一种分子标签的制备方法与流程
本发明涉及DNA变异检测领域,尤其涉及一种分子标签的制备方法。
背景技术:
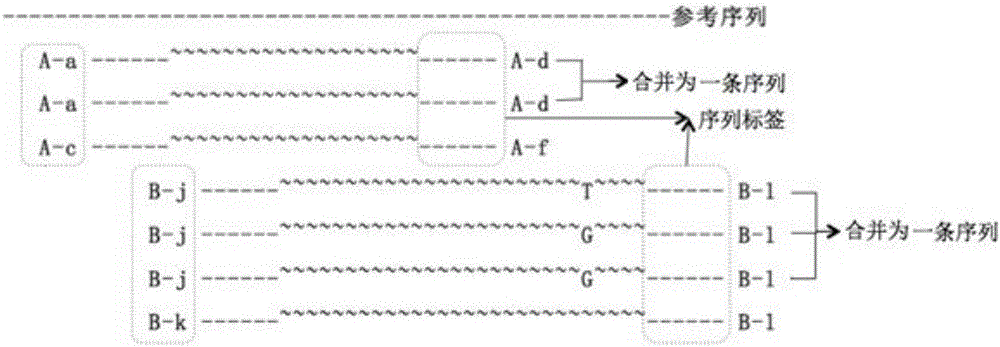
:液态活检技术,相比于传统的检测方法,因具有副作用小、操作简单、能重复取样等特点,是目前肿瘤精准医疗领域炙手可热的技术,其主要检测对象包括:循环肿瘤细胞(CTC)、循环肿瘤DNA(ctDNA)以及肿瘤外泌体等,可以提示肿瘤发展进程及抗药性等信息,指导个体化的用药治疗。ctDNA是一种存在于血浆或血清、脑脊液等体液中的细胞外游离的单链或双链DNA,主要来自于坏死或凋亡的肿瘤细胞、肿瘤细胞分泌的外排体及循环肿瘤细胞。ctDNA片段大小通常为160-180bp,在血液中半衰期2小时,携带有点突变、插入缺失、拷贝数、融合基因等突变信息,在人体血液循环系统中不断流动,可实时反映肿瘤患者当前的信息。然而,在大部分实体瘤的早期和部分实体瘤的中晚期,血浆中的ctDNA含量极低,利用高通量测序技术(NGS)获得的有效数据率偏低,同时对实验要求较高,限制实验样本用量与实验次数。其次ctDNA样本中含有大量的基因组DNA,导致高通量测序背景噪声很高,用常规建库测序方法时导致肿瘤信号完全淹没在背景噪声中,只能通过增加测序深度来提高ctDNA的检测准确性,这不仅增加了测序成本,同时不可避免的存在实验过程中PCR引入的扩增错误。由于在文库构建、簇生成及边合成边测序过程中,存在PCR(聚合酶链式反应)聚合酶的扩增错误,导致NGS测序每个碱基错误率在5×10-4到10-2。因此,低于该频率的变异检测将无法与实验错误区分出来。科学家通过计算算法和分子实验技术来改善NGS检测的错误率。CAPP-seq(深度测序癌症个性化谱)系统利用统计模型来区分真正的变异,但仅限于已知的变异位点。目前,一种称为UID(唯一标识)技术,也被称为分子标签技术,用于NGS错误纠正。该技术是在PCR扩增前将每条分子引入了一个可识别的惟一序列编码(barcode),通过该惟一序列编码来区分哪些是PCR扩增引入的分子序列,哪些是样本中原始的分子序列,从而提高测序数据有效率;同时也可以通过该惟一序列编码来区分真正的变异与实验错误,以提高测序准确性,尤其对ctDNA样本的低频突变,意义重大。目前采用的UID技术,主要有两种方法。第一种方法,在双链DNA分子模板上引入双链barcode序列,最为典型的例子是双重测序技术,具体方法如下:(1)合成两条单链接头序列,其中一条带有12个碱基的DuplexTag(双面标签)序列和4个GTCA固定序列,两条接头序列进行退火互补;(2)通过DNA聚合酶延伸,合成互补碱基;(3)通过DNA聚合酶在双链3′端加上A碱基。在建库过程中,每条DNA片段化分子都分别标记了两个不同tag序列(分别用α和β表示),通过PCR扩增之后,形成两种PCR产物,即αβ家族和βα家族。只有在所有αβ家族和βα家族reads中,且双链DNA片段中都存在的变异位点才是真正的变异位点。随后又出现了对双重测序技术的改进(NatureProtocols,2014,9,2586-2606),将加barcode序列的Y字型接头从3′端A碱基改为了3′端T碱基,提高了建库过程中DNA片段与接头的连接效率。第二种方法是在通过在扩增引物上增加标签来识别每条DNA序列,然后再进行扩增子的高通量测序。随着技术的不断改进,又出现了iDES方法(集成数字错误抑制)CAPP-seq技术检测ctDNA(NatureBiotechnology,2016),检测极限达到0.0025%。该技术结合了以上两种分子标签方法,利用双重测序方法引入了插入序列编码,同时通过PCR引入了指数序列编码,从而进一步提高了检测准确性。