亮氨酸脱氢酶突变体、编码基因、载体、工程菌及其应用的制作方法
本发明涉及亮氨酸脱氢酶突变体,和产亮氨酸脱氢酶基因工程菌及其构建方法和应用,属于基因工程领域。
(二)技术背景
亮氨酸脱氢酶(Leucine dehydrogenase,简称Leu DH,EC 1.4.1.9),能够催化α-丁酮酸转化为L-2-氨基丁酸,是医药生产领域重要的生物催化剂之一,尤其是在生物转化制备L-2-氨基丁酸的工艺中具有关键的作用。
L-2-氨基丁酸(L-ABA)是一种非天然的手性α-氨基酸,是重要的化工原料和医药中间体。例如,用于抗癫痫药左乙拉西坦和抗结核药物盐酸乙胺丁醇的生产。
目前已经有亮氨酸脱氢酶用于生产L-2-氨基丁酸的报道,而且从目前的结果来看,亮氨酸脱氢酶也是该生产工艺中的限制因素。因此突变出新型的高活力的亮氨酸脱氢酶基因,并通过基因工程技术构建高表达的基因工程菌在L-ABA的制备中具有重要的意义。
(三)
技术实现要素:
本发明目的是提供一种亮氨酸脱氢酶突变体、编码基因、重组载体、基因工程菌以及在催化α-丁酮酸制备L-2-氨基丁酸中的应用。
本发明采用的技术方案是:
本发明提供一种亮氨酸脱氢酶突变体,所述突变体是将SEQ ID NO.12所示氨基酸序列(编码基因核苷酸序列如SEQ ID NO.1所示)第41位、61位、77位、347位和358位进行单、双或三位点突变而成。
进一步,所述单突变是将第41位亮氨酸突变为甘氨酸(L41G)、第77位亮氨酸突变为半胱氨酸(L77C)、第61位丙氨酸突变为苏氨酸(A61T)、第347位蛋氨酸突变为甘氨酸(M347G)或第358位谷氨酰胺突变为苏氨酸(Q358T)。所述双突变为M347G/Q358T、M347G/L41G、M347G/L77C;所述三位点突变为M347G/Q358T/L41G、M347G/Q358T/L77C。
更进一步,所述突变体氨基酸序列为下列之一:SEQ ID NO.13,SEQ ID NO.14,SEQ ID NO.15,SEQ ID NO.16,SEQ ID NO.17,SEQ ID NO.18,SEQ ID NO.19,SEQ ID NO.20,SEQ ID NO.21和SEQ ID NO.22。
由于氨基酸序列的特殊性,任何含有SEQ ID NO.12,SEQ ID NO.13,SEQ ID NO.14,SEQ ID NO.15,SEQ ID NO.16,SEQ ID NO.17,SEQ ID NO.18,SEQ ID NO.19,SEQ ID NO.20所示氨基酸序列的多肽的片段或其变体,如其保守性变体、生物活性片段或衍生物,只要该多肽的片段或多肽变体与前述氨基酸序列同源性在95%以上,均属于本发明保护范围之列。具体的,所述改变可包括氨基酸序列中氨基酸的缺失、插入或替换;其中,对于变体的保守性改变,所替换的氨基酸具有与原氨基酸相似的结构或化学性质,如用亮氨酸替换异亮氨酸,变体也可具有非保守性改变,如用色氨酸替换甘氨酸。
本发明还提供一种所述亮氨酸脱氢酶突变体编码基因,所述基因核苷酸序列为下列之一:SEQ ID NO.2,SEQ ID NO.3,SEQ ID NO.4,SEQ ID NO.5,SEQ ID NO.6,SEQ ID NO.7,SEQ ID NO.8,SEQ ID NO.9,SEQ ID NO.10和SEQ ID NO.11。
