甲苯:1,4-二噁烷:水的3:1:1混合物(500mL)中的混合物充氮气1小时。添加反式Pd(PPh3)2Cl2(54mg,0.077mmol,0.05当量),并且使反应混合物回流16小时。将反应混合物用二氯甲烷(200mL)稀释。将有机层用温水(2×100mL)洗涤,经硫酸钠干燥,并且在减压下浓缩,得到10-甲氧基-6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.527g,97%产率)。
J.合成6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-10-醇:
在-78℃下逐滴添加1M于二氯甲烷中的三溴化硼(7.5mL,7.5mmol,5当量)到10-甲氧基-6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.527g,1.5mmol,1当量)于二氯甲烷(100mL)中的溶液中。使反应物升温到室温,并且搅拌16小时。将反应混合物小心地倾入冰水(150mL)中,并且将所得固体过滤并且依序用水(30ml)和庚烷(10mL)洗涤,得到6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-10-醇(0.47g,93%产率)。
K.合成10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-6-苯基-3,4-二氢-二苯并[b,ij]咪唑并[2,1,5-de]喹嗪:
将2-溴-9-(4-异丙基吡啶-2-基)-9H-咔唑(0.528g,1.446mmol,1当量)、6-苯基-3,4-二氢二苯并[b,ij]咪唑并[2,1,5-de]喹嗪-10-醇(0.486g,1.446mmol,1当量)、磷酸钾(1.67g,7.23mmol,5当量)、碘化铜(I)(0.138g,0.723mmol,0.5当量)和吡啶甲酸(0.445g,3.62mmol,2.5当量)于DMSO(50mL)中的混合物在150℃下加热4.5小时。在冷却到室温之后,将反应混合物倾入水(300mL)中,并且用乙酸乙酯(4×100mL)萃取。将经合并的有机层经硫酸钠干燥,并且在减压下浓缩。通过柱色谱法纯化粗产物,得到呈茶色固体状的10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-6-苯基-3,4-二氢-二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(0.55g,61%产率)。
L.合成化合物108:
向10-((9-(4-异丙基吡啶-2-基)-9H-咔唑-2-基)氧基)-6-苯基-3,4-二氢-二苯并[b,ij]咪唑并[2,1,5-de]喹嗪(350mg,0.564mmol,1当量)于冰乙酸(60mL)中的溶液充氩气40分钟。添加K2PtCl4(234mg,0.564mmol,1当量),并且使反应混合物回流16小时。在冷却到室温之后,将黄绿色沉淀物过滤,并且依序用水(4×15mL)和庚烷(2×10mL)洗涤,并且在真空下在20℃下干燥18小时。将粗产物溶解于二氯甲烷(500mL)中,并且使其通过硅胶塞(10g)以去除残余K2PtCl4。在减压下去除溶剂。将残余物用二氯甲烷和庚烷的1比1混合物(20mL)湿磨,过滤,并且用二氯甲烷(2×3mL)洗涤,得到化合物108(40mg,产率8.7%产率,83.2%)。
讨论:以下展示金属配位的咪唑并菲啶配位体的一个实施例的通式结构。计算研究中所关注的键是四种碳-氮(C-N)单键。其标记为针对具有三种C-N单键的氮的C-N1、C-N2、C-Nph和针对配位到金属的氮的C-Nm。
