一种磁性多孔分子印迹聚合物的制备方法与流程
本发明涉及一种印迹聚合物的制备方法,尤其涉及一种制备磁性多孔分子印迹吸附剂的制备方法,属环境功能材料制备技术领域。
背景技术:
三氟氯氰菊酯(LC)是一种广谱性且低毒高效的杀虫剂,被广泛应用于农业中。然而使用过程中无意识的释放及残存在自然环境中的LC,会持续的污染河流及地下水。自然环境中检测到的LC在进入人体后会对神经系统、基因、免疫系统等造成不同程度的影响,且具有一定的内分泌干扰效应。因此,需要一种高效快速的方法来去除LC。
吸附是去除环境中残留的有机农药最简单有效的方法,然而在实际应用过程中选择性有待提高。分子印迹技术是一种用于制备分子印迹聚合物(Molecular Imprinted Polymers,MIPs)的技术,这种MIPs能对目标物有选择性地快速有效去除,并且能够重复利用。通过传统方法,例如沉淀聚合,通过将功能单体,模板分子,交联剂,引发剂溶解在惰性溶剂中引发聚合后,洗脱模板分子制备的MIPs由于大多结合位点包埋过深,印迹分子进入困难,导致吸附能力差,吸附亲和力低,这限制的MIPs的使用范围。而多孔材料突出的孔结构和超大的比表面积在吸附方面有极高的应用价值,制备多孔MIPs用于快速吸附分离LC具有极佳的应用前景。
Pickering高内相乳液模板法是制取多孔材料的理想的方法。同时使用固体粒子和乳化剂来稳定Pickering高内相乳液,制备的多孔材料机械性能强,溶液渗透率高。将 Pickering高内相乳液模板法与分子印迹技术相结合制备出分子印迹多孔聚合物用于吸附,具有吸附效率高,吸附速率快,可重复使用等优点。Pan等(Pan J.M., Ma Y., Zeng J., Niu X.H.et al. Facile assembly of hollow polydopamine capsules onto macroporous poly(glycidyl methacrylate) foams for simultaneous removal of λ-cyhalothrin and copper ions. Chem. Eng. J. 2016,302:670-681. )基于Pickering高内相乳液模板法,采用中空多巴胺纳米粒子作为稳定粒子制备了聚甲基丙烯酸缩水甘油酯分子印迹聚合物用于同时去除LC和铜离子。然而,这些分子印迹聚合物通常需要通过离心或者过滤来分离回收,过程繁琐耗时。而具有磁性的分子印迹聚合物,往往可以通过外加磁场来分离,方便快捷。使用具有磁性的纳米粒子作为稳定粒子来稳定Pickering高内相乳液制备具有磁性的分子印迹多孔聚合物的工作还未见报道。
因此,本发明通过使用磁性纳米颗粒作为Pickering高内相乳液的稳定粒子,制备了具有磁性的多孔分子印迹聚合物(MMIPs),可以通过外加磁场快速分离,并以此作为吸附剂用于选择吸附分离三氟氯氰菊酯。
技术实现要素:
本发明的目的在于克服现有技术中印迹吸附剂分离回收繁琐耗时,限制了使用效率的缺陷,而提供一种磁性多孔分子印迹聚合物的制备方法。
本发明首先将Fe3O4纳米粒子分散在三氯甲烷和油酸的混合溶液中,搅拌形成油酸修饰的Fe3O4纳米粒子(Fe3O4-OA),以调整Fe3O4纳米粒子表面润湿性。