由于核苷酸序列的特殊性,任何SEQ ID NO.2,SEQ ID NO.3,SEQ ID NO.4,SEQ ID NO.5,SEQ ID NO.6,SEQ ID NO.7,SEQ ID NO.8,SEQ ID NO.9,SEQ ID NO.10、SEQ ID NO.11所示多核苷酸的变体,只要其与该多核苷酸具有90%以上同源性,均属于本发明保护范围之列。所述多核苷酸的变体是指一种具有一个或多个核苷酸改变的多核苷酸序列。此多核苷酸的变体可以使生的等位变异体或非生的变异体,包括取代变异体、缺失变异体和插入变异体。如本领域所知的,等位变异体是一个多核苷酸的替换形式,它可能是一个多个核苷酸的取代、缺失或插入,但不会从实质上改变其编码的氨基酸的功能。
本发明还涉及一种由所述亮氨酸脱氢酶突变体编码基因构建的重组载体。
本发明还涉及一种由所述重组载体转化制备的重组工程菌。
本发明提供一种所述亮氨酸脱氢酶突变体在催化2-丁酮酸异构化制备L-2-氨基丁酸中的应用,具体所述的应用为:以含亮氨酸脱氢酶突变体编码基因的重组工程菌经诱导培养获得的湿菌体分离纯化的纯酶为酶源,以2-丁酮酸为底物,以NADH和氨水为助剂,以pH=7.5、0.02M的磷酸缓冲液为反应介质构成反应体系,在10-50℃、150r/min条件下反应,反应完全后,将反应液分离纯化,获得L-2-氨基丁酸。
进一步,所述反应体系中,底物2-丁酮酸初始浓度为5-200g/L,优选10g/L,酶源用量为500~1000U/L,优选818.7U/L,所述NADH质量用量为5~100g/L,优选70g/L,所述氨水体积终浓度为1~10%,优选1%。
进一步,所述酶源按如下步骤制备:将含亮氨酸脱氢酶突变体编码基因的重组工程菌接种至含终浓度40μg/mL卡那霉素的LB液体培养基,37℃、150r/min培养至OD600=0.8~1.0,获得种子液;将种子液以体积浓度2%的接种量转入含终浓度40μg/mL卡那霉素的LB液体培养基中,于37℃、150r/min条件下培养OD600=0.4~0.8,添加终浓度0.5mM IPTG,28℃、150r/min诱导培养10h,获得诱导培养液,将诱导培养液离心,收集湿菌体,用0.85%生理盐水润洗后,按100g菌体/L生理盐水的比例溶解菌体,振荡均匀后,超声破碎,12000r/min,20min离心,上清为粗酶液,经过镍柱纯化,收集得到纯酶液即为酶源;所述LB液体培养基组成:胰蛋白胨10g/L,酵母粉5g/L,NaCl 10g/L,溶剂为水,pH值为7.0。
本发明还提供一种所述亮氨酸脱氢酶突变体编码基因在制备亮氨酸脱氢酶中的应用,所述的应用为:构建含有所述亮氨酸脱氢酶突变体基因的重组载体,将所述重组载体转化至大肠杆菌中,获得的重组基因工程菌进行诱导培养,取培养液分离得到含亮氨酸脱氢酶突变体的菌体细胞。
更进一步,本发明所述亮氨酸脱氢酶突变体在催化α-丁酮酸制备L-2-氨基丁酸中的应用方法为:
(1)重组菌的筛选:所述重组表达质粒pET-28b-leudh上带有卡那霉素抗性基因,如果重组质粒已经转化到大肠杆菌中,重组菌株具有卡那霉素抗性,可在含有50μg/mL卡那霉素的平板上生长;挑取阳性转化子,即为亮氨酸脱氢酶重组菌BL21/pET-28b-leudh。
(2)亮氨酸脱氢酶突变体表达菌株的构建
以重组菌BL21/pET-28b-leudh的质粒为模板,连续进行两轮突变PCR,将第347位甲硫氨酸突变成了甘氨酸,将第358位谷氨酰胺突变成了苏氨酸,突变质粒经Dpn I酶切模板后转入大肠杆菌中,重组菌株具有卡那霉素抗性,可在含有40μg/mL卡那霉素的平板上生长,挑取阳性转化子测序,序列显示成功突变347位点和358位点,即为亮氨酸脱氢酶突变体菌BL21/pET-28b-mut-leudh。
(3)重组突变体菌经培养表达亮氨酸脱氢酶
