用于检测甲基化状态的核酸和方法与流程
本申请要求于2015年11月16日提交的美国临时申请号62/255,947的权益,所述临时申请特此以引用的方式整体并入本文。
发明领域
本发明涉及用于确定尤其是受试者是否具有对于与核酸的甲基化状态相关联的疾病状态的易感性的系统和方法。本发明还涉及用于诊断受试者中的疾病或病状的系统和方法,其中所述疾病或病状与核酸的甲基化状态相关联。本发明还涉及用于检测受试者中的疾病或病状的状态的系统和方法,其中所述疾病或病状与核酸的甲基化状态相关联。
发明背景
个体的基因组的甲基化状态可能与和甲基化状态相关联的疾病或病状相关。然而,当前的方法耗费劳力且昂贵。例如,全基因组亚硫酸氢盐测序(wgbs)需要大量输入dna用于文库制备,并且还需要深度测序用于高水平的基因组覆盖。可替代地,经靶向的dna甲基化方法需要引入偏差的广泛多重设计并且具有受限的基因组覆盖宽度。此外,当前的方法提供仅甲基化状态的状况,从而忽视其他参数,诸如突变图谱和基因组不稳定性,所述参数可组合使用来对个体进行诊断或确定个体是否易感疾病或病状。因此,需要开发经济的、有效的并且具有高度灵敏性和特异性的全面基因组和表观基因组测试。
发明概述
一些实施方案是:
1.一种确定受试者中的核酸的甲基化状态的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的所述目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;以及
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,以便确定核酸的甲基化状态。
2.一种确定受试者是否具有对于与核酸的甲基化状态相关联的疾病或病状的易感性的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来确定受试者是否易感所述疾病或病状。
3.一种诊断受试者的疾病或病状的方法,所述疾病或病状与核酸的甲基化状态相关联,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来诊断所述受试者中的所述疾病或病状。
4.一种检测受试者的疾病或病状的状态的方法,所述疾病或病状与核酸的甲基化状态相关联,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来检测所述受试者中的所述疾病或病状的状态。
5.一种区分来源于受试者和一个或多个另外个体的核酸种类的方法,所述受试者和所述一个或多个另外个体具有核酸的不同甲基化状态,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品,所述血液样品包含来源于所述受试者和所述一个或多个另外个体的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有所述核酸的不同甲基化状态的个体分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来区分来源于所述受试者和所述一个或多个另外个体的核酸种类。
6.如实施方案5所述的方法,其中所述受试者是妊娠女性并且所述一个或多个另外个体是未出生胎儿。
7.如实施方案5或6所述的方法,其中所述血液样品是母体血浆或母体血清。
8.如实施方案5所述的方法,其中所述受试者是组织移植物接受者。
9.如实施方案8所述的方法,其中所述一个或多个另外个体是组织移植物供体。
10.一种区分来源于受试者的第一组织和一个或多个另外组织的核酸种类的方法,所述第一组织和所述一个或多个另外组织具有核酸的不同甲基化状态,所述方法包括:
a)获得从来自所述受试者的无细胞体液样品分离的核酸样品,所述无细胞体液样品包含来源于所述第一组织和所述一个或多个另外组织的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有所述核酸的不同甲基化状态的参考组织分离的参考核酸样品计算的多个参考比率进行比较;
h)基于在步骤g)中的所述比较来区分来源于所述第一组织和所述一个或多个另外组织的核酸种类。
11.如实施方案10所述的方法,其中所述体液样品是血液样品。
12.如实施方案10或11所述的方法,其中所述方法还包括:(i)确定特定组织类型对于所述核酸的贡献百分比;(ii)将所述特定组织类型的贡献百分比与基于从具有疾病或病状的参考受试者分离的参考核酸样品计算的所述组织类型的参考贡献百分比进行比较;以及(iii)基于在步骤(ii)中的所述比较检测所述受试者中的所述疾病或病状的状态。
13.如实施方案10-12中任一项所述的方法,其中所述第一组织选自由以下组成的组:肝脏、肾、子宫、卵巢、胎盘、胰腺、结肠、胃、肺以及膀胱。
14.一种确定受试者中的核酸的甲基化状态并且检测拷贝数变异的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的所述目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,以便确定核酸的甲基化状态;以及
f)确定在步骤d)测序的所述独特mip扩增子的数量;至少部分地基于独特mip扩增子序列的所述数量确定读取密度;以及通过将所述读取密度与基于从参考受试者分离的参考核酸样品计算的多个参考读取密度进行比较来检测拷贝数变异。
15.如实施方案14所述的方法,其还包括使用所述靶向性分子标签来去除重复以改进分析。
