四苯乙烯双冠醚和氟硼二吡咯衍生物的自组装超分子的制作方法
本发明属于超分子自组装技术领域,具体的一种基于四苯乙烯结构的双冠醚和氟硼二吡咯衍生物自组装的超分子。
背景技术:
超分子化学作为一门有着高度交叉的学科,已经涉及到化学的各个领域。形成许多新的学科,如超分子工程、超分子光化学、纳米超分子化学等,其涵盖了相比分子自身更为复杂的化学物种的化学、物理和生物学特征。随着超分子概念的提出,主-客体化学的发展,其在分子机器中应用逐渐成为化学工作者研究热门,其应用在多功能荧光分子开关,分子马达、分子传感器、分子运输等,以此来用于光电信号处理、分子机器与器件的构筑、纳米粒子的可控聚集、药物分子的运输和控释、环境响应型材料等方面。
超分子化学是“超越分子的化学”,它基于分子间的非共价键相互作用而形成的分子聚集体的化学。相较于传统化学所研究的共价键,超分子化学的研究对象是一些较弱的非共价键之间作用,例如氢键、金属配位、π-π堆积、疏水相互作用、范德华力以及静电相互作用等。通过这些非共价键相互作用,构筑基元按照一定的方式可以自组装形成结构复杂而有序,具有精细微观结构和特定功能的分子聚集体。这些有序的超分子体系表现出单个分子所不具备的特有性质,在生物医学、能量传递、物质传输、电子转移及光、电、磁和机械运动等方面展示了多种新颖特性。
经过几十年来,人们不断的提高聚合技术,虽然传统的高分子化学合成技术已经能够实现利用不同的聚合方法来制备人类所需的聚合物材料,但是由于不同的聚合方法之间存在技术上的不兼容性等特征,所以传统的高分子化学合成方法就难以高效而又便捷的将各类不同的聚合物模块链接在同一个母体上,而超分子主客体相互作用这一特性很好的弥补了传统聚合物合成方法上的缺陷。
技术实现要素:
本发明的目的在于提供一种结构新颖、性能优异的超分子材料及其制备方法,并能够发生共振能量转移,起到研发光学材料作用。
实现本发明目的的技术解决方案为:
一种自组装超分子材料,其主体分子结构如下:
其客体分子结构如下:
上述的自组装超分子材料主体分子制备方法,包括如下步骤:
步骤一:将化合物A、过氧化二苯甲酰和N-溴代丁二酰亚胺,加入溶剂四氯化碳,加热搅拌,制得中间体A;
步骤二:将化合物B置于容器中,加入溶剂四氢呋喃和甲醇的混合溶剂,加入硼氢化钠,搅拌过夜,制得中间体B;
步骤三:将步骤一得到的中间体A和步骤二得到的中间体B置于容器中,加入溶剂无水四氢呋喃,冰浴条件下加氢化钠,搅拌过夜,制得主体分子化合物。
步骤一中,化合物A、过氧化二苯甲酰和N-溴代丁二酰亚胺的摩尔比为1:1:2。
步骤二中,化合物B和硼氢化钠的摩尔比为1:3。
步骤三中,中间体A、中间体B和氢化钠的摩尔比为1:2:20。
上述的自组装超分子材料客体分子制备方法,包括如下步骤:
步骤一:将化合物C和2,4-二甲基吡咯置于容器中,加入干燥处理后的二氯甲烷,滴加催化剂三氟乙酸,常温下搅拌6小时以上,加入2,3-二氯-5,6-二氰对苯醌,搅拌6小时以上,滴加三氟化硼乙醚和三乙胺,搅拌过夜,反应得到中间体C;
步骤二:将化合物D和苄胺置于容器中,加入干燥处理后的二氯甲烷,搅拌过夜,在冰浴的情况下,加入硼氢化钠,制得中间体D;
步骤三:将步骤一得到的中间体C和步骤二得到的中间体D置于容器中,加入四氢呋喃和水的混合物,加入五水硫酸铜和抗坏血酸钠,搅拌过夜,制得客体分子化合物。
