一种含芴紫外吸收剂及其制备方法与流程
【技术领域】
本发明属于紫外吸收剂及其制备领域,特别涉及一种具有紫外光吸收能力的芴类衍生物及其制备方法。
背景技术:
紫外线是指波长为200nm~400nm范围的射线,近年来,随着大气层污染的日益加重,致使地球臭氧层遭到严重破坏,到达地面的紫外线具有高能特性,长时间暴露在紫外线下,高聚物会老化、有机物会分解、皮肤会发生病变。因此,紫外线吸收剂成为人们研究的热点材料之一。紫外线吸收剂是一类能够强烈的选择性地吸收高能量紫外光,并进行能量转换,以热能形式或无害的低辐射将能量释放或消耗的物质。
目前主要有无机和有机紫外线吸收剂。无机紫外线吸收剂主要有二氧化钛、氧化锌等,品种较少。有机紫外线吸收剂由于其原料来源广泛,多品种,成为目前发展的主要方向。目前广泛使用的有机紫外吸收剂基本以水杨酸酯类、苯并三唑类和三嗪类等有机化合物。它们都有各自的优缺点,以水杨酸酯类为例,其优点是成本低廉、与树脂等高分子材料有良好的相容性,缺点是对紫外光的吸收性能较差,效率不高,而且只有窄的吸收波段(340nm以下),紫外线对其又有明显的老化作用,光照后分子内部结构会重排,重排之后的化合物对光有显著的吸收,使制品带色。因此从一种新的分子角度合成一种新型吸光材料,对填补已有材料的空缺实现优势互补。
芴类化合物具有较高的光热稳定性,芴上2,7号位以及9号位碳上氢具有较高的反应活性,可方便地进行反应,通过结构修饰引入多种官能团得到一系列芴类衍生物。目前国内外已开始出现芴类化合物在紫外吸收剂方面的应用,例如中国发明cn102558555b公开了一种含芴聚三唑耐高温紫外吸收剂及其制备方法。本发明提供一种新的芴类衍生物。
技术实现要素:
本发明所要解决的技术问题是提供一种含芴紫外吸收剂能在保持吸光性能的同时猝灭荧光,保持产品无色透明的特性。
本发明提供一种含芴紫外吸收剂,其化学式为:9,9’-二正丁基-2,7-二(2-溴噻吩)芴,分子式为:c29h28br2s2,结构式为:
其中x1=br,x2=br。
本发明进一步地公开了一种含芴紫外吸收剂的制备方法,步骤包括:
1、设计产物合成路线:
2、合成中间产物:
芴与过量的98%纯度的一溴丁烷在由50%质量分数的氢氧化钠水溶液提供的碱性环境中在二甲基亚砜溶剂中进行烷基化反应,室温搅拌7小时;
当反应完全后将反应液用分析纯级别的乙酸乙酯萃取三次,将所得有机层用饱和食盐水洗涤三次后,加入过量无水硫酸钠干燥,用倾析法倒出有机溶液,加入硅胶粉,旋转蒸发仪蒸干;
随后用硅胶装层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到白色固体产物9,9’-二正丁基芴,产率为86%;
将9,9’-二正丁基芴与n-溴代琥珀酰亚胺(nbs)(在分析纯的氯仿溶剂中以1:2.1的摩尔量比例投料进行溴化反应,保持冰浴0oc下搅拌约1小时,然后升至室温搅拌7小时,薄层层析板(tlc)监测反应,当反应完全后,加入硅胶粉,将反应液用旋转蒸发仪蒸干,随后用硅胶层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到白色固体产物9,9’-二正丁基-2,7-二溴芴,产率为92%;
将9,9’-二正丁基-2,7-二溴芴和2-硼酸噻吩按照1:3的摩尔比,溶解在分析纯thf中,并加入10%摩尔比的四三苯基膦钯和2mk2co3水溶液,在氮气的气氛下加热搅拌至70℃,进行铃木偶合反应48h,用tlc板监测反应程度,直至反应完全,将最后所得反应液用分析纯的乙酸乙酯萃取,饱和食盐水洗涤,浓缩蒸发,以正己烷为淋洗剂,硅胶层析柱分离提纯,得到固体中间产物9,9’-二正丁基-2,7-二噻吩芴(c1,分子式:c29h30s2),产率为69%)。
3、溴代产物的合成
将中间产物c1和0.114g0.64mmolnbs分别按照摩尔比1:2投入分析纯的氯仿中,保持冰浴0oc下搅拌约1小时,然后升至室温搅拌7小时,薄层层析板(tlc)监测反应,当反应完全后,加入硅胶粉,将反应液用旋转蒸发仪蒸干,随后用硅胶层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到双溴代的产物9,9’-二正丁基-2,7-二(2-溴噻吩)芴(c2,分子式:c29h28br2s2),产率为89.6%。