LB液体培养基(g/L)组成:蛋白胨10,酵母粉5,NaCl 10,溶剂为水,用1M NaOH将pH调至7.0;LB固体培养基添加20g/L琼脂;高压灭菌;使用前添加终浓度50μg/mL卡那霉素。
将亮氨酸脱氢酶突变体菌接种至终浓度50μg/mL卡那霉素的LB液体培养基,250mL三角瓶,装液量为50mL,培养温度37℃、摇床转速150r/min,培养至OD600=0.8~1.0,获得种子液;将种子液以体积浓度2%的接种量转入装有100mL终浓度50μg/mL卡那霉素的发酵培养基(LB液体培养基)的500mL三角瓶中,于37℃、150r/min条件下培养2~3h(OD600=0.4~0.8),添加终浓度24μg/mL IPTG,28℃、150r/min诱导培养10h,获得诱导培养液。
(4)亮氨酸脱氢酶酶活测定
反应体系为:50mM丁酮酸、50mM氨(以氨水的形式添加,10mL体系添加33μL)0.4mM NADH、dehy经镍柱纯化后的酶液,pH=7.5、0.02M的磷酸缓冲液;将底物、氨水和NADH在35℃预热10min,然后加3mL于石英比色皿中,并迅速加入经镍柱纯化后的酶液10μL,立即使用分光光度计测定OD340值并定为反应0时刻的OD340值。实时监测其OD340下降值。1U酶活定义为35℃下每分钟催化消耗1μmoL NADH所需的酶量。
本发明所述亮氨酸脱氢酶突变体能够在底物浓度为360g/L、反应温度35℃时酶活达到414.3U/g(野生型亮氨酸脱氢酶酶活为104.4U/g),转化率达到99%。
本发明所用氨水通常指质量浓度25-28%的氨水水溶液。
与现有技术相比,本发明有益效果主要体现在:
本发明构建了一株高效表达亮氨酸脱氢酶突变体的重组大肠杆菌,提高了亮氨酸脱氢酶的酶活;应用该菌种生产亮氨酸脱氢酶具有产量高、工艺简单、便于工业化应用等优点,该菌株可以用于L-2-氨基丁酸的生产,发酵结束后在35℃条件下对α-丁酮酸的总酶活达到414.3U/g,转化率达到99%,这为L-2-氨基丁酸的大规模生产以及其作为重要的医药中间体提供了良好的技术支持。
(四)附图说明
图1 leudh基因片段核酸电泳图,泳道2mark,泳道1,3leudhPCR扩增产物。
图2质粒pET-28b-leudh物理图谱。
图3野生型菌SDS-PAGE电泳图,泳道1,2破碎液沉淀中蛋白,泳道3,4破碎液上清中的蛋白。
图4温度对M347G/Q358T活力的影响。
图5金属离子对M347G/Q358T活力的影响。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例1:亮氨酸脱氢酶的基因合成
亮氨酸脱氢酶的基因来源于中间型嗜热放线菌(Thermoactinomyces intermedius)的全基因组序列。为了使该基因连接到载体pET-28b后可以表达带有His-tag的蛋白,切除其终止密码子,并以B.subtilis 168的密码子偏好性为参照对其序列和常用限制性内切酶识别位点BamH I、Xho I、Pst I、Hind III和Nco I进行序列优化,新设计的亮氨酸脱氢酶基因(leudh)的序列如SEQ ID NO.1所示,编码氨基酸的序列为SEQ ID NO.12所示,基因合成工作委托上海旭冠生物科技发展有限公司完成,基因合成后连接到克隆载体pET28b中。
实施例2:亮氨酸脱氢酶重组大肠杆菌的构建