16.如实施方案14所述的方法,其还包括确定限定区域中的独特mip扩增子序列的数量来确定所述读取密度。
17.一种确定受试者的甲基化年龄的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来确定所述受试者的甲基化年龄。
18.一种确定受试者的组织的甲基化年龄的方法,所述方法包括:
a)获得从来自所述受试者的无细胞体液样品分离的核酸样品,所述无细胞体液样品包含来源于所述组织的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从参考组织分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来确定所述组织的甲基化年龄。
19.如实施方案18所述的方法,其中所述体液样品是血液样品。
20.如实施方案18或19所述的方法,其中所述受试者是妊娠女性。
21.如实施方案18-20中任一项所述的方法,其中所述组织是胎盘组织,并且所述胎盘组织的甲基化年龄是未出生胎儿的胎龄的指标。
22.如实施方案1-4、10、11、14和17中任一项所述的方法,其中所述受试者是妊娠女性。
23如实施方案22所述的方法,其中所述核酸样品包括母体和胎儿核酸。
24.如实施方案1-23中任一项所述的方法,其中所述核酸样品是dna或rna。
25.如实施方案24所述的方法,其中所述核酸样品是基因组dna。
26.如实施方案1-9、11-17和19-25中任一项所述的方法,其中所述血液样品是全血样品、血浆样品或血清样品。
27.如实施方案24所述的方法,其中所述血液样品是血浆样品。
28.如实施方案1-27中任一项所述的方法,其中所述第一靶向性多核苷酸臂的长度在14与30个碱基之间。
29.如实施方案1-28中任一项所述的方法,其中所述第二靶向性多核苷酸臂的长度在14与30个碱基之间。
30.如实施方案1-29中任一项所述的方法,其中所述靶向性多核苷酸臂中的每一个具有在45℃与66℃之间的解链温度。
31.如实施方案1-30中任一项所述的方法,其中所述靶向性多核苷酸臂中的每一个具有在10%与40%之间的gc含量。
32.如实施方案1-31中任一项所述的方法,其中所述第一独特分子标签的长度在4与15个碱基之间。
33.如实施方案1-32中任一项所述的方法,其中所述第二独特分子标签的长度在4与15个碱基之间。
34.如实施方案1-33中任一项所述的方法,其中所述多核苷酸接头与所述受试者的任何基因组区域基本上不互补。
35.如实施方案1-34中任一项所述的方法,其中所述多核苷酸接头具有在14与50个碱基之间的长度。
36.如实施方案1-35中任一项所述的方法,其中所述多核苷酸接头具有在45℃与85℃之间的解链温度。
37.如实施方案1-36中任一项所述的方法,其中所述多核苷酸接头具有在30%与66%之间的gc含量。
38.如实施方案1-37中任一项所述的方法,其中所述多核苷酸接头包含至少一个扩增引物结合位点。
39.如实施方案38所述的方法,其中所述多核苷酸接头包含正向扩增引物结合位点。
40.如实施方案39所述的方法,其中所述正向扩增引物的序列包含ccgtaatcgggaagctgaag(seqidno:1)的核苷酸序列。
41.如实施方案1-29中任一项所述的方法,其中反向扩增引物的序列包含gcacgatccgacggtagtgt(seqidno:2)的核苷酸序列。
42.如实施方案1-41中任一项所述的方法,其中所述核苷酸接头包含cttcagcttcccgattacgggcacgatccgacggtagtgt(seqidno:3)的核苷酸序列。
43.如实施方案1-42中任一项所述的方法,其中所述第一靶向性多核苷酸臂包含cactacactccaacctaa(seqidno:4)或ttctcctacctcaacctc(seqidno:5)的核苷酸序列。
44.如实施方案1-43中任一项所述的方法,其中所述第二靶向性多核苷酸臂包含caaaaaactaaaacaaaa(seqidno:6)或ccaaactaaaatacaata(seqidno:7)的核苷酸序列。
45.如实施方案1-44中任一项所述的方法,其中所述mip包含cactacactccaacctaa(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)caaaaaactaaaacaaaa(seqidno:8)或ttctcctacctcaacctc(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)ccaaactaaaatacaata(seqidno:9)的核苷酸序列,其中(n1-10)表示所述第一独特分子标签并且(n11-20)表示所述第二独特分子标签。
46.如实施方案1-45中任一项所述的方法,其中所述mip群体具有在10fm与100nm之间的浓度。
47.如实施方案1-46中任一项所述的方法,其中在步骤c)中获得的所述mip复制子中的每一个通过以下来产生:
i)所述第一靶向性多核苷酸臂和所述第二靶向性多核苷酸臂分别与所述核酸样品中的分别所述第一重复区域和所述第二重复区域杂交,其中所述第一重复区域和所述第二重复区域侧接目标靶序列;以及
ii)在所述杂交之后,使用连接/延伸混合物来延伸并连接所述两个靶向性多核苷酸臂之间的空位区域以形成单链环状核酸分子。