有益效果:
本发明在芴的9号位上接上两个烷基可提高分子在有机溶剂中的溶解,同时可改善材料的旋涂成膜性能,引入溴原子后依托溴的重原子效应来猝灭荧光,达到仅吸收紫外光而不吸收可见光,可很好地保持无色透明的特性。
【附图说明】
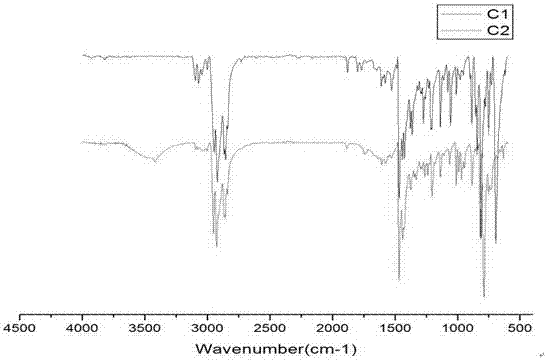
图1为中间产物c1和最终产物c2傅里叶红外光谱图;
图2为中间产物c1质谱图;
图3为最终产物c2质谱图;
图4为最终产物c2的核磁共振氢谱图;
图5a为中间产物c1和最终产物c2二氯甲烷溶液紫外可见光吸收光谱图;
图5b为中间产物c1和最终产物c2薄膜紫外可见光吸收光谱图;
图6a为为中间产物c1和最终产物c2二氯甲烷溶液分子荧光发射光谱图;
图6b为为中间产物c1和最终产物c2薄膜分子荧光发射光谱图。
【具体实施方式】
下面结合附图与具体实施例对本发明作进一步详细说明,但本发明保护范围不局限于所述内容。实施例中使用的试剂、仪器和方法,如无特殊说明,均采用常规试剂、仪器和使用常规方法。
实施例:
1、设计产物合成路线:
2、合成中间产物:
0.1g(0.62mmol)的芴与过量的98%纯度的一溴丁烷1ml在由50%质量分数的氢氧化钠水溶液2ml提供的碱性环境中在二甲基亚砜溶剂中进行烷基化反应,室温搅拌7小时;
当反应完全后将反应液用分析纯级别的乙酸乙酯萃取三次,将所得有机层用饱和食盐水洗涤三次后,加入过量无水硫酸钠干燥,用倾析法倒出有机溶液,加5g200--300目的硅胶粉,旋转蒸发仪蒸干;
随后用30g200--300目的硅胶装层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到白色固体产物9,9’-二正丁基芴;
将9,9’-二正丁基芴与n-溴代琥珀酰亚胺(nbs)(98%纯度,0.198g,约1.1mmol)在分析纯的氯仿溶剂中以1:2.1的摩尔量比例投料进行溴化反应,保持冰浴0oc下搅拌约1小时,然后升至室温搅拌7小时,薄层层析板(tlc)监测反应,当反应完全后,加入5g200--300目的硅胶粉,将反应液用旋转蒸发仪蒸干,随后用200-300目的硅胶层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到白色固体产物9,9’-二正丁基-2,7-二溴芴;
将9,9’-二正丁基-2,7-二溴芴和2-硼酸噻吩(0.19g,约1.5mmol)按照1:3的摩尔比,溶解在50ml的分析纯thf中,并加入0.06g10%摩尔比的四三苯基膦钯和10ml2mk2co3水溶液,在氮气的气氛下加热搅拌至70℃,进行铃木偶合反应48h,用tlc板监测反应程度,直至反应完全,将最后所得反应液用分析纯的乙酸乙酯萃取,饱和食盐水洗涤,浓缩蒸发,以正己烷为淋洗剂,硅胶层析柱分离提纯,得到固体中间产物9,9’-二正丁基-2,7-二噻吩芴(c1,分子式:c29h30s2)。
3、溴代产物的合成
将中间产物c1和0.114g0.64mmolnbs分别按照摩尔比1:2投入分析纯的氯仿中,保持冰浴0oc下搅拌约一小时,然后升至室温搅拌七小时,薄层层析板(tlc)监测反应,当反应完全后,加入5g200--300目的硅胶粉,将反应液用旋转蒸发仪蒸干,随后用200-300目的硅胶层析柱,以分析纯的正己烷为淋洗剂进行分离纯化,得到双溴代的产物9,9’-二正丁基-2,7-二(2-溴噻吩)芴(c2,分子式:c29h28br2s2)。
4、产物的红外光谱分析:
将中间产物c1和最终产物c2与溴化钾研磨压片后,测定各自的红外光谱,得到的谱图如图1所示,图中波长3001cm‒1附近的峰可以归属为噻吩环及苯环上c‒h的伸缩振动,2858‒2950cm‒1之间的峰应是烷基链上ch2或者ch3的c‒h伸缩振动,1467‒1605cm‒1应该归属于噻吩环或苯环上c꞊꞊c对称伸缩振动及c꞊꞊c不对称伸缩振动,1200cm‒1左右的峰对应的是噻吩环上c‒h的面内弯曲振动,850cm‒1附近的峰应该是由噻吩环上c‒h的面外弯曲振动引起。c‒br的伸缩振动峰应该位于约600cm‒1处,但由于噻吩环及苯环上取代基所致指纹区振动峰的覆盖,所以红外光谱图中对比不太明显。