在实施例1的基础上,利用PCR技术,以合成的核苷酸序列(SEQ ID NO.1所示)为模板,以leudh-F:5’-CGTACGCTTCCGAANNNGAGGCGATTGAAG-3’和leudh-R:5’-CTTCAATCGCCTCNNNTTCGGAAGCGTACG-3’为引物,对亮氨酸脱氢酶基因(leudh)进行扩增,并在其5'端和3'分别引入Nco I和Xho I限制性酶切位点。PCR反应体系(50μL)为:10×Taq polymerase buffer 5μL,dNTP Mixture 4μL;模板DNA 1μL;上下游引物各0.5μL;Taq polymerase DNA聚合酶0.5μL;无菌水38.5μL。PCR反应的程序为94℃预变性5min;94℃变性45s,50℃退火45s,72℃延伸1min(30个循环);72℃延伸10min。以1%琼脂糖凝胶电泳验证并以0.8%琼脂糖凝胶电泳回收PCR扩增产物,结果扩增到与SEQ ID No.1序列大小相符的leudh基因片段(图1中泳道1、3)。
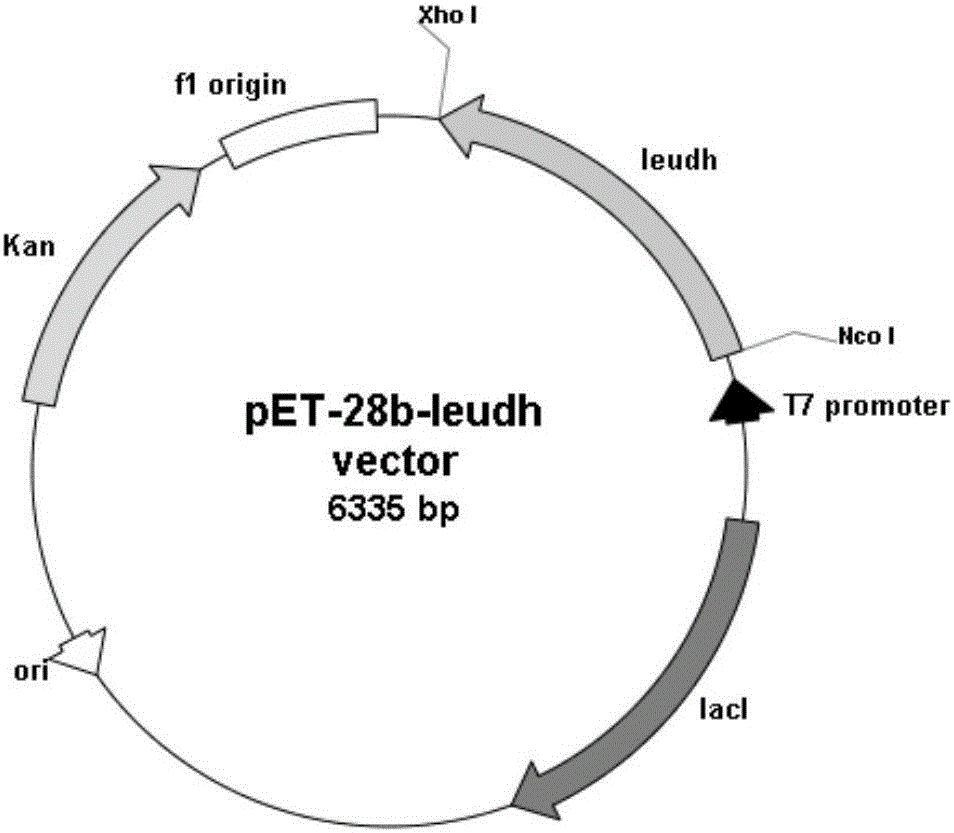
用限制性内切酶NdeI和Bgl II双酶切纯化后的leudh基因(即回收的PCR扩增产物)和载体pET-28b(+),用T4DNA连接酶于16℃过夜连接。连接反应体系为(20μL):目的基因片段12μL,载体DNA 5μL,10×T4DNA连接酶buffer 2μL,T4 DNA连接酶1μL。连接产物转化大肠杆菌BL21感受态细胞,转化方法为:取连接产物5~10μL,加入到100μL大肠杆菌BL21感受态细胞中,充分混匀后,冰浴30min;将装有混合物的Eppendorf管置于42℃水浴热休克90s,然后立即转移到冰上冷却2min;向管中加入600μL LB液体培养基,置于37℃、150r/min恒温摇床上培养2~4h,然后涂布于含有50μg/mL卡那霉素的LB平板上,37℃培养12~18h。挑取阳性克隆,37℃、150r/min振荡培养过夜,提取质粒,测序验证后,得到正确构建的原核表达质粒pET-28b-leudh,其物理图谱见图2。含有质粒pET-28b-leudh的阳性克隆为正确构建的重组大肠杆菌BL21/pET-28b-leudh,命名为LEUDH。
实施例3:亮氨酸脱氢酶单位点突变体的构建
将实施例2中构建的重组大肠杆菌BL21/pET-28b-leudh提取质粒,以质粒pET-28b-leudh作为模板,根据亲本序列设计六对定点突变的突变引物。
L41G-F:5′-CCCTGGGTCCGGCGGTTGGTGGTATGCGTATG-3′和
L41G-R:5′-CATACGCATACCACCACCAGCCGGACCCAGGG-3′;
A61T-F:5′-GATTGAAGATGCTCTACTCTGGGTCGTGGCATGAC-3′和
A61T-R:5′-GTCATGCCACGACCCAGAGTCAGAGCATCTTCAATC-3′;
L77C-F:5’-GCTGCAGGTCTGAACTGTGGTGGCGGCAAAACC-3’和
L77C-R:5’-GGTTTTGCCGCCACCACAGTTCAGACCTGCAGG-3’;
M347G-F:5’-GAACGTATCGAAATGGGTCGTAAGACCCGCAGAAAGGTG-3’和
M347G-R:5’-GTGCTGCGGGTCTTACGNNNCAACCCGATACGATACGTTC-3’;
Q358T-F:5’-CACCTTTCTGCAGGACACTCGTAACCTGATCAAC-3’和