48.如实施方案1-47中任一项所述的方法,其中所述mip复制子中的每一个是单链环状核酸分子。
49.如实施方案1-48中任一项所述的方法,其中所述测序步骤d)包括下一代测序方法。
50.如实施方案49所述的方法,其中所述下一代测序方法包括大规模平行测序方法。
51.如实施方案1-50中任一项所述的方法,其中所述方法包括在所述测序步骤d)之前进行扩增所述mip复制子的pcr反应以用于测序。
52.如实施方案51所述的方法,其中所述pcr反应是索引pcr反应。
53.如实施方案52所述的方法,其中所述索引pcr反应向所述mip扩增子中的每一个引入以下组件:用于多重测序的包含独特分子标签的一对测序衔接子。
54.如实施方案53所述的方法,其中所述带条形码的mip扩增子依次包含以下组件:
第一测序衔接子-第一测序引物结合位点-第一独特靶向性分子标签-第一靶向性多核苷酸臂-捕获的核酸-第二靶向性多核苷酸臂-第二独特靶向性分子标签-第二测序引物结合位点-独特样品条形码-第二测序衔接子。
55.如实施方案1-54中任一项所述的方法,所述方法还包括从所述一个或多个mip群体获得测序信息。
56.如实施方案55所述的方法,其中所述测序信息分别用于确定受试者中的核酸的甲基化状态、或用于确定所述受试者是否具有对于所述疾病或病状的易感性、或用于诊断所述疾病或病状、或用于检测所述疾病或病状的状态、或用于区分来源于所述受试者和所述一个或多个另外个体的所述核酸种类、或用于区分来源于所述第一组织和所述一个或多个另外组织的所述核酸种类、或用于确定所述受试者中的核酸的甲基化状态并且检测拷贝数变异、或用于确定所述受试者的甲基化年龄、或用于确定所述组织的甲基化年龄。
57.一种确定受试者中的核酸的甲基化状态的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;以及
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,以便确定核酸的甲基化状态。
58.一种确定受试者是否具有对于与核酸的甲基化状态相关联的疾病或病状的易感性的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来确定所述受试者是否易感所述疾病或病状。
59.一种诊断受试者的疾病或病状的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来诊断所述受试者中的所述疾病或病状。
60.一种检测受试者的疾病或病状的状态的方法,所述疾病或病状与核酸的甲基化状态相关联,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来检测所述受试者中的所述疾病或病状的状态。
61.一种区分来源于受试者和一个或多个另外个体的核酸种类的方法,所述受试者和所述一个或多个另外个体具有核酸的不同甲基化状态,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品,所述血液样品包含来源于所述受试者和所述一个或多个另外个体的核酸;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来区分来源于所述受试者和所述一个或多个另外个体的核酸种类。
62.如实施方案61所述的方法,其中所述受试者是妊娠女性并且所述一个或多个另外个体是未出生胎儿。
63.如实施方案61或62所述的方法,其中所述血液样品是母体血浆或母体血清。
64.如实施方案61所述的方法,其中所述受试者是组织移植物接受者。
65.如实施方案64所述的方法,其中所述一个或多个另外个体是组织移植物供体。
66.一种区分来源于受试者的第一组织和一个或多个另外组织的核酸种类的方法,所述第一组织和所述一个或多个另外组织具有核酸的不同甲基化状态,所述方法包括:
a)获得来自所述受试者的无细胞体液样品的dna样品,所述无细胞体液样品包含来源于所述第一组织和所述一个或多个另外组织的核酸;
b)进行所述dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有所述核酸的不同甲基化状态的参考组织分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来区分来源于所述第一组织和所述一个或多个另外组织的核酸种类。
67.如实施方案66所述的方法,其中所述体液样品是血液样品。
68.如实施方案66或67所述的方法,其中所述方法还包括:(i)确定特定组织类型对于所述核酸的贡献百分比;(ii)将所述特定组织类型的贡献百分比与基于从具有疾病或病状的参考受试者分离的参考核酸样品计算的所述组织类型的参考贡献百分比进行比较;以及(iii)基于在步骤(ii)中的所述比较检测所述受试者中的所述疾病或病状的状态。
69.如实施方案66-68中任一项所述的方法,其中所述第一组织选自由以下组成的组:肝脏、肾、子宫、卵巢、胎盘、胰腺、结肠、胃、肺以及膀胱。
70.一种确定受试者中的核酸的甲基化状态并且检测拷贝数变异的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,以便确定核酸的甲基化状态;以及
i)使用所述大规模平行测序方法来i)进一步确定独特mip扩增子的数量;ii)至少部分地基于独特mip扩增子序列的所述数量确定第一读取密度;以及iii)将所述第一读取密度与基于从参考受试者分离的参考核酸样品计算的多个参考读取密度进行比较;以及iv)基于步骤iii)中的所述比较来检测拷贝数变异。