双重测序技术采用12bp的随机引物,以及4bp或5bp的固定序列,导致DNA测序数据中前16-17bp的数据不可用,降低了数据有效率;CAGT固定序列对于区分DNA分子序列无帮助,导致测序质量降低;该方法中限于所有αβ家族和βα家族reads中,且双链DNA片段中都存在的变异位点才是真正的变异位点;而双重测序技术的改进方法,通过内酶切方法在3′端引入T碱基,反应时间16h,操作周期长,酶切反应效率较低。鉴于上述缺陷,本设计人积极加以研究创新,以期创设一种新型分子标签的制备方法,使其更具有产业上的利用价值。技术实现要素:为解决上述技术问题,本发明的目的是提供一种分子标签的制备方法,该分子标签制备方法通过改变barcode长度,去除固定序列,barcode不限于两种固定序列,减少测序中数据的浪费;使用DNA聚合酶在接头末尾引入胸腺嘧啶T,提高反应速率;用增加的DNA序列识别DNA扩增中同一DNA模板产生的多个副本,有效区分DNA扩增过程中的错误和真实的DNA变异,更正确地评估DNA扩增的偏好性。本发明的分子标签的制备方法包括以下步骤:(1)在DNA分子模板的5′接头端引入barcode序列;(2)用DNA聚合酶延伸3′接头端,与barcode序列互补,得到补齐序列;(3)向步骤(2)得到的补齐序列中加入DNA聚合酶,在补齐序列的3′接头端引入胸腺嘧啶,得到改造的接头;(4)将待测DNA破碎成DNA短片段,用DNA连接酶连接DNA短片段与改造的接头,得到DNA连接产物;(5)聚合酶链式反应扩增DNA连接产物,得到扩增产物;(6)对扩增产物进行测序,然后比对到人类参考基因组,在多个DNA模板副本中选取碱基错误率最低的DNA序列,即为分子标签。进一步的,在步骤(1)中,barcode序列的长度为2-20bp。进一步的,在步骤(1)中,barcode序列可以是已知的或未知的序列。进一步的,在步骤(2)中,DNA聚合酶为TAQDNA聚合酶或Klenow酶。进一步的,在步骤(3)中,DNA聚合酶为TAQDNA聚合酶或Klenow酶。进一步的,在步骤(4)中,DNA短片段的长度为150-300bp。进一步的,在步骤(4)中,DNA连接酶为T4DNA连接酶或E·coliDNA连接酶。进一步的,在步骤(6)中,用增加的DNA序列识别PCR扩增中同一DNA模板产生的多个副本。进一步的,在步骤(6)中,对扩增产物进行双端测序或单端测序。进一步的,在步骤(6)中,将DNA序列比对到参考基因组时,DNA片段在参考基因组上的位置的起点与终点相同的序列分为一组,比较同组DNA序列两端的barcode序列,barcode序列相同的DNA序列认为来自于同一DNA模板,来自同一DNA模板的多条类似或相同序列合并为或保留其中一条序列。进一步的,在步骤(6)中,由同一DNA模板产生的多个DNA副本的序列,通过计算每个位置碱基出错的概率,选取碱基错误率最低的DNA序列,即为分子标签。进一步的,分子标签用于基因变异检测。借由上述方案,本发明具有以下优点:Barcode序列为随机序列,最小长度为2bp,能够减少测序数据量的浪费,且barcode序列不限于固定序列;接头末尾引入碱基T,构成粘性末端,提高了反应速率;使用随机序列标记来源于同一DNA模板的测序序列,2个及以上DNA模板共同支持的变异即为真实变异;可有效识别DNA扩增过程中同一DNA模板产生的多个副本,有效区分DNA扩增过程中的错误和真实的DNA变异,更正确地评估DNA扩增的偏好性。上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。附图说明图1是本发明接头改造过程示意图;图2是本发明将DNA对比到人类参考基因组的过程示意图。具体实施方式下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。