Q358T-R:5’-GTGCTGCGGGTCTTACGAGTCATTTCGATACGTTC-3’(下划线为突变碱基)利用PCR技术进行突变。PCR反应体系为:2× GC Buffer 25μL,dNTPs(各2.5mmol/L)4μL,正向引物(10μM)1μL,反向引物(10μM)1μL,模板DNA 1μL, HS DNA Polymerase(2.5U/μL)0.5μL,加入双蒸水至50μL。PCR扩增程序为:98℃预变性3min;随后进行30个循环(98℃ 10s,58℃ 15s,72℃ 7min);72℃延伸10min;最后4℃保温。PCR产物经0.8%琼脂糖凝胶电泳验证,结果扩增到与目的载体大小相符的基因片段。
PCR产物经过Dpn I酶切模板3h后,转化大肠杆菌BL21(DE3)感受态细胞(具体过程见实施例2),感受态细胞在LB固体培养基(含有卡那霉素40μg/mL)培养过夜后,挑单克隆进行菌落PCR验证,验证为阳性克隆的菌接种于LB液体培养基(含有卡那霉素40μg/mL)中培养8h后,提取质粒,送上海桑尼生物科技有限公司测序,测序结果显示正确,构建的单突变体亮氨酸脱氢酶分别命名为LeuDH-L41G,LeuDH-L77C,LeuDH-A61T,LeuDH-M347G,LeuDH-Q358T。
实施例4:亮氨酸脱氢酶双位点突变体的构建
将实施例2中构建的重组大肠杆菌BL21/pET-28b-leudh提取质粒,以质粒pET-28b-leudh作为模板,根据亲本序列设计定点突变的突变引物M347G-F:5’-GAACGTATCGAAATGGGTCGTAAGACCCGCAGAAAGGTG-3’和M347G-R:5’-GTGCTGCGGGTCTTACGNNNCAACCCGATACGATACGTTC-3’,(下划线为突变碱基)利用快速PCR技术进行第一轮突变。再以引入Q358T、L41G、L77C突变点为模板进行第二轮突变,引物为Q358T-F:5’-CACCTTTCTGCAGGACACTCGTAACCTGATCAAC-3’和Q358T-R:5’-GTGCTGCGGGTCTTACGAGTCATTTCGATACGTTC-3’;L41G-F:5′-CCCTGGGTCCGGCGGTTGGTGGTATGCGTATG-3′和L41G-R:5′-CATACGCATACCACCACCAGCCGGACCCAGGG-3′;L77C-F:5’-GCTGCAGGTCTGAACTGTGGTGGCGGCAAAACC-3’和L77C-R:5’-GGTTTTGCCGCCACCACAGTTCAGACCTGCAGG-3’。PCR反应体系为:2× GC Buffer 25μL,dNTPs(各2.5mmol/L)4μL,正向引物(10μM)1μL,反向引物(10μM)1μL,模板DNA 1μL, HS DNA Polymerase(2.5U/μL)0.5μL,加入双蒸水至50μL。PCR扩增条件为:98℃预变性3min;随后进行30个循环(98℃ 10s,58℃ 15s,72℃ 7min);72℃延伸10min;最后4℃保温。PCR产物经0.8%琼脂糖凝胶电泳验证,结果扩增到与目的载体大小相符的基因片段。
PCR产物经过Dpn I酶切模板3h后,转化大肠杆菌BL21(DE3)感受态细胞(具体过程见实施例2),感受态细胞在LB固体培养基(含有卡那霉素40μg/mL)培养过夜后,挑单克隆进行菌落PCR验证,验证为阳性克隆的菌接种于LB液体培养基(含有卡那霉素40μg/mL)中培养8h后,提取质粒,送上海桑尼生物科技有限公司测序,测序结果显示正确,构建的双突变体亮氨酸脱氢酶命名为LeuDH-M347G/Q358T、LeuDH-M347G/L41G、LeuDH-M347G/L77C。
实施例5:亮氨酸脱氢酶三位点突变体的构建