步骤一中,化合物C、2,4-二甲基吡咯、三氟乙酸、2,3-二氯-5,6-二氰对苯醌、三氟化硼乙醚和三乙胺的摩尔比为10:20:1:10:20:20。
步骤二中,化合物D、苄胺和硼氢化钠的摩尔比为1:2:2。
步骤三中,中间体C、中间体D、五水硫酸铜和抗坏血酸钠的摩尔比为2:3:3:3。
上述的自组装超分子材料制备方法,包括如下步骤:
在THF/H2O=1:9(V/V)配制的主体分子溶液中滴加客体分子得到所述的自组装超分子材料。
本发明与现有技术相比,具有以下优点:
(1)制备过程简单,原料易得,成本较低。(2)该发明成功解决了自组装体的识别自组装成超分子的过程。(3)该发明成功解决两个化合物间的荧光共振能量转移这一技术难题。
附图说明
图1为本发明以氟硼二吡咯衍生物作为客体分子,四苯乙烯双冠醚作为主体分子,两个分子自组装形成的超分子的荧光共振能量转移其转移机制过程示意图。
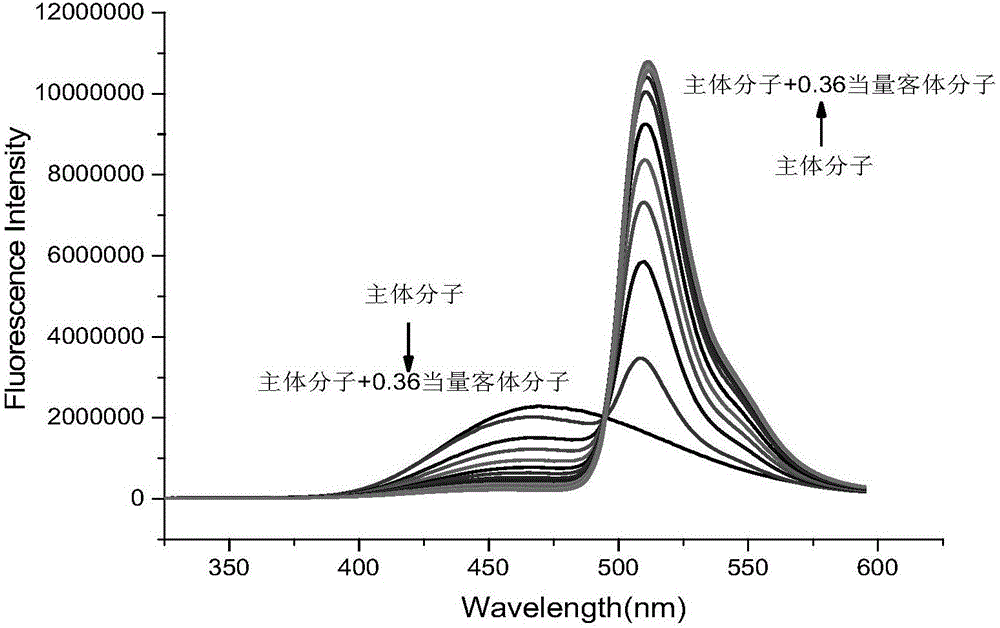
图2为本发明主体分子化合物待测液中梯度滴加客体分子待测液(0.03eq,0.06eq,0.09eq,0.12eq,0.15eq,0.18eq,0.21eq,0.24eq,0.27eq,0.30eq,0.33eq,0.36eq)分别测试每次的荧光发射光谱,得到一系列的荧光滴定发射光谱图。
具体实施方式
(一)自组装超分子材料主体分子合成路线如下所示
(二)自组装超分子材料客体分子合成路线如下所示
(三)以氟硼二吡咯衍生物作为客体分子溶于四氢呋喃与水的溶液中,四氢呋喃与水的体积比为1:9,配成溶液浓度为10-2mol/L,四苯乙烯双冠醚作为主体分子溶于四氢呋喃与水的溶液中,四氢呋喃与水的体积比为1:9,配成溶液浓度为10-5mol/L,两个分子自组装形成的超分子材料,其过程见图1。
实例1:主体分子的合成
(1)取烧瓶,将其放入装有丙酮的玻璃器皿中,将三口烧瓶进行抽真空处理,然后向里面充满氩气,反复进行三次,确保体系没有氧气。向三口烧瓶中加入物质0.1mol 4-甲基二苯甲酮,缓缓加入0.25mol锌粉,再向里面加入处理过的无水四氢呋喃,搅拌溶解。然后向装有丙酮的器皿中加入液氮,直至玻璃器皿里面的丙酮溶液完全结块。向烧瓶中缓缓加入30ml的四氯化钛,加完后继续保持低温搅拌30分钟,然后移去低温体系,转入回流加热搅拌过夜。12小时后停止反应,自然冷却至室温,反应液用抽滤装置进行抽滤,得到滤液通过旋转蒸发仪除去溶剂,拌硅胶,柱层析提纯(展开剂:石油醚),最后得到白色固体,本步骤产率88.1%。
(2)取烧瓶,进行抽真空,氩气保护,向烧瓶内加入15mmol步骤(1)所得化合物,30mmol溴代丁亚酰亚胺,15mmol过氧化二苯甲酰,并加入约250mL四氯化碳,搅拌溶解,此时整个反应液呈现出淡黄色。随后,反应体系升温至80℃搅拌回流,过夜反应。12小时后停止反应,反应体系冷却至室温,静置后几分钟后反应体系出现分层现象,有固体生成,过滤除去固体,得到滤液进行旋转蒸发除去溶剂,柱层析提纯,展开剂为石油醚和二氯甲烷的混合溶剂,得到白色片状固体产物,本步骤产率37.1%。