71.如实施方案70所述的方法,其还包括使用所述靶向性分子标签来去除重复以改进分析。
72.如实施方案70所述的方法,其还包括确定限定区域中的独特mip扩增子序列的数量来确定所述读取密度。
73.一种确定受试者的甲基化年龄的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来确定所述受试者的甲基化年龄。
74.一种确定受试者的组织的甲基化年龄的方法,所述方法包括:
a)获得来自所述受试者的无细胞体液样品的dna样品;
b)进行所述dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从参考组织分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来确定所述组织的甲基化年龄。
75.如实施方案74所述的方法,其中所述体液样品是血液样品。
76.如实施方案74或75所述的方法,其中所述受试者是妊娠女性。
77.如实施方案74-76中任一项所述的方法,其中所述组织是胎盘组织,并且所述胎盘组织的甲基化年龄是未出生胎儿的胎龄的指标。
78.如实施方案57-65、67-73和75-77中任一项所述的方法,其中所述血液样品是全血样品、血浆样品或血清样品。
79.如实施方案78所述的方法,其中所述血液样品是血浆样品。
80.如实施方案57-79中任一项所述的方法,其中所述第一靶向性多核苷酸臂的长度在14与30个碱基之间。
81.如实施方案57-80中任一项所述的方法,其中所述第二靶向性多核苷酸臂的长度在14与30个碱基之间。
82.如实施方案57-81中任一项所述的方法,其中所述靶向性多核苷酸臂中的每一个具有在45℃与80℃之间的解链温度。
83.如实施方案57-82中任一项所述的方法,其中所述靶向性多核苷酸臂中的每一个具有在10%与50%之间的gc含量。
84.如实施方案57-83中任一项所述的方法,其中所述第一独特分子标签的长度在4与15个碱基之间。
85.如实施方案57-84中任一项所述的方法,其中所述第二独特分子标签的长度在4与15个碱基之间。
86.如实施方案57-85中任一项所述的方法,其中所述多核苷酸接头与所述受试者的任何基因组区域基本上不互补。
87.如实施方案57-86中任一项所述的方法,其中所述多核苷酸接头具有在20与1,000个碱基之间的长度。
88.如实施方案57-87中任一项所述的方法,其中所述多核苷酸接头具有在45℃与80℃之间的解链温度。
89.如实施方案57-88中任一项所述的方法,其中所述多核苷酸接头具有在30%与80%之间的gc含量。
90.如实施方案57-89中任一项所述的方法,其中所述多核苷酸接头包含至少一个扩增引物结合位点。
91.如实施方案90所述的方法,其中所述多核苷酸接头包含正向扩增引物结合位点。
92.如实施方案91所述的方法,其中所述正向扩增引物的序列包含ccgtaatcgggaagctgaag(seqidno:1)的核苷酸序列。
93.如实施方案57-92中任一项所述的方法,其中反向扩增引物的序列包含gcacgatccgacggtagtgt(seqidno:2)的核苷酸序列。
94.如实施方案57-93中任一项所述的方法,其中所述核苷酸接头包含cttcagcttcccgattacgggcacgatccgacggtagtgt(seqidno:3)的核苷酸序列。
95.如实施方案57-94中任一项所述的方法,其中所述第一靶向性多核苷酸臂包含cactacactccaacctaa(seqidno:4)或ttctcctacctcaacctc(seqidno:5)的核苷酸序列。
96.如实施方案57-95中任一项所述的方法,其中所述第二靶向性多核苷酸臂包含核苷酸序列gaggctgaggcaggagaa(seqidno:10)或ccaaactaaaatacaata(seqidno:7)。
97.如实施方案57-96中任一项所述的方法,其中所述mip包含cactacactccaacctaa(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)caaaaaactaaaacaaaa(seqidno:8)或ttctcctacctcaacctc(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)ccaaactaaaatacaata(seqidno:9)的核苷酸序列,其中(n1-10)表示所述第一独特分子标签并且(n11-20)表示所述第二独特分子标签。
98.如实施方案57-97中任一项所述的方法,其中所述mip群体具有在10fm与100nm之间的浓度。
99.一种从用于检测受试者的甲基化的多个候选mip选择分子倒置探针(mip)方法,所述方法包括:
a)接收所述多个候选mip的核酸序列,其中所述多个候选mip中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
b)对于所述多个候选mip中的每个相应的mip,
i)计算预测在所述结合臂序列上没有错配的情况下由所述相应的mip捕获的独特cpg位点的第一数量(a);
ii)计算预测在所述结合臂序列上有一个错配的情况下由所述相应的mip捕获的独特cpg位点的第二数量(c);
iii)计算预测在所述结合臂序列上没有错配的情况下在整个基因组上由所述相应的mip捕获的独特位点的第三数量(e);