在高斯(Gaussian)09软件包中使用混合B3LYP泛函与CEP-31g有效核势基组对所有络合物和配位体执行几何优化。除非另外陈述,否则在结果和讨论中所有结果都使用此方法。
通过使一键断裂以形成双自由基物质来计算咪唑并菲啶配位体上的键强度。断键的双自由基物质经计算为三重态,因为其能量通常比双自由基单重态低,并且因此产物更可能在断键事件中形成。在B3LYP/6-31g(d)水平下执行计算,并且针对基态单重态→断键的三重态和最低能量三重态(激发态)→断键的三重态进行热力学报道。
最低三重态激发态(T1)的计算TD-DFT值还在B3LYP/CEP-31g理论水平下执行,但包括使用THF作为溶剂的CPCM连续溶剂场,其已经显示更好地匹配实验结果。
对以下化合物执行键强度计算:
计算的键强度展示于表1中。
表1:
表1展示了一系列比较实例和本发明化合物1的计算的键强度。在两个数值见于同一单元格中时,上面的数值表示激发态三重态→断键的三重态之间的热力学差异。下面的数值表示基态单重态→断键的三重态。如果单元格中仅存在一个数值,那么其表示三重态→三重态键强度(T→T)。对于所有比较化合物1-4,C-N1键都显示为最弱键。发现键强度在激发三重态下与基态单重态相比更弱。这是由于具有激发态的能量的络合物可用作起始点,到通常更高能量的断键的状态。在一些情况下,如关于比较化合物2和3所展示,断键的状态能量比起始三重态低。因此,断键事件可以视为热力学上有利的或放热的。发现当芳基取代基添加于C-N1键碳原子时,键强度降低,如将比较化合物1与比较化合物2和3比较所见。此影响可能是由于断键位点处自由基物质的共振稳定化,所述自由基物质通过芳基取代而稳定化。
弱C-N1键的稳定化可以通过键联取代来实现,所述键联取代使C-N1碳键联到相邻稠合芳基环上的碳,如式(1a)中的“A”所描绘。此键联基团优选由如下元素组成,其提供恰当结构几何构型以形成跨菲啶环系统的两个碳的桥,提供必需刚度以使C-N1键稳定化,同时不会降低所得配位体和络合物的三重态能量。
稳定化键联基团的作用关于本发明化合物1展示于表1中。此处,三重态C-N1键强度已经从类似比较化合物1的11.81kcal/mol大大提高到本发明化合物的35.38kcal/mol,热力学键强度有>20kcal/mol的增加。两个碳键联取代基阻止配位体获得CN1断键的状态的适当松弛的几何构型。重要地,三重态能量并不受此取代影响,因为本发明化合物1和比较化合物1两者都具有468nm的计算相同三重态能量。
比较实例1的最小化非断键和断键几何构型展示于图3a和3b中。可以看出,断键的几何构型使咪唑并菲啶配位体的稠合环系统的环应力松弛。如关于本发明化合物1所展示,系栓取代抑制松弛的断键的几何构型,因此增加C-N1键的热力学键强度。
C-N1键的薄弱的其它实验证据由基质辅助激光解吸电离质谱分析(MALDI-MS)展示。MALDI-MS可以用以探测分子的激发态下的键的薄弱性。据相信,作为光化学稳定性的量度,MALDI-MS可以模拟存在充电和激发态两者的OLED装置内所见的一些条件。图3展示了比较化合物3的以负模式获取的MALDI-MS。母体离子的峰值鉴别为1529amu下。然而,最高强度峰值见于1275amu下。此质量对应于比较化合物3的片段,其中咪唑环已经失去两个碳的质量和联三苯取代。所提出的片段的结构展示于图3中。同位素模式证实此片段含有铱并且与所提出的片段的化学式一致。如图4中所示,关于1083amu下的配位体损失和1020amu下两种配位体的咪唑环分解,鉴别其它片段。数据表明,主要片段的形成需要C-N1键断裂,所述键通过计算经预测为弱键。
本发明化合物的光物理性质