5、质谱图和核磁共振氢谱图分析:
c1、c2的质谱图如图2、图3所示,c2的核磁共振氢谱图如图4所示,c1的质谱ms(maldi-tof):m/z442.1787[m+h]+.c2的质谱ms(quadrupole):m/z600.99[m+h]+.c2的核磁共振图谱:1hnmr(cdcl3):δ(ppm)7.6637-7.6835(d,2h,ar),7.5122-7.5162(d,2h,ar),7.4965-7.4962(d,2h,ar),7.0530-7.0626(d,2h,thiophene),7.119-7.129(d,2h,thiophene),0.611-0.695(m,10h,butyl),1.063-1.099(m,4h,butyl),1.976-2.017(m,4h,butyl)。
6、产物紫外吸收光谱分析:
将4.4mg的c1和6.0mg的c2分别溶解于100ml分析纯二氯甲烷中,分别获得了c1和c2约10-5m的溶液,并在室温下测定了相应的紫外可见光吸收光谱,归一化处理后如图5a所示。化合物c1的吸收范围最大347–360nm之间,推断该吸收峰可能归属于π-π*的跃迁吸收。其中c2的最大紫外吸收峰出现在361nm,由两曲线对比可知当c1引入了两个br原子后,c2的紫外吸收光谱发生了轻微的红移现象,并且最大吸收峰收窄。当把化合物溶于二氯甲烷中,并将溶液涂抹于石英玻璃片后烘干,即制备得到相应的化合物薄膜,其归一化处理后的紫外可见光吸收光谱如图5b所示。c1的薄膜对紫外光吸收集中300nm–400nm之间,当引入溴原子后,200–280nm之间出现了一个吸收带,而在320–350nm之间出现了强吸收峰,这些吸收峰可能是来自化合物本身π-π*的电子跃迁。这些吸收范围覆盖了uvb和uva两个波段的紫外光区,而且吸收光谱局限在420nm以内,表明了这两个化合物仅对紫外光有良好的吸收,但对可见光的吸收很少,因此,这两个固体薄膜基本为无色透明材料。
7、分子荧光发射光谱分析:
按照上述紫外可见光吸收光谱的方法制备化合物的二氯甲烷溶液(浓度一致约为10-5m)及薄膜,室温下以356nm紫外光为激发波长,得到的光致发光光谱图。如图6a所示,c1的二氯甲烷溶液有强烈的紫蓝色荧光发射,最大发射峰位于407nm,在395nm处还有一个强发射峰,可归属为π*-π的跃迁发射。当引入两个溴原子后,c2的荧光发射强度就剧烈下降,在同等浓度下,其发光相对强度还不及c1的10%,证明了溴原子这类重原子的存在能有效淬灭目标化合物的荧光发射。在薄膜状态下,以365nm紫外光的激发波长激发,化合物的荧光强度出现了减小的现象,最大荧光发射峰出现在约418nm处,而且分子结构中噻吩环的相对振动被抑制,所以仅保留一个从350–460nm之间的发射峰,因此推断其荧光颜色仍为紫蓝色如图6b。在引入溴原子进行荧光猝灭后,c2的相对荧光强度基本都低于c1的10%,说明在薄膜状态下,溴原子的引入也很好地猝灭了荧光。
如上述实施例所述,通过对芴基两侧接上噻吩基团后的产物利用双光束紫外可见光分光光度计分析,结果表明其紫外吸收波段覆盖了大部分uva波段,最大吸收峰吸收范围在347–360nm之间,引入溴原子后最大紫外吸收峰出现在361nm,相对中间产物有红移,且最大吸收峰收窄。溶液和薄膜的紫外表征结果表明所合成的产物是理想的吸收uva波段紫外光的材料。对未上溴及上双边溴之后的最终产物利用荧光分光光度计测试其荧光产率,表明引入溴原子进行修饰后,在保持其紫外光吸收的强度和范围的同时,大幅减弱了其荧光的发射强度,上双边溴之后的最大荧光发射效率不及未上溴前的10%,表明对芴基材料进行溴化后,其吸光性能得到保持的同时溴原子的存在很好地猝灭了荧光。
虽然以上描述了本发明的具体实施方式,但是熟悉本技术领域的技术人员应当理解本案所描述的具体实施例只是说明性的,而不是用于对本发明的范围的限定,熟悉本领域的技术人员在依照本发明的精神所作的等效的修饰以及变化,都应当涵盖在本发明的权利要求所保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!