iv)计算预测在所述结合臂序列上有一个错配的情况下在整个所述基因组上由所述相应的mip捕获的独特位点的第四数量(g);
v)计算预测在所述结合臂序列上没有错配的情况下在整个所述基因组上由所述相应的mip捕获的非独特位点的第五数量(f);
vi)计算预测在所述结合臂序列上有一个错配的情况下在整个所述基因组上由所述相应的mip捕获的非独特位点的第六数量(h);
vii)计算存在于所述第一靶向性多核苷酸臂上的cpg位点的第七数量(i);
viii)计算存在于所述第二靶向性多核苷酸臂上的cpg位点的第八数量(j);
ix)至少部分地基于所述第一、第二、第三、第四、第五、第六、第七和第八数量来计算所述相应的mip的性能度量;
c)至少部分地基于针对所述多个候选mip中的每个mip在步骤b)ix)中计算的所述性能度量来选择mip。
100.如实施方案99所述的方法,其中选择步骤c)的所述mip使得所述第七数量(i)和所述第八数量(j)的总和小于所述候选mip的剩余集合的对应总和。
101.如实施方案99或100所述的方法,其中:
第一总和是所述第一数量(a)和所述第二数量(c)的总和;
第二总和是所述第三数量(e)、所述第四数量(g)、所述第五数量(f)、和所述第六数量(h)的总和;并且
选择步骤c)的所述mip使得所述第一总和与所述第二总和之间的比率大于所述候选mip的剩余集合的比率。
102.如实施方案99-101中任一项所述的方法,其中:
第三总和是所述第三数量(e)和所述第四数量(g)的总和;
第四总和是所述第三数量(e)、所述第四数量(g)、所述第五数量(f)、和所述第六数量(h)的总和;并且
选择步骤c)的所述mip使得所述第三总和与所述第四总和之间的比率大于所述候选mip的剩余集合的比率。
103.如实施方案99-102中任一项所述的方法,其中步骤c)的所述mip基于一个错配位点的平均捕获系数(k1)和零个错配位点的平均捕获系数(k0)的比率(ke)来选择:
并且其中所述比率(ke)通过实验来估算。
104.如实施方案99-103中任一项所述的方法,其中步骤b)ix的所述性能度量包括对应于所述第一数量(a)和所述第二数量(c)的加权总和的因数。
105.如实施方案104所述的方法,其中所述加权总和对应于a+ke×c。
106.如实施方案99-015中任一项所述的方法,其中步骤b)ix的所述性能度量包括对应于所述第三数量(e)和所述第四数量(g)的加权总和的因数。
107.如实施方案106所述的方法,其中所述加权总和对应于e+ke×g。
108.如实施方案99-107中任一项所述的方法,其中选择步骤c)的所述mip使得第一加权总和a+ke×c与第二加权总和e+ke×g之间的乘积大于所述候选mip的剩余集合的乘积。
109.一种核酸分子,其包含cactacactccaacctaa(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)caaaaaactaaaacaaaa(seqidno:8)或ttctcctacctcaacctc(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)ccaaactaaaatacaata(seqidno:9)的核苷酸序列,其中(n1-10)表示第一独特分子标签并且(n11-20)表示第二独特分子标签。
110.如实施方案109所述的核酸,其中所述第一独特分子标签的长度在4与15个碱基之间。
111.如实施方案109或110所述的核酸,其中所述第二独特分子标签的长度在4与15个碱基之间。
112.如实施方案2-5、10、17、18、58、59-61、66、73和74中任一项所述的方法,其中步骤e)还包括确定在步骤d)测序的所述mip扩增子的子集内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,其中所述子集先前被确定有差异地甲基化。
113.如实施方案112所述的方法,其中所述子集在不同个体之间或在不同组织之间或在测试样品与参考样品之间或在疾病样品与健康对照样品之间有差异地甲基化。
附图简述
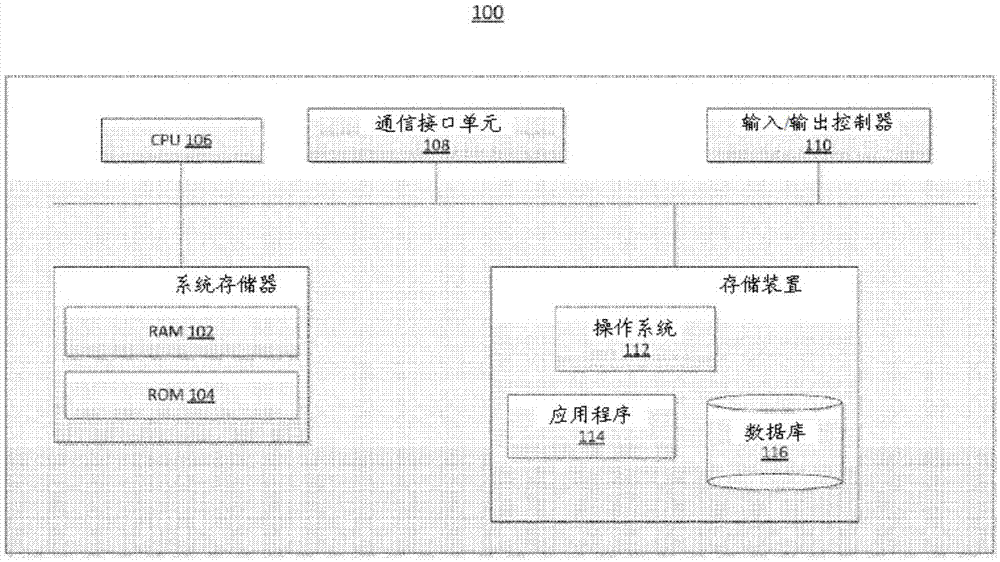
图1是用于进行如根据本发明的方法描述的过程中的任一种的计算设备的说明性实施方案。
图2是根据本发明的方法用于设计并选择探针的代表性过程流程图。
图3是根据本发明的方法用于预测测试受试者的甲基化状态的代表性过程流程图。
图4是根据本发明的方法用于预测测试受试者的疾病状态的另一个代表性且更详细的过程流程图。
图5描绘示例性分子倒置探针(mip)。
图6描绘通过其mip可捕获目标靶序列的过程。
图7描绘通过其捕获的目标靶序列进行索引和扩增的过程。
图8是描绘从甲基化状态测定获得的数据的图形表示的圆环图。
图9描绘对应于两个dna样品的各种百分比混合的重复甲基化评分的数据。
图10提供如何在多个样品中跨多个cpg位点计算甲基化评分的实例。
图11a和图11b描绘通过其mip可捕获亚硫酸氢盐转化的目标靶序列,接着进行创建复制子的延伸和连接反应的过程。复制子随后通过pcr进行扩增以产生包含目标靶序列的pcr产物。
图12描绘用于扩增来自图11b的用于进一步扩增的pcr产物的过程。
图13是用于处理来自图12的用于进一步甲基化分析的扩增产物的代表性过程流程图。
图14示出描绘从结肠直肠样品获得的甲基化数据的图形表示的两个圆环图。