本发明化合物的所测量的光物理性质报道于下表2中。在77K下和在室温下在2-甲基四氢呋喃溶剂中在高稀释浓度下测量络合物。使用配备有氙气灯、积分球和C10027型号光子多通道分析仪的滨松(Hamamatsu)C9920系统,以1重量%于聚甲基丙烯酸甲酯(PMMA)固态基质或0.4重量%聚苯乙烯(PS)固态基质中测量光致发光量子产率(PLQY,ΦPL)。通过时间相关单光子计数法,使用与IBH数据站中心集成的堀场乔宾伊冯(Horiba Jobin Yvon)Fluorolog-3,使用335nm纳米LED作为激发源,进行PL瞬态测量(τ)。
表2:
化合物35经测量具有深蓝色发射,在77K下的最高能量峰值是451nm,然而,络合物的PLQY是仅5%。化合物49展现对配位体的改性可以如何用以提高PLQY。咪唑环上的甲基取代已经发现可提高非乙基桥连菲啶咪唑类似物的PLQY。另外,外部苯环上的甲基取代通过计算显示可影响配位体咬角,这是由于相邻芳基环上的甲基取代基和质子的空间影响。此空间影响使得菲啶咪唑多环系统几何构型更接近非桥连配位体的几何构型,其中配位位点可以更紧密连接到金属。配位体的几何构型的此细微变化使得金属与中性配位的氮之间的相互作用更强,提高金属-氮键强度。据相信,更强金属-氮键强度可以通过减小金属-氮断键非-辐射衰变而改进络合物的发射性。因此,两个甲基取代基都可能引起与化合物35相比提高化合物49的PLQY。化合物49经测量为在PMMA基质中具有62%的PLQY,其极接近非桥连类似物比较化合物6的PLQY值,所述比较化合物经测量为具有68%的PLQY。另外,与化合物35的5.1微秒的激发态寿命相比,化合物49经测量在77K下具有2.9微秒的短得多的激发态寿命。这进一步表明,甲基取代基改进化合物49的辐射性质。
具有苯基吡唑配位体(ppz)的杂配实例化合物48和化合物50经测量具有深蓝色发射,但具有低PLQY。然而,非桥连参考化合物比较化合物8也测量为具有14%的低PLQY。据相信,低效率可能是由于吡唑配位体的弱金属-氮键。为了进一步支持此假设,三Ir(ppz)3已经在文献中显示为在室温溶液中是非发射的,但在77K下是高度发射的。室温下的非发射性归因于弱金属氮键。
具有桥连菲啶咪唑配位体的铂络合物也被发现以深蓝色高度发射。化合物105和比较化合物7两者都测量为在光学惰性聚苯乙烯基质中具有分别为85%和87%的高PLQY值。铂络合物可能不需要如关于铱类似物化合物49所描述进行配位体改性来提高PLQY,这是由于与铱相比相对更强的铂-氮键强度。
本领域的技术人员应了解,可以在不脱离上文所展示并且描述的例示性实施例的广泛发明构思情况下改变所述例示性实施例。因此,应理解,本发明不限于所展示并且描述的例示性实施例,但希望涵盖在如由权利要求书所界定的本发明的精神和范围内的修改。举例来说,例示性实施例的特定特征可以是或可以不是所要求的发明的一部分并且可以将所公开的实施例的特征组合。本文中除非特定阐述,否则术语“一个(a/an)”和“所述”不限于一个要素,而是应理解为意指“至少一个”。
应理解,已经将本发明的图式和描述中的至少一些简化以集中于与本发明的清楚理解相关的要素,同时为清楚起见,排除本领域普通技术人员将了解也可以构成本发明的一部分的其它要素。然而,因为此类要素在本领域中熟知并且因为它们不必促进更好地理解本发明,所以本文中未提供此类要素的描述。
此外,本发明的任何方法不依赖于本文中阐述的特定步骤次序到一定程度以便,特定步骤次序不应理解为对权利要求的限制。针对此类方法的权利要求不应限于以所写次序执行其步骤,并且本领域技术人员可以容易了解,步骤可以变化并且仍保持在本发明的精神和范围内。
本文所引用的所有参考文献,包括公开案、专利申请和专利特此以引用的方式并入,其引用程度就如同每一参考文献单独地并且特定地指示为以引用的方式并入并且全文阐述于本文中一般。
- 还没有人留言评论。精彩留言会获得点赞!