将实施例2中构建的重组大肠杆菌BL21/pET-28b-leudh提取质粒,以质粒pET-28b-leudh作为模板,根据亲本序列设计定点突变的突变引物M347G-F:5’-GAACGTATCGAAATGGGTCGTAAGACCCGCAGAAAGGTG-3’和M347G-R:5’-GTGCTGCGGGTCTTACGNNNCAACCCGATACGATACGTTC-3’,(下划线为突变碱基)利用快速PCR技术进行第一轮突变。再以引入Q358T突变点为模板进行第二轮突变,引物为Q358T-F:5’-CACCTTTCTGCAGGACACTCGTAACCTGATCAAC-3’和Q358T-R:5’-GTGCTGCGGGTCTTACGAGTCATTTCGATACGTTC-3’。再以引入L41G、L77C突变点为模板进行第三轮突变,引物为L41G-F:5′-CCCTGGGTCCGGCGGTTGGTGGTATGCGTATG-3′和L41G-R:5′-CATACGCATACCACCACCAGCCGGACCCAGGG-3′;L77C-F:5’-GCTGCAGGTCTGAACTGTGGTGGCGGCAAAACC-3’和L77C-R:5’-GGTTTTGCCGCCACCACAGTTCAGACCTGCAGG-3’。PCR反应体系为:2× GC Buffer 25μL,dNTPs(各2.5mmol/L)4μL,正向引物(10μM)1μL,反向引物(10μM)1μL,模板DNA 1μL, HS DNA Polymerase(2.5U/μL)0.5μL,加入双蒸水至50μL。PCR扩增条件为:98℃预变性3min;随后进行30个循环(98℃ 10s,58℃ 15s,72℃ 7min);72℃延伸10min;最后4℃保温。PCR产物经0.8%琼脂糖凝胶电泳验证,结果扩增到与目的载体大小相符的基因片段。
PCR产物经过Dpn I酶切模板3h后,转化大肠杆菌BL21(DE3)感受态细胞(具体过程见实施例4),感受态细胞在LB固体培养基(含有卡那霉素40μg/mL)培养过夜后,挑单克隆进行菌落PCR验证,验证为阳性克隆的菌接种于LB液体培养基(含有卡那霉素40μg/mL)中培养8h后,提取质粒,送上海桑尼生物科技有限公司测序,测序结果显示正确,构建的三突变体亮氨酸脱氢酶命名为LeuDH-M347G/Q358T/L41G、LeuDH-M347G/Q358T/L77C。
实施例6:野生型重组大肠杆菌的表达
将实施例2中构建的野生型亮氨酸脱氢酶LeuDH菌株接种至50mL LB液体培养基中(含有卡那霉素40μg/mL),37℃、150r/min振荡培养到OD600=0.8~1.0;培养液以2%(v/v)接种量接种到新鲜的含有40μg/mL卡那霉素的100mL LB液体培养基中,37℃、150r/min振荡培养至菌体浓度OD600=0.6~0.8,加入终浓度0.5mM IPTG,28℃、150r/min诱导培养10h,收集菌体细胞,获得野生型亮氨酸脱氢酶湿菌体LeuDH。取一定量菌体,用0.85%(w/v)生理盐水润洗一遍菌体后,按100g菌体/L生理盐水的比例溶解菌体,振荡均匀后,超声破碎,12000r/min,20min离心,上清为粗酶液,经过镍柱纯化,收集得到纯酶液。
诱导培养10h后,取20μL菌液,加20μL SDS缓冲液混合,沸水浴加热5min,取8μL进行SDS-PAGE电泳分析,得到一条分子量约为40kDa的蛋白条带(见图3)。
实施例7亮氨酸脱氢酶突变体工程菌的构建