其次,以三氟氯氰菊酯为模板分子,以油酸修饰的Fe3O4纳米粒子为稳定粒子,苯乙烯(St)为骨架单体,二乙烯苯(DVB)为交联剂,甲基丙烯酸(MAA)、丙烯酰胺(AM)为功能单体,偶氮二异丁腈(AIBN)为引发剂形成有机相,并加入一定量的表面活性剂Hypermer 2296,水相是二水氯化钙(CaCl2·2H2O)的水溶液,形成Pickering高内相乳液;制备出分子的印迹聚合物泡沫,并将其应用于水溶液中的高效氯氟氰菊酯(LC)选择性吸附与分离。
具体的,本发明采用的技术方案是:
(1)油酸修饰四氧化三铁纳米颗粒的制备:
首先,参考方法(Ikem, V.O., Menner, A., Horozov, T.S., and Bismarck, A.Highly permeable macroporous polymers synthesized from Pickering medium and high internal phase emulsion templates. Adv. Mater. 2010,22:3588-3592.)对Fe3O4纳米颗粒进行表面修饰,将0.5-1.5g 亲水性的Fe3O4纳米颗粒分散在氯仿与油酸混合溶液中搅拌1.5-4.5h,随后在120℃中干燥24h,得到油酸修饰Fe3O4纳米颗粒(Fe3O4-OA)。其中三氯甲烷和油酸体积比为1mL:1-3mL。
(2)磁性多孔分子印迹聚合物的制备:
首先,在机械搅拌下,将苯乙烯、二乙烯苯、 甲基丙烯酸、丙烯酰胺、三氟氯氰菊酯和偶氮二异丁腈按照比例添加到圆底烧瓶中超声分散后通氮气并且在黑暗中静置,让丙烯酰胺、甲基丙烯酸和三氟氯氰菊酯自组装;搅拌混匀并向有机相中加入步骤(1)中得到的Fe3O4-OA以及表面活性剂Hypermer 2296得到油相Pickering;持续不断搅拌,向油相中缓慢逐滴加入氯化钙水溶液,搅拌后形成W/O型的Pickering高内相乳液;将形成的W/O型高内相乳液转移到安陪瓶中,加热聚合反应后得到产物MMIPs;接着使用索氏提取器对产物进行洗涤纯化去除残余的表面活性剂和未反应的物质;再用甲醇/冰醋酸的混合溶液(9: 1,V/ V)作为洗脱溶液对MMIPs洗涤以去除LC,最后在120℃真空干燥MMIPs直至质量恒定。
其中所述的苯乙烯、二乙烯苯、 甲基丙烯酸、丙烯酰胺、三氟氯氰菊酯和
偶氮二异丁腈的比例为1.0-2.0mL :0.2mL :0.04mL :0.04g :0.05 g :0.06g;所述超声分散时间为30min;所述自组装12 h;
所述加入的Hypermer 2296和Fe3O4-OA的比例为0.5-1.0 mL:0-0.4g;
所述加入的氯化钙水溶液的浓度为0.27 mol/L;
所述加入氯化钙水溶液体积为7.5-9.0mL;
所述聚合为在70℃下聚合24 h;
所述纯化的MMIPs干燥条件为120℃下干燥24 h;
所述加入步骤(1)中得到的Fe3O4-OA以及表面活性剂Hypermer 2296时搅拌转速为400 rpm;
所述逐滴加入氯化钙水溶液时搅拌转速为500 rpm。
作为对比,不加LC制取磁性多孔非印迹聚合物(MNIPs)。
与现有技术相比较,本发明的有益效果体现在如下方面:
传统的印迹聚合物通常采用沉淀聚合等聚合方法,这导致了印迹位点包埋过深,目标分子难以被吸附分离。此外,现有的印迹聚合物通常需要通过离心或者过滤来分离回收,过程繁琐耗时,效率低下,难以适应实际应用。因此,本发明首先以油酸作为修饰剂,对亲水性的Fe3O4纳米颗粒进行修饰调整其表面润湿性,其次,以少量Hypermer 2296为辅助乳化剂,制备的稳定的Pickering HIPEs模板,在连续相中加入引发剂偶氮二异丁腈、功能单体丙烯酰胺和甲基丙烯酸、模板分子LC后,热引发自由基聚合得到了高度通透的磁性多孔分子印迹聚合物(MMIPs)并以LC为模板型污染物研究了选择性吸附分离的行为和机理,最大吸附量为404.4 μmol/g。选择性实验结果表明MMIPs对LC具有特异性识别和选择性吸附的能力,并且可以通过外加磁场加以直接分离,且磁性Fe3O4纳米颗粒的引入可显著增强MMIPs的机械性能和热稳定性。
该方法印迹位点处于聚合物孔道表面,解决了印迹位点包埋过深的问题,并且采用修饰过的Fe3O4纳米颗粒作为稳定粒子,形成 Pickering高内相乳液模板法制取磁性多孔分子印迹聚合物。使用固体颗粒取代乳化剂来稳定Pickering高内相乳液,减少了乳化剂的用量,节约了合成成本,对环境友好。常见的固体颗粒一般为无机或聚合物颗粒,通常仅仅用来稳定Pickering高内相乳液,并且,制备的磁性分子迹聚合物能够被外加磁场快速分离,提高了重复使用效率,同时扩大了吸附剂的应用范围。
附图说明
图1 为实施例1制备流程图,图A是油水相分离图;图B是W/O的Pickering 高内相乳液图;图C整块的磁性多孔分子印迹聚合物(MMIPs);图D 是Pickering 高内相乳液的显微镜图片。
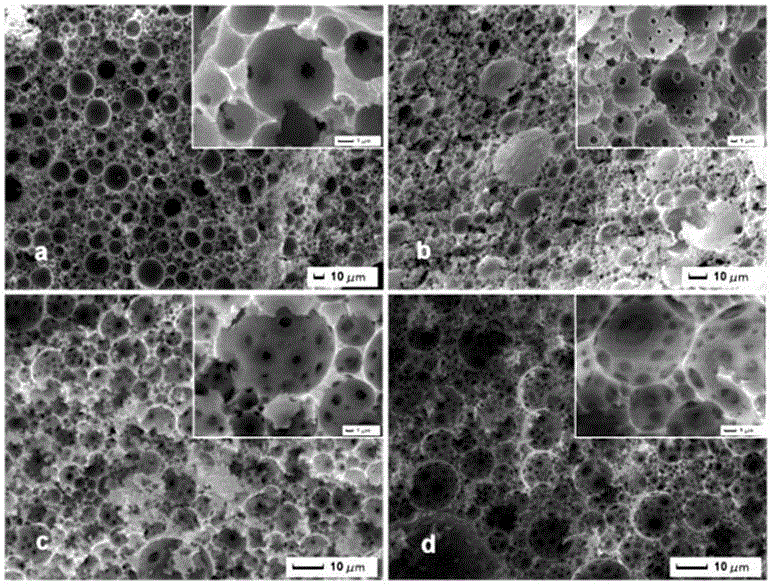
图2 为实施例1,2,3, 4中不同比例水相的MMIPs的SEM图,图(a)75%;图(b)80%;图(c)85%;图(d)90%。
图3为实施例1中制得的Fe3O4 (a), Fe3O4-OA (b), MMIPs (c) and MNIPs (d)的红外光谱图。
图4为实施例1中MMIPs的磁滞回线图,内部为MMIPs对磁铁的响应图。
图5为实施例1中 Fe3O4(a), Fe3O4-OA(b), MMIPs(c) and MNIPs (d)的热重图。
图6 为实验例3中 MMIPs,MNIPs对邻苯二甲酸二乙酯(DEP)、氰戊菊酯(FL)和三氟氯氰菊酯(LC)的吸附能力图。
具体实施方式
本发明具体实施方式中识别性能评价按照下述方法进行:利用静态吸附实验完成。将10mL一定浓度的LC溶液加入到离心管中,加入一定量的磁性多孔分子印迹纳米颗粒吸附剂MMIPs,放在25℃恒温水域中静置若干小时,吸附后LC含量用紫外可见分光光度计测定,并根据结果计算出吸附容量;饱和吸附后,磁性多孔分子印迹吸附剂通过磁铁聚集回收,选择几种结构和性质类似的菊酯类化合物,作为竞争吸附物,参与研究MMIPs聚合物的选择性识别性能。
下面结合具体实施实例对本发明做进一步说明。
实施例1:
(1)油酸修饰四氧化三铁纳米颗粒的制备:
首先,将0.5g亲水性的Fe3O4纳米颗粒分散在1mL氯仿与1mL油酸混合溶液中搅拌1.5h,随后在120℃中干燥24h,得到油酸修饰Fe3O4纳米颗粒(Fe3O4-OA)。
(2)磁性多孔分子印迹聚合物(MMIPs)的制备:
首先,在机械搅拌下,将1.0mL苯乙烯(St)、0.2mL二乙烯苯(DVB)、0.04 mL 甲基丙烯酸(MAA)、0.04g丙烯酰胺(AM)、0.05 g 三氟氯氰菊酯(LC)和0.06g 偶氮二异丁腈(AIBN)添加到100 mL的圆底烧瓶中超声分散30 min后在有机相中通氮气并且在黑暗中静置,让AM、MAA和LC自组装12h。在400 rpm转速下搅拌并向有机相中加入0.1 g Fe3O4-OA以及0.5 mL表面活性剂Hypermer 2296得到油相。随后,以500 rpm转速持续不断搅拌,向油相中缓慢逐滴加入7.5mL 0.27 mol/L的氯化钙水溶液,振荡后形成W/O型的Pickering高内相乳液。将形成的W/O型Pickering高内相乳液转移到安陪瓶中,在70℃下聚合24 h得到产物MMIPs。紧接着使用索氏提取器对产物进行洗涤纯化去除残余的表面活性剂和未反应的物质。再用甲醇/冰醋酸的混合溶液作为洗脱溶液(9: 1,V/ V)对MMIPs洗涤以去除LC,此操作重复多次,直至在洗脱液中无法检测到LC。最后,在120℃下真空干燥直至MMIPs质量恒定。作为对比,用以上操作,不加LC制取磁性多孔分子非印迹聚合物(MNIPs)。
图1 为实施例1制备流程图,图A是油水相分离图,油酸修饰的Fe3O4纳米颗粒聚集在油水界面上,修饰成功;图B是W/O的Pickering 高内相乳液图,稳定性很高;图C整块的磁性多孔分子印迹聚合物(MMIPs),表面可以观察到明显的孔道;图D是Pickering 高内相乳液的显微镜图片,可以发现乳液液滴为球形并且多分散,直径大小约为10 μm。
图3为实施例1中制得的Fe3O4 (a), Fe3O4/OA (b), MMIPs (c) and MNIPs (d)的红外光谱图。曲线a和b对比可知,在573cm-1处的强的Fe-O吸收峰以及出现在1716cm-1,2927cm-1,2859cm-1处新的吸收峰表面Fe3O4纳米颗粒修饰成功。而对比曲线c和d,在1635cm-1处C=C的双键吸收峰和3440cm-1处OH宽的吸收峰的消失表明MMIPS成功形成了印迹位点。
图4为实施例1中MMIPs的磁滞回线图,内部插图为MMIPs对磁铁的响应图。表明MMIPs壳层成功沉积在 Fe3O4/OA纳米粒子表面,并且能够被外加磁场成功从溶液中分离,内部的响应图直接表明了制备的MMIPs对磁场具有极佳的响应性。
图5为实施例1中 Fe3O4(a), Fe3O4-OA(b), MMIPs(c) and MNIPs (d)的热重曲线图。不同的阶段的质量变化表明Fe3O4-OA纳米粒子被成功修饰,而磁性多孔印迹聚合物MMIPs也被成功制备。
实施例2:
(1)油酸修饰四氧化三铁纳米颗粒的制备:
首先,将1.0 g亲水性的Fe3O4纳米颗粒分散在1mL氯仿与2mL油酸混合溶液中搅拌3h,随后在120℃中干燥24h,得到油酸修饰Fe3O4纳米颗粒(Fe3O4-OA)。
(2)磁性多孔分子印迹聚合物(MMIPs)的制备:
首先,在机械搅拌下,将1.5mL苯乙烯(St)、0.2mL二乙烯苯(DVB)、0.04 mL 甲基丙烯酸(MAA)、0.04g丙烯酰胺(AM)、0.05 g 三氟氯氰菊酯(LC)和0.06g 偶氮二异丁腈(AIBN)添加到100 mL的圆底烧瓶中超声分散30 min后在有机相中通氮气并且在黑暗中静置,让AM、MAA和LC自组装12h。在400 rpm转速下搅拌并向有机相中加入0.2 g Fe3O4-OA以及0.75 mL表面活性剂Hypermer 2296得到油相。随后,以500 rpm转速持续不断搅拌,向油相中缓慢逐滴加入8.0 mL 0.27 mol/L的氯化钙水溶液,振荡后形成W/O型的Pickering高内相乳液。将形成的W/O型Pickering高内相乳液转移到安陪瓶中,在70℃下聚合24 h得到产物MMIPs。紧接着使用索氏提取器对产物进行洗涤纯化去除残余的表面活性剂和未反应的物质。再用甲醇/冰醋酸的混合溶液作为洗脱溶液(9: 1,V/ V)对MMIPs洗涤以去除LC,此操作重复多次,直至在洗脱液中无法检测到LC。最后,在120℃下真空干燥直至MMIPs质量恒定。作为对比,用以上操作,不加LC制取磁性多孔分子非印迹聚合物(MNIPs)。