(3)取烧瓶,加入0.532mol三缩二乙二醇,加入50mL四氢呋喃,然后缓缓滴加冷却好的0.2mol氢氧化钠水溶液,滴加完成后搅拌数分钟,在冰浴情况下慢慢滴加0.134mol对甲基磺酰氯溶液,一小时滴完,滴加完成后撤掉冰水浴,转为常温反应,5小时后停止反应,除去溶剂,得到白色或浅黄色液体,乙酸乙酯溶解,饱和氯化钠水溶液洗涤数次,合并有机相无水硫酸镁干燥。过滤,除去溶剂,得到无色或浅黄色油状物,本步骤产率88.3%。
(4)取烧瓶,进行抽真空,氩气保护,加入24.8mmol步骤(3)所得化合物,11.3mmol邻苯二酚,34.5mmol碳酸钾,大约100mL乙腈,85℃回流反应过夜。反应完成后冷却至室温,过滤,滤液除去溶剂,浓缩得到红色或褐色油状物,加入烧瓶中,再加入40mL四氢呋喃,在冰浴情况下慢慢加入冷却好的30mmol四氢呋喃溶液,保持在冰浴情况下慢慢滴加30mmol对甲基磺酰氯溶液,半小时滴加完成后转为常温反应,一天后停止反应,二氯甲烷多次萃取,有机相用饱和的氯化钠水溶液进行洗涤,合并有机相,无水硫酸镁干燥,过滤,柱层析,得到黄色油状物,本步骤产率产率66.1%。
(5)取烧瓶,抽真空,氩气保护,加入11.72mmol步骤(4)所得化合物,24.8mmol碳酸铯,11.72mmol 3,4-二羟基苯甲醛,200mL处理过的无水N,N-二甲基甲酰胺,110℃搅拌4天后停止反应,冷却至室温,过滤,滤液减压旋蒸除去溶剂,得到黑色固体物质,用二氯甲烷进行溶解,拌硅胶,干法上样柱层析,得到白色固体,本步骤产率37.2%。
(6)取烧瓶,加入2.94mmol步骤(5)所得化合物,加入四氢呋喃和甲醇的混合溶剂,在冰浴情况下分批慢慢加入10mmol硼氢化钠,加入完成后搅拌数十分钟后撤掉冰水浴转为常温反应,搅拌过夜。反应结束加入少量水淬灭反应,二氯甲烷萃取多次后,水洗,无水硫酸镁干燥,过滤,滤液减压蒸馏除去溶剂得到白色固体,本步骤产率90%。
(7)取烧瓶,加入1.0mmol步骤(6)所得化合物,溶于处理过的无水四氢呋喃中,在冰水浴条件下慢慢加入20mmol氢化钠,搅拌均匀,将0.42mmol步骤(2)所得化合物溶于20mL处理过的无水四氢呋喃中,缓缓滴加至三口烧瓶中,滴加结束后保持40min分钟冰水浴,然后撤掉冰水浴,加热回流搅拌12小时后冷却至室温,加入少量水进行淬灭反应,二氯甲烷萃取多次后水洗,加入无水硫酸镁干燥溶剂,过滤,除去溶剂,柱层析分离得到白色或浅黄色固体,本步骤产率34.2%。
白色或浅黄色固体,产率34.2%。
1H NMR(500MHz,CDCl3)δ(TMS,ppm):7.08(d,J=6.5,3.0Hz,10H),7.00(s,8H),6.87(t,J=2.6Hz,10H),6.82(s,4H),4.41(d,J=3.0Hz,8H),4.14(d,J=5.5,3.0Hz,16H),3.91(m,J=2.2Hz,16H),3.83(s,16H).
13C NMR(126MHz,CDCl3)δ(TMS,ppm):148.97,148.50,148.33,143.68,143.12,140.73,136.29,134.48,131.35,127.67,127.63,127.21,126.44,121.47,121.05,119.89,114.18,113.98,113.82,113.05,77.40,77.14,76.89,69.91,69.90,69.53,69.38,64.96.