图15a和图15b示出描绘与疾病相关联的基因组不稳定性的结肠直肠癌样品中在整个基因组上的拷贝数改变(cna)的两个曲线图。
图16示出在正常组织相对于肿瘤组织中在整个基因组上具有在cna位点处的甲基化不足的cpg位点的百分比。
图17是用于基于甲基化状态确定来源组织的代表性过程流程图。
图18示出基于甲基化模式的血液来源dna(b)与肝脏dnal)之间的分离。
图19是示出基于甲基化的10种不同组织类型的聚类的树状图。
图20是示出总体甲基化随着受试者的年龄降低的图表。
图21是示出总体甲基化随着胎儿的胎龄降低的图表。
发明详述
本发明提供一种用于检测疾病或病状的系统和方法。需要促进人类疾病的改善的诊断、预后和监控的信息式非侵入性工具。包括癌症发生的若干种复杂疾病在特定基因座处并且更广泛地在整个基因组上显示改变的cpg甲基化。靶向和全基因组亚硫酸氢盐测序方法两者均已用于辨别组织中和细胞-游离dna中的这些变化,但是具有显著的成本和复杂性缺点。为此,发明人已开发用于对来自低至200pg的组织或循环遗传物质的dna输入的简便文库进行测序的单一探针捕获方法。此方法跨基因组同时评估>200,000个位点,所述位点可同时被分析以确定甲基化状态、基因组不稳定性(例如,拷贝数变异)或突变图谱中的一种或多种。此外,发明人已开发识别甲基化的区域和模式、基因组不稳定性的区域、以及在相同类型之间显著不同的突变图谱的方法。
提出以下详细描述以帮助理解各种实施方案。
除非本文另外定义,否则本申请使用的科学和技术术语应具有由本发明所属的领域的普通技术人员通常所理解的含义。通常,与本文所述的细胞和组织培养、分子生物学、细胞生物学、癌症生物学、神经生物学、神经化学、病毒学、免疫学、微生物学、遗传学、蛋白质和核酸化学、化学以及药理学结合使用的命名以及其技术是本领域众所周知和常用的那些命名和技术。本文描述的本发明的每个实施方案可单独采用或与本发明的一个或多个其他实施方案组合采用。
除非另外指示,否则各种实施方案的方法和技术通常是根据本领域众所周知及如本说明书整篇引用和论述的各种一般性和更特定参考文献中所述的分子生物学、细胞生物学、生物化学、微阵列和测序技术来进行。参见例如,motulsky,“intuitivebiostatistics”,oxforduniversitypress,inc.(1995);lodish等,“molecularcellbiology,第4版”,w.h.freeman&co.,newyork(2000);griffiths等,“introductiontogeneticanalysis,第7版”,w.h.freeman&co.,n.y.(1999);gilbert等,“developmentalbiology,第6版”,sinauerassociates,inc.,sunderland,ma(2000)。
本文使用的化学术语根据本领域中的常规用法来使用,如由“themcgraw-hilldictionaryofchemicalterms”,parkers.编,mcgraw-hill,sanfrancisco,c.a.(1985)所示例的。
以上所有内容以及本申请中提及的任何其他出版物、专利和公开的专利申请均以引用的方式明确并入本文。当发生冲突时,以本说明书(包括其特定定义)为准。
在本说明书通篇,词语“包括/包含(comprise)”或变型(诸如“包括/包含(comprises)”或“包括/包含(comprising)”)应理解为暗示包括所述整体(或组件)或整体(或组件)组,而非排除任何其他整体(或组件)或整体(或组件)组。
除非上下文另外明确指示,否则单数形式“一个/种(a)”、“一个/种(an)”以及“所述(the)”包括复数。
术语“包括(including)”被用于意指“包括但不限于”。“包括”和“包括但不限于”可互换使用。
本文提供以下术语和定义。
定义
如本文所用,术语“dna甲基化”是指向cpg双核苷酸之间的胞嘧啶残基的5’碳添加甲基(即,5-甲基胞嘧啶)。dna甲基化可在其他情况下发生在胞嘧啶中,例如chg和chh,其中h是腺嘌呤、胞嘧啶或胸腺嘧啶。胞嘧啶甲基化还可呈5-羟甲基胞嘧啶的形式。dna甲基化可包括非胞嘧啶甲基化,诸如n6-甲基腺嘌呤。
如本文所用,术语“甲基化状态(methylationstate)”或“甲基化状态(methylationstatus)”是指核酸分子或核酸分子的群体的关于某些核苷酸的甲基化的状态。例如,基因组dna在胞嘧啶核苷酸处的某些位点(例如,cpg位点)处甲基化。因此,核酸的甲基化状态可以是指甲基化的基因组中cpg位点的比率或非甲基化的基因组中cpg位点的比率。
如本文所用,术语“甲基化评分”是指根据在cpg位点处观察到的胞嘧啶位点(c)的数量计算的比率。它还可被称为“测试比率”或“参考比率”。甲基化评分提供通常在一个区域上或整个基因组上在一个或多个cpg位点处的未转化(即,甲基化)胞嘧啶核苷酸的比率。甲基化评分可使用以下比率计算:
在cpg位点处的甲基化c/(在cpg位点处的甲基化c+在cpg位点处的非甲基化c)。