实施例3:
(1)油酸修饰四氧化三铁纳米颗粒的制备:
首先,将1.5g亲水性的Fe3O4纳米颗粒分散在1mL氯仿与3mL油酸混合溶液中搅拌4.5h,随后在120℃中干燥24h,得到油酸修饰Fe3O4纳米颗粒(Fe3O4-OA)。
(2)磁性多孔分子印迹聚合物(MMIPs)的制备:
首先,在机械搅拌下,将2.0 mL苯乙烯(St)、0.2 mL二乙烯苯(DVB)、0.04 mL 甲基丙烯酸(MAA)、0.04g丙烯酰胺(AM)、0.05 g 三氟氯氰菊酯(LC)和0.06g 偶氮二异丁腈(AIBN)添加到100 mL的圆底烧瓶中超声分散30 min后在有机相中通氮气并且在黑暗中静置,让AM、MAA和LC自组装12h。在400 rpm转速下搅拌并向有机相中加入0.4 g Fe3O4-OA以及1.0 mL表面活性剂Hypermer 2296得到油相。随后,以500 rpm转速持续不断搅拌,向油相中缓慢逐滴加入8.5 mL 0.27 mol/L的氯化钙水溶液,振荡后形成W/O型的Pickering高内相乳液。将形成的W/O型Pickering高内相乳液转移到安陪瓶中,在70℃下聚合24 h得到产物MMIPs。紧接着使用索氏提取器对产物进行洗涤纯化去除残余的表面活性剂和未反应的物质。再用甲醇/冰醋酸的混合溶液作为洗脱溶液(9: 1,V/ V)对MMIPs洗涤以去除LC,此操作重复多次,直至在洗脱液中无法检测到LC。最后,在120℃下真空干燥直至MMIPs质量恒定。作为对比,用以上操作,不加LC制取磁性多孔分子非印迹聚合物(MNIPs)。
实施例4:
(1)油酸修饰四氧化三铁纳米颗粒的制备
首先,将1.5g亲水性的Fe3O4纳米颗粒分散在1mL氯仿与3mL油酸混合溶液中搅拌4.5h,随后在120℃中干燥24h,得到油酸修饰Fe3O4纳米颗粒(Fe3O4-OA)。
(2)磁性多孔分子印迹聚合物(MMIPs)的制备:
首先,在机械搅拌下,将2.0 mL苯乙烯(St)、0.2 mL二乙烯苯(DVB)、0.04 mL 甲基丙烯酸(MAA)、0.04g丙烯酰胺(AM)、0.05 g 三氟氯氰菊酯(LC)和0.06g 偶氮二异丁腈(AIBN)添加到100 mL的圆底烧瓶中超声分散30 min后在有机相中通氮气并且在黑暗中静置,让AM、MAA和LC自组装12h。在400 rpm转速下搅拌并向有机相中加入0.4 g Fe3O4-OA以及1.0 mL表面活性剂Hypermer 2296得到油相。随后,以500 rpm转速持续不断搅拌,向油相中缓慢逐滴加入9.0 mL 0.27 mol/L的氯化钙水溶液,振荡后形成W/O型的Pickering高内相乳液。将形成的W/O型Pickering高内相乳液转移到安陪瓶中,在70℃下聚合24 h得到产物MMIPs。紧接着使用索氏提取器对产物进行洗涤纯化去除残余的表面活性剂和未反应的物质。再用甲醇/冰醋酸的混合溶液作为洗脱溶液(9: 1,V/ V)对MMIPs洗涤以去除LC,此操作重复多次,直至在洗脱液中无法检测到LC。最后,在120℃下真空干燥直至MMIPs质量恒定。作为对比,用以上操作,不加LC制取磁性多孔分子非印迹聚合物(MNIPs)。