实施例11、接头(adapter)的合成与退火(1)各取序列为5'-TACACTCTTTCCCTACACGACGCTCTTCCGATCT-3'和5’-TCTTCTACAGTCANNNNNNAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3’的测序adapterDNA一管,其中序列中的NNNNNN为6bp的随机barcode,这里的N表示A/T/G/C中任何一个碱基都可以。即这管adapterDNA中包含的序列最多是4的6次方种,这些序列的差异取决于N为何种碱基。在4℃下,20000g离心10min,保证adapter粉末聚集在管底部,标号分别为adapter-1、adapter-2。(2)开启管盖,此时需注意不要让adapter粉末从管中飘出。分别向adapter-1、adapter-2中加入Nuclease-Freewater(无核酸酶水)溶解粉末,使其浓度为100nmol/mL,即100μM;然后在-20℃保存,若长期不使用在-80℃下保存。(3)各取25μLadapter-1(100μM)与25μLadapter-2(100μM),均匀混合,得到50μL浓度为50μM的母液。在PCR仪(ABI9700)上进行梯度温度降温退火,退火程序选择如表1所示。表1退火程序选择(4)将退火后的母液置于-20℃保存,取用时再根据需要稀释至工作液浓度。2、退火后Adapter质控抽取退火后的母液,用水稀释到1μM(1:49加水稀释),测Aglient2100,峰值在90-100bp。3、Adapter补齐实验使用TAKARAPrimeSTARHSDNAPolymerase(R010Q)补齐接头,将50μL接头分两管,配制50μLPCR反应液体系反应,每管组分如表2所示。可在室温下配制PCR反应液,配制反应液时,将各组份放于冰上。配制完成后,在68℃下孵育30min。表2各管中的反应体系组分体积(μL)终浓度5×PrimeSTARBuffer(Mg2+Plus)101xdNTPMixture(2.5mMeach)4200μMeachTemplate(接头)25~82.5ngPrimeSTARHSDNAPolymerase(2.5U/μl)0.51.25U/50μL灭菌蒸馏水10.5Total504、Adapter补齐后纯化然后采用乙醇沉淀法纯化补齐后的adapter后,步骤如下:(1)加入30μL醋酸钠和250μL的无水乙醇,混匀后-20℃放置一夜;(2)4℃下离心,16000rcf离心30min;(3)弃掉上清液,然后加750μL冷冻的80%乙醇,4℃下离心,16000rcf离心5min;(4)弃掉上清液,瞬时离心到1000rcf,然后停止;(5)吸干液体,室温下开盖,晾干5min;(6)加30μLNuclease-Freewater,彻底溶解沉淀,混匀离心,瞬时离心到1000rcf,然后停止。5、Adapter加“T”使用TAKARAExTaqDNAPolymerase(RR001Q)、TAKARAdTTP(4029Q),反应体系50μL,各组分如表3所示。将反应体系在72℃下孵育2h。表3adapter加“T”反应体系组分体积(μL)终浓度10×ExTaqBuffer(Mg2+Plus)51xdTTP(20μmol100mM)12mMTemplate(adapter)31~82.5ngTaKaRaExTaq(5U/μl)15U灭菌蒸馏水12Total506、Adapter加“T”后纯化使用乙醇沉淀法对引入了胸腺嘧啶的adapter进行纯化,具体步骤如下:(1)加入30μL醋酸钠和125μL的无水乙醇,混匀后于-80℃下放置2h;(2)4℃离心,16000rcf离心30min;(3)弃掉上清液,加750μL冷冻的80%乙醇,4℃离心16000rcf5min;(4)弃掉上清液,瞬时离心到1000rcf,然后停止;(5)吸干液体,室温下开盖晾干5min;(6)加30μLNuclease-Freewater,溶解沉淀,混匀离心,瞬时离心到1000rcf,然后停止,将其保存于-20℃下。