虽然核酸分子中的单个cpg位点可甲基化或非甲基化,但是更普遍并且临床上可用的是,确定细胞群体的甲基化状态,其中每个细胞含有独特的二倍体基因组。在一些实施方案中,本文所述的组合物和方法提供所选样品内的总二倍体基因组的子集的甲基化评分。因此,单个cpg位点的集合的二元甲基化状态在细胞群体上进行求和以得出样品中在许多cpg位点上的甲基化评分。在一些实施方案中,一个区域(例如,嵌段、基因、染色体、或整体上)的甲基化评分可计算为个体位点比率的中值、平均值(mean)或平均值(average)。甲基化评分还可表达为比率或百分比。在采用测序读出诸如下一代测序的一些实施方案中,甲基化评分根据包括cpg位点的测序读取中含有的核酸序列信息来计算。因此,甲基化评分可被认为是显示cpg位点处的甲基化的序列读取相对于覆盖cpg位点(无论甲基化或非甲基化)的读取的总数量的比例。在一些实施方案中,如果单个读取包括多个cpg位点,则所述单个读取可生成多个计数。例如,在图10中,被测定覆盖的cpg位点的数量不一定决定甲基化评分。如果是这种情况,如图10所示,样品1和样品2两者中的三个cpg位点将均为甲基化的并且甲基化指数对于两个样品将均为100%。在一些实施方案中,甲基化评分可用于确定单个cpg位点或一系列个体cpg位点的甲基化状态。在一些实施方案中,cpg位点可从分析选择性地过滤掉或者cpg位点可进行分组并且可计算甲基化密度。甲基化评分还可以是个体cpg位点处的甲基化评分的平均值,以便针对在每个个体位点处的不同测序覆盖进行校正。例如,在没有进行平均的情况下,具有100x覆盖的cpg在甲基化评分中可能比具有20x覆盖的位点加权更多。然而,通过对个体胞嘧啶处的甲基化评分进行平均,具有甲基化评分的每个胞嘧啶被给予甲基化评分,无论测序覆盖如何。
在一些实施方案中,甲基化评分可表达为“甲基化密度”,它是限定区域中的cpg位点(例如,特定cpg位点、cpg岛内的cpg位点、或较大区域诸如嵌段)的甲基化评分。例如,人类基因组中1mb仓的甲基化密度可根据显示cpg甲基化的计数的数量除以1mb区域中覆盖cpg位点的计数的总数量来确定。此分析还可针对其他仓大小进行,例如50kb、100kb、200kb、250kb、300kb、400kb、500kb、750kb等。
在一些实施方案中,甲基化评分可表达为“甲基化胞嘧啶的比例”,它包括区域中cpg环境以外的胞嘧啶。
在一些实施方案中,甲基化评分可表达为“整体甲基化评分”或“整体甲基化指数”。整体甲基化指数是指由本文所述的组合物和方法询问的所有cpg位点的甲基化评分,它包括整个基因组上的cpg位点(例如,分布在整个基因组上的大于50,000、60,000、70,000、80,000、100,000、150,000、200,000、300,000、400,000、500,000个或更多个cpg位点);从而允许使用单个测定生成整体甲基化指数。技术人员将理解,并不需要研究基因组中的每个cpg位点来确定整体甲基化指数。例如,cpg位点的子集的甲基化评分可确定为整个基因组的整体甲基化状态的指标,并且作为“整体甲基化指数”给出。
在一些实施方案中,针对测试受试者、样品、组织或其部分确定甲基化评分,在这种情况下,它被称为“测试甲基化评分”或“测试比率”。测试比率可与来自对应的已知(参考)受试者、样品或组织的“参考甲基化评分”或“参考比率”进行比较。例如,可确定来自细胞群体(例如,来自肿瘤、或来自特定组织类型)、多个或混合细胞群体(例如,母体和胎儿细胞)、或多个受试者(例如,吸烟者相对于非吸烟者)的甲基化评分并且与对应的良好表征的参考样品或受试者进行比较。
在一些实施方案中,在测试样品与参考样品之间具有甲基化差异的区域被称为“差异甲基化区域”(dmr),所述区域是具有不同的甲基化评分的区域或嵌段。差异甲基化区域(例如,嵌段、染色体、基因、岛等)通过跨足够数量的样品在测试样品与参考样品之间的甲基化评分的差异被识别为显著的。
如本文所用,术语“位点”是指单个位点,它可以是单个碱基位置或相关碱基位置的组,例如cpg位点;而“嵌段”或“区域”是指包括多个位点的基因组部分。嵌段可包括一个或多个cpg岛、基因、染色质区域诸如大的组织染色质赖氨酸修饰、或核组织区域诸如核纤层蛋白相关结构域。嵌段可包含一个或多个重复元件。本文所述的组合物和方法提供用于识别疾病诸如癌症中的甲基化失调的大规模现象的改进的方法。并非在特定位点处的甲基化变化的特异性靶向,所述的组合物和方法能够测定被认为对于细胞分化和疾病在病理上是重要的基因组的区域。此外,在癌症的情况下,有证据显示表观遗传失调在癌症早期发生-甚至在完全癌症发展之前发生(参见timp等genomemedicine2014,6:61)并且更可能在驻留在alu重复元件中的cpg位点处发生(参见luo等biomedresearchinternational2014);从而为本文所述的方法和组合物增加临床实用性。
如本文所用,术语“甲基化组”是指在细胞群体内在不同位点或区域处的甲基化的量或模式。因此,甲基化组可被认为是特定细胞群体的甲基化评分。例如,疾病状态可具有甲基化组,诸如健康肝脏相对于坏死肝脏。组织类型可具有甲基化组,诸如肝脏甲基化组相对于血液甲基化组。细胞表型可具有甲基化组,诸如衰老细胞相对于分裂细胞。甲基化组可对应于所有基因组、基因组的子集(例如,基因组中的重复元件)、或子集的一部分(例如,发现与疾病相关联的那些区域)。“胎儿甲基化组”对应于胎儿或妊娠女性的甲基化组。胎儿甲基化组可使用各种胎儿组织或胎儿dna的来源(包括胎盘组织和母体血浆中的细胞-游离胎儿dna)来确定。“肿瘤甲基化组”对应于生物体(例如,人类)的肿瘤的甲基化组。肿瘤甲基化组可使用肿瘤组织或细胞-游离肿瘤dna来确定。在某些实施方案中,细胞-游离肿瘤dna存在于血浆中。胎儿甲基化组和肿瘤甲基化组是目标甲基化组的实例。目标甲基化组的其他实例是可将dna贡献到体液(例如,血浆、血清、汗液、唾液、尿液、生殖器分泌物、精液、粪便液、腹泻液、脑脊液、胃肠道的分泌物、腹水、胸膜液、眼内液、来自水囊肿的(例如睾丸的)液体、来自包囊的液体、胰腺分泌物、肠分泌物、痰、泪液、来自乳腺和甲状腺的抽吸液等)中的器官的甲基化组(例如,肝脏、肺、前列腺、胃肠道、膀胱等的甲基化组)。所述器官可以是移植器官。来自血浆的甲基化组可被称为“血浆甲基化组”。血浆甲基化组是细胞-游离甲基化组的实例,因为血浆和血清包含细胞-游离dna(cfdna)。血浆甲基化组也是混合群体甲基化组的实例,因为它是来源自不同组织或器官的胎儿/母体甲基化组或肿瘤/非肿瘤甲基化组或dna的混合物。