图2 为实施例1,2,3, 4中不同比例水相的MMIPs的SEM图,图(a)75%;图(b)80%;图(c)85%;图(d)90%。 可以观察到水相比例的改变并没有影响到MMIPs聚合的孔径大小,平均孔径为10 μm,孔喉尺寸为0.5-2μm 。
试验例1:取10mL初始浓度分别为10、30、50、80、100mg/L的LC溶液,LC溶解在乙醇和水的混合液体中(乙醇: 水=5: 5,V/ V)加入到两组离心管中,一组离心管中分别加入10mg实施例1中制备的磁性多孔分子印迹聚合物吸附剂(MMIPs)作为测试液,同样的,另一组离心管加入磁性多孔分子非印迹聚合物吸附剂(MNIPs)作为对比。
把测试液和对比液放在25℃的水浴中静置12h后,使用磁铁聚集后,上层清液用高速离心机分离收集,未吸附的LC分子浓度用紫外可见分光光度计测定,并根据结果计算出吸附容量,结果表明,达到吸附平衡时磁性多孔分子印迹聚合物(MMIPs)的最大吸附容量是404.4 μmol/g,磁性多孔分子非印迹聚合物(MNIPs)最大吸附容量是272.96 μmol/g,在相同温度下比磁性多孔分子非印迹聚合物(MNIPs)要高,说明MMIPs是一种有效识别和去除LC的良好的吸附剂。
试验例2:取10mL初始浓度为100mg/L的三氟氯氰菊酯(LC)溶液分别加入到两组离心管中,一组中分别加入10mg实施例1中制备的的磁性多孔分子印迹聚合物(MMIPs),同样的,另一组离心管加入磁性多孔分子非印迹聚合物(MNIPs)作为对比。把测试液放在25℃的水浴振荡器中,分别在5、15、30、60、120、240、360、480、720 min的时候取出;通过磁铁聚集将印迹吸附剂和溶液分离开,再使用孔径为0.45mm的微孔硝酸纤维素膜对溶液进行过滤去除悬浮的粒子。滤液中的LC浓度由紫外分光光度计在278nm的波长下测定,并根据结果计算出吸附容量;结果表明,在最初的60 min,MMIPs和MNIPs的吸附容量快速增加,说明模板分子能很容易地扩散进入多孔的吸附剂。而且MMIPs的吸附效率明显要比MNIPs更快,对LC的吸附容量也比MNIPs要大,说明在MMIPs表面有大量空的印迹位点。在快速吸附后,由于LC浓度的下降以及结合位点数量的减少,吸附速率急剧下降并且在2.0 h时达到平衡,表面化学结合过程是吸附速率的控制步骤。
试验例3:选择对邻苯二甲酸二乙酯(DEP)、氰戊菊酯(FL)为三氟氯氰菊酯(LC)竞争吸附的酯基类化合物,配置LC,DEP, FL的单组分水溶液,三种物质的浓度都为100mg/L,分别取10mL配置好的溶液分加入到三个离心管中,分别加入10mg实施例1中的MMIPs吸附剂,把测试液放在25℃的水浴中分别静置12h,静置时间完成后,上层清液用高速离心分离收集,并且分别在紫外波长278.5nm,275nm,277.5nm处测定剩余浓度。MMIPs对DEP,FL和LC的吸附容量分别为11.25 μmol/g,100.00 μmol/g和151.06μmol/g。表明MMIPs对LC有显著的专一识别性,吸附容量高于其它酯类化合物。
图6为实验例3中MMIPs,MNIPs对邻苯二甲酸二乙酯(DEP)、氰戊菊酯(FL)和三氟氯氰菊酯(LC)的吸附能力图及DEP、FL、LC的结构图。表明MMIPs对LC吸附量大于DEP和FL,具有很强的吸附选择性。
- 还没有人留言评论。精彩留言会获得点赞!