Adapter制备好后,按Illumina常规建库方法对DNA进行建库,即将待检测DNA用DNA破碎仪破碎成长度为200bp的DNA短片段,用T4DNA连接酶连接DNA短片段与改造的adapter,得到DNA连接产物;PCR扩增得到的DNA连接产物,得到扩增产物;用IlluminaHiseq4000测序仪对扩增产物进行双端测序。然后用bwa比对软件将测序得到的DNA序列比对到人类参考基因组hg19,具体如下:如图2所示,序列标签中“~”代表从样本中扩增到的DNA序列,“~”两端的“-”代表adapter中引入的序列标签(barcode序列),两端的“-”可以一样也可以不一样,按照比对后DNA序列在hg19参考基因组上的位置将序列分成A、B两组,小写字母表示barcode序列名称。将DNA序列比对到参考基因组时,DNA片段在参考基因组上的位置的起点与终点相同的序列分为一组,如在图2中,A-a----A-d和B-j-----B-l序列分别有多条,分别是同一DNA模板的多个副本,将多条类似或相同序列合并为或保留其中一条序列,形成新的DNA待检测数据;其中如果同一DNA模板的多个副本之间序列有差异,这些有差异的位置的碱基将通过计算联合错误概率得到错误率最低的碱基型,得到处理后的序列,即为分子标签,如图中B-j------B-l序列,处理后的序列可能与原来序列中某一条相同,也可能与原来的序列都不同。本专利制备的分子标签用于识别DNA或者cDNA文库制备中PCR步骤产生的DNA副本,提高基因变异检测的准确性,2个及以上DNA模板共同支持的变异即为真实变异。实施例21、接头(adapter)的合成与退火(1)各取序列为5'-TACACTCTTTCCCTACACGACGCTCTTCCGATCT-3'和5’-TCTTCTACAGTCANNAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3’的测序adapterDNA一管,其中序列中的NN为2bp的随机barcode,这里的N表示A/T/G/C中任何一个碱基都可以。在4℃下,20000g离心10min,保证adapter粉末聚集在管底部,标号分别为adapter-1、adapter-2。(2)开启管盖,此时需注意不要让adapter粉末从管中飘出。分别向adapter-1、adapter-2中加入Nuclease-Freewater(无核酸酶水)溶解粉末,使其浓度为100nmol/mL,即100μM;然后在-20℃保存,若长期不使用在-80℃下保存。(3)各取25μLadapter-1(100μM)与25μLadapter-2(100μM),均匀混合,得到50μL浓度为50μM的母液。在PCR仪(ABI9700)上进行梯度温度降温退火,退火程序选择如表1所示。表1退火程序选择(4)将退火后的母液置于-20℃保存,取用时再根据需要稀释至工作液浓度。2、退火后Adapter质控抽取退火后的母液,用水稀释到1μM(1:49加水稀释),测Aglient2100,峰值在90-100bp。3、Adapter补齐实验使用TAKARAPrimeSTARHSDNAPolymerase(R010Q)补齐接头,将50μL接头分两管,配制50μLPCR反应液体系反应,每管组分如表2所示。可在室温下配制PCR反应液,配制反应液时,将各组份放于冰上。配制完成后,在68℃下孵育30min。