将实施例3-5中构建的亮氨酸脱氢酶突变体菌株分别接种至50mL LB液体培养基中(含有卡那霉素40μg/mL),37℃、150r/min振荡培养到OD600=0.8~1.0;培养液以2%(v/v)接种量接种到新鲜的含有40μg/mL卡那霉素的100mL LB液体培养基中,37℃、150r/min振荡培养至菌体浓度OD600=0.6~0.8,加入终浓度0.5mM IPTG,28℃、150r/min诱导培养10h,于4℃、9000r/min离心10min,收集湿菌体,取一定量菌体,用0.85%(w/v)生理盐水润洗一遍菌体后,按100g菌体/L生理盐水的比例溶解菌体,振荡均匀后,超声破碎,12000r/min,20min离心,上清为粗酶液,经过镍柱纯化,收集得到纯酶液,分别为亮氨酸脱氢酶突变体工程菌LeuDH-L41G,LeuDH-L77C,LeuDH-A61T,LeuDH-M347G,LeuDH-Q358T,LeuDH-M347G/Q358T、LeuDH-M347G/L41G、LeuDH-M347G/L77C,LeuDH-M347G/Q358T/L41G、LeuDH-M347G/Q358T/L77C。
实施例8:亮氨酸脱氢酶及其突变体的酶活测定
取实施例6中野生型亮氨酸脱氢酶LeuDH纯酶液,实施例7中单突变体亮氨酸脱氢酶LeuDH-L41G,LeuDH-L77C,LeuDH-A61T,LeuDH-M347G,LeuDH-Q358T,实施例7中双突变体亮氨酸脱氢酶LeuDH-M347G/Q358T、LeuDH-M347G/L41G、LeuDH-M347G/L77C,实施例7中三突变体亮氨酸脱氢酶LeuDH-M347G/Q358T/L41G、LeuDH-M347G/Q358T/L77C纯酶液,测定该重组菌及突变体菌对α-丁酮酸的酶活。
酶活测定方法为:反应于1.5mL Ep管中进行,1mL反应体系:0.01g丁酮酸,0.07gNADH,10μL氨水,加0.02M的磷酸缓冲液(pH=7.5)至900μL,100μL纯酶液,温度35℃,转数200rpm,反应1h,加20μL 6M HCl终止反应,8 000rpm离心10min,0.22μm滤膜过滤后用氨基酸分析仪方法检测L-ABA的含量。
酶活单位定义:在35℃,pH 7.5条件下,每分钟产生1μmol L-2-氨基丁酸所需的酶蛋白量,即为一个亮氨酸脱氢酶活力单位,以U表示。亮氨酸脱氢酶的比酶活以每g湿菌体所含有的酶活力单位表示(U/g),结果见表1。亮氨酸脱氢酶酶突变体LeuDH-Q358T活力是野生型LeuDH的2倍,达到965.7U/g。
表1 亮氨酸脱氢酶酶及其突变体酶活力
实施例9:亮氨酸脱氢酶及其突变体的动力学常数测定
取实施例6野生型亮氨酸脱氢酶LeuDH纯酶,实施例7中单突变体亮氨酸脱氢酶LeuDH-L41G,LeuDH-L77C,LeuDH-A61T,LeuDH-M347G,LeuDH-Q358T,实施例7中双突变体亮氨酸脱氢酶LeuDH-M347G/Q358T、LeuDH-M347G/L41G、LeuDH-M347G/L77C,实施例7中三突变体亮氨酸脱氢酶LeuDH-M347G/Q358T/L41G、LeuDH-M347G/Q358T/L77C纯酶液用于催化底物丁酮酸测定动力学常数。测定仪器选用酶标仪,反应在96黑孔板中进行,因此体系较小为100μL体系,固定NADH/氨水溶液浓度为0.5M,用pH=7.5、0.02M的磷酸缓冲液配浓度为0.25、0.2、0.1、0.05、0.04、0.025、0.02、0.01M的丁酮酸溶液。每个孔中分别加入不同浓度的丁酮酸溶液和固定浓度的NADH/氨水溶液50μL,加完后酶标仪中震荡均匀,迅速加入5μL亮氨酸脱氢酶或其突变体纯酶液,震荡后开测。提前设定好程序,测10分钟的荧光值。