ESI-MS m/z=1335.80[M+Na+].
实例2:客体分子的合成
(1)取烧瓶,加入0.01mol羟基苯甲醛,0.05mol 1,2-二溴乙烷,0.03mol碳酸钾,溶于20ml N,N-二甲基甲酰胺,加热搅拌回流,5小时后点板跟踪反应进度。反应完成后,先过滤,滤液旋蒸除去溶剂N,N-二甲基甲酰胺,残留物用乙酸乙酯溶解,水洗多次,乙酸乙酯萃取合并有机相,无水硫酸镁干燥,过滤,除去溶剂,拌硅胶,柱层析提纯得到白色固体中间物质,本步骤产率80%。
(2)取烧瓶,加入6.5mmol步骤(1)所得化合物,8.0mmol叠氮化钠,溶入经干燥处理后的5.0ml N,N-二甲基甲酰胺,室温搅拌一天。反应完成后,反应液里加水,再加入二氯甲烷进行萃取,合并有机相,有机层用无水硫酸镁进行干燥。过滤,所得滤液经减压旋蒸除去溶剂,剩余物质为无色液体,得到产物,本步骤产率90%。
(3)取烧瓶,加入1mol对羟基苯甲醛和2mol溴丙炔置于烧瓶中,加入N,N-二甲基甲酰胺,加入3mol碳酸钾,加热搅拌过夜,得到化合物,本步骤产率80%。
(4)取烧瓶,加入1mol步骤(3)得到化合物和2mol苄胺置于烧瓶中,加入干燥处理后的二氯甲烷,搅拌过夜,在冰浴的情况下,加入硼氢化钠,制得化合物,本步骤产率80%。
(5)取烧瓶,抽真空,严格氩气保护条件下,加入0.021mol步骤(2)所得化合物和3.99g 2,4-二甲基吡咯溶于200mL处理过的无水二氯甲烷中搅拌,使原料完全混合在一起,然后缓慢滴加2~3滴三氟乙酸,此时反应液的颜色逐渐加深至酒红色,室温搅拌过夜。在氩气保护条件下加入0.02mmol四氯苯醌,反应12小时,再加入20mL三乙胺,继续搅拌15分钟,最后加入20mL三氟化硼乙醚溶液,反应约半天。反应结束后,将反应液用水洗涤,二氯甲烷萃取,合并有机相,无水硫酸镁干燥,硅胶柱层析得到红色产物,本步骤产率10%。
(6)取烧瓶,加入1mmol步骤(4)所得产物,1mmol步骤(5)所得产物,四氢呋喃与水的混合溶剂,400mg五水硫酸铜,640mg抗坏血酸钠,此反应在室温下搅拌进行半天,反应结束后,过滤,滤液用二氯甲烷萃取,水洗多次,无水硫酸镁干燥,过滤,除去溶剂,拌硅胶,干法上样柱层析,得到紫色产物,本步骤产率56%。
紫色固体,产率56%。
1H NMR(500MHz,DMSO-d6)(TMS,ppm):8.33(s,1H),7.40–7.18(m,9H),7.09(d,J=8.7Hz,2H),6.99(d,J=8.6Hz,2H),6.16(s,2H),5.14(s,2H),4.82(t,J=5.0Hz,2H),4.48(t,J=5.1Hz,2H),3.65(d,J=23.6Hz,4H),2.44(s,6H),1.37(s,6H).
13C NMR(126MHz,DMSO-d6)δ154.01,142.05,130.40,128.57,128.53,127.45,127.36,125.98,124.49,120.61,114.63,113.72,60.36,51.26,50.76,48.41,38.80,38.38,13.56.
ESI-MS:m/z=661.24[M+H+].
实例3:自组装超分子荧光测试
(1)用移液枪吸取3mL主体分子化合物的待测液(溶剂为四氢呋喃和水,体积比为1:9,1.0×10-5mol/L)置于3mL比色皿中,通过查阅文献我们预先设置305nm波长激发,激发狭缝宽度和发射狭缝宽度都选择3nm,来测定主体分子化合物的荧光发射谱图,见图2。
(2)然后向上述比色皿中,梯度滴加客体分子待测液(1.0×10-2mol/L)(0.03eq,0.06eq,0.09eq,0.12eq,0.15eq,0.18eq,0.21eq,0.24eq,0.27eq,0.30eq,0.33eq,0.36eq)分别测试每次的荧光发射光谱,这样就可以得到一系列的荧光滴定发射光谱图,见图2。
- 还没有人留言评论。精彩留言会获得点赞!