表2各管中的反应体系组分体积(μL)终浓度5×PrimeSTARBuffer(Mg2+Plus)101xdNTPMixture(2.5mMeach)4200μMeachTemplate(接头)25~82.5ngPrimeSTARHSDNAPolymerase(2.5U/μl)0.51.25U/50μL灭菌蒸馏水10.5Total504、Adapter补齐后纯化然后采用乙醇沉淀法纯化补齐后的adapter后,步骤如下:(1)加入30μL醋酸钠和250μL的无水乙醇,混匀后-20℃放置一夜;(2)4℃下离心,16000rcf离心30min;(3)弃掉上清液,然后加750μL冷冻的80%乙醇,4℃下离心,16000rcf离心5min;(4)弃掉上清液,瞬时离心到1000rcf,然后停止;(5)吸干液体,室温下开盖,晾干5min;(6)加30μLNuclease-Freewater,彻底溶解沉淀,混匀离心,瞬时离心到1000rcf,然后停止。5、Adapter加“T”使用TAKARAExTaqDNAPolymerase(RR001Q)、TAKARAdTTP(4029Q),反应体系50μL,各组分如表3所示。将反应体系在72℃下孵育2h。表3adapter加“T”反应体系组分体积(μL)终浓度10×ExTaqBuffer(Mg2+Plus)51xdTTP(20μmol100mM)12mMTemplate(adapter)31~82.5ngTaKaRaExTaq(5U/μl)15U灭菌蒸馏水12Total506、Adapter加“T”后纯化使用乙醇沉淀法对引入了胸腺嘧啶的adapter进行纯化,具体步骤如下:(1)加入30μL醋酸钠和125μL的无水乙醇,混匀后于-80℃下放置2h;(2)4℃离心,16000rcf离心30min;(3)弃掉上清液,加750μL冷冻的80%乙醇,4℃离心16000rcf5min;(4)弃掉上清液,瞬时离心到1000rcf,然后停止;(5)吸干液体,室温下开盖晾干5min;(6)加30μLNuclease-Freewater,溶解沉淀,混匀离心,瞬时离心到1000rcf,然后停止,将其保存于-20℃下。Adapter制备好后,按Illumina常规建库方法对DNA进行建库,即将待检测DNA用DNA破碎仪破碎成长度为200bp的DNA短片段,用T4DNA连接酶连接DNA短片段与改造的adapter,得到DNA连接产物;PCR扩增得到的DNA连接产物,得到扩增产物;用IlluminaHiseq4000测序仪对扩增产物进行双端测序。然后用bwa比对软件将测序得到的DNA序列比对到人类参考基因组hg19,具体如下:如图2所示,序列标签中“~”代表从样本中扩增到的DNA序列,“~”两端的“-”代表adapter中引入的序列标签(barcode序列),两端的“-”可以一样也可以不一样,按照比对后DNA序列在hg19参考基因组上的位置将序列分成A、B两组,小写字母表示barcode序列名称。将DNA序列比对到参考基因组时,DNA片段在参考基因组上的位置的起点与终点相同的序列分为一组,如在图2中,A-a----A-d和B-j-----B-l序列分别有多条,分别是同一DNA模板的多个副本,将多条类似或相同序列合并为或保留其中一条序列,形成新的DNA待检测数据;其中如果同一DNA模板的多个副本之间序列有差异,这些有差异的位置的碱基将通过计算联合错误概率得到错误率最低的碱基型,得到处理后的序列,即为分子标签,如图中B-j------B-l序列,处理后的序列可能与原来序列中某一条相同,也可能与原来的序列都不同。本专利制备的分子标签用于识别DNA或者cDNA文库制备中PCR步骤产生的DNA副本,提高基因变异检测的准确性,2个及以上DNA模板共同支持的变异即为真实变异。以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1