表2 亮氨酸脱氢酶及其突变体酶动力学常数
实施例10:亮氨酸脱氢酶及其突变体催化制备L-2-氨基丁酸最适温度测定
以野生型LeuDH作为对照,对双突变体LeuDH-M347G/Q358T催化制备L-2-氨基丁酸进行探索。将实施例6中6野生型亮氨酸脱氢酶LeuDH纯酶,实施例7中双突变体LeuDH-M347G/Q358T纯酶液作为转化用酶,以丁酮酸为底物,进行转化反应制备L-2-氨基丁酸。具体操作如下:在1mL反应体系条件下,0.01g丁酮酸,0.07gNADH,10μL氨水,加0.02M的磷酸缓冲液(pH=7.5)至900μL,在20℃、25℃、30℃、35℃、40℃、45℃、50℃水浴锅中预热5分钟,再向里面加入100μL(8178U/L)野生型酶或突变酶的酶液。在不同反应温度20℃、25℃、30℃、35℃、40℃、45℃、50℃下,150rpm反应8h,测定酶的活力。将最适反应温度下的酶活设为100%。
实施例11:亮氨酸脱氢酶及其突变体热稳定性测定
以野生型LeuDH作为对照,对双突变体LeuDH-M347G/Q358T催化制备L-2-氨基丁酸进行探索。将实施例6中野生型亮氨酸脱氢酶LeuDH纯酶,实施例7中双突变体LeuDH-M347G/Q358T纯酶液作为转化用酶,以丁酮酸为底物,进行转化反应制备L-2-氨基丁酸。具体操作如下:在1mL反应体系条件下,0.01g丁酮酸,0.07gNADH,10μL氨水,加0.02M的磷酸缓冲液(pH=7.5)至900μL,将原始酶和突变酶的纯酶液(8178U/L)分别保温在30℃、40℃、50℃水浴中,并且每隔24h取100μL酶液测定其残余酶活。通过计算得到亮氨酸脱氢酶原始菌株在30℃、40℃和50℃保存条件下的半衰期分别为12.83d,16.00d和1.76d;突变菌株M347G/Q358T在30℃、40℃和50℃保存条件下的半衰期分别为17.02d,19.69d和2.01d。实验结果表明在40℃条件下,亮氨酸脱氢酶的热稳定性要好于30℃的保存条件,但在50℃的保存条件下又变的非常的不稳定。通过此实验还发现突变体的热稳定性相比于原始菌株提高了32%。
实施例12:二价金属离子对亮氨酸脱氢酶及其突变体的影响
以野生型LeuDH作为对照,对双突变体LeuDH-M347G/Q358T催化制备L-2-氨基丁酸进行探索。将实施例6野生型亮氨酸脱氢酶LeuDH纯酶,实施例7双突变体LeuDH-M347G/Q358T纯酶液作为转化用酶,以丁酮酸为底物,进行转化反应制备L-2-氨基丁酸。具体操作如下:在0.02M、pH 7.5的磷酸缓冲液中1mL反应体系:0.01g丁酮酸,0.07gNADH,10μL氨水,加缓冲液至880μL,再加100μL原始酶或突变酶的酶液(8178U/L),20μL已配好的浓度为100mM的Cu2+,Co2+,Ni2+,Mn2+,Ca2+,Mg2+,Ba2+,Zn2+,Fe3+,Fe2+,EDTA水溶液,在35℃水浴摇床中,反应8h,测定酶的活力。将未添加金属离子所得到的酶活设为100%。金属离子和表面活性剂对亮氨酸脱氢酶酶活的正影响变化不大,无论是突变前还是突变后。Cu2+对原始酶及突变酶的酶活有较轻微的抑制作用,而Zn2+对突变后的酶活具有抑制作用。
由上述实验结果可知,将本发明亮氨酸脱氢酶突变体LeuDH-M347G,TLeuDH-Q358T,LeuDH-M347G/Q358T基因转化大肠杆菌得到的重组大肠杆菌都具有较强的产L-2-氨基丁酸能力。
- 还没有人留言评论。精彩留言会获得点赞!