如本文所用,术语“读取”是指测序系统(诸如大规模平行测序)的原始或处理输出。在一些实施方案中,本文所述的方法和组合物的输出是读取。在一些实施方案中,这些读取可能需要进行修整、过滤和对齐,从而产生原始读取、修整读取、对齐读取。如本文所用,术语“计数”是指目标靶序列内的独特对齐读取。在甲基化评分的情况下,计数对应于从在cpg位点处的读取(甲基化或非甲基化)检索的信息。因此,如果读取涵盖多个cpg位点,则此读取可产生多个计数。
在某些实施方案中,所述方法可用于检测拷贝数变异。如本文所用,“拷贝数变异”(cnv)通常是遗传变异或染色体畸变的一个种类或类型。在一些情况下,拷贝数变异是指生殖系细胞中拷贝数的变化,而拷贝数改变/畸变(cna)是指已在体细胞组织中(例如,在肿瘤细胞中)出现的拷贝数的变化。如本文所用,拷贝数变异包括拷贝数改变/畸变。拷贝数变异可以是缺失(例如微缺失)、重复(例如微重复)、或插入(例如微插入)。在某些实施方案中,如本文所用,前缀“微”可以是指长度小于5个碱基对的核酸区段。拷贝数变异可包括染色体区段的一个或多个缺失(例如微缺失)、重复和/或插入(例如微重复、微插入)。在某些实施方案中,重复包括插入。在某些实施方案中,插入是重复。在某些实施方案中,插入不是重复。例如,一部分中的序列重复增加发现重复的部分的计数。通常一部分中的序列重复增加高度或水平。在某些实施方案中,在构成第一高度或水平的部分中存在的重复相对于其中不存在重复的第二高度或水平增加高度或水平。在某些实施方案中,插入增加部分的计数,并且表示插入的序列在同一部分内的另一个位置处存在(即重复)。在某些实施方案中,插入不显著增加部分的计数或者高度或水平,并且插入的序列不是同一部分内的序列的重复。在某些实施方案中,插入未被检测或被表示为重复,并且表示插入的重复序列不存在于同一部分中。在一些实施方案中,拷贝数变异是胎儿拷贝数变异。通常,胎儿拷贝数变异是胎儿基因组中的拷贝数变异。在一些实施方案中,拷贝数变异是母体和/或胎儿拷贝数变异。在某些实施方案中,母体和/或胎儿拷贝数变异是妊娠女性(例如,怀孕的女性受试者)、已生产的女性受试者或能够怀孕的女性的基因组内的拷贝数变异。拷贝数变异可以是杂合拷贝数变异,其中所述变异(例如,重复或缺失)存在于基因组的一个等位基因上。拷贝数变异可以是纯合拷贝数变异,其中所述变异存在于基因组的两个等位基因上。在一些实施方案中,拷贝数变异是杂合或纯合的胎儿拷贝数变异。在一些实施方案中,拷贝数变异是杂合或纯合的母体和/或胎儿拷贝数变异。拷贝数变异有时存在于母体基因组和胎儿基因组中,母体基因组中而不是胎儿基因组中,或者胎儿基因组中而不是母体基因组中。
如本文所用,术语“非整倍性”是指特征在于染色体数量的异常变异的染色体异常,例如染色体数量不是染色体单倍体数量的确切倍数。例如,整倍体个体将具有等于2n的染色体数量,其中n为单倍体个体中的染色体数量。在人类中,单倍体数量为23因此,二倍体个体将具有46条染色体。非整倍体个体可能含有染色体的额外拷贝(所述染色体三体性)或缺少染色体的拷贝(所述染色体单体性)。异常变异是关于每个单独的染色体。因此,具有三体性和单体性的个体尽管具有46条染色体但为非整倍体。非整倍性疾病或病状的实例包括但不限于唐氏综合征(21号染色体三体性)、爱德华氏综合征(18号染色体三体性)、帕陶综合征(13号染色体三体性)、特纳综合征(女性x染色体单体性)和克兰费尔特综合征(男性x染色体的额外拷贝)。另外,非非整倍体染色体异常包括易位(其中染色体的一个区段已转移到另一条染色体)、缺失(其中染色体的一片已经丢失)以及其他类型的染色体损伤(例如脆性x综合征,其由异常地易感损伤的x染色体引起)。
如本文所用,术语“受试者”和“患者”是指任何动物,诸如狗、猫、鸟、家畜,并且特别是哺乳动物,并且优选人类。术语“参考受试者”和“参考患者”是指表现出已知基因型(例如,已知的整倍性或非整倍性)、表型或衰老的任何受试者或患者,或者是指已知患有疾病或病状、或已知未患有疾病或病状、或已知具有疾病或病状的特定状态、或已知具有对于疾病或病状的易感性、或已知经受药物、毒素、特定饮食、或怀疑导致甲基化变化的药剂或条件的受试者或患者。技术人员将理解,受试者可以是任何人类。在某些实施方案中,受试者是妊娠女性。在这些实施方案中,血液样品可以是母体血浆或血清样品。在某些实施方案中,受试者时器官移植物接受者,并且受试者的甲基化状态可指示器官排斥。在某些实施方案中,具有目标靶序列的群体的(例如,胎儿、肿瘤或疾病起源的)甲基化状态可在具有目标靶序列的背景(例如,母体、非肿瘤或无疾病起源)中确定。背景目标靶序列可充当参考,其中与参考的差异指示疾病或病状,或识别核酸种类。
如本文所用,术语“突变图谱”是通常跨越基因组的突变的集合的累积频率。构成突变图谱的突变类型包括但不限于单核苷酸变异、缺失和插入,并且突变类型还告知给定的突变图谱或模式。与疾病或病状相关联的特定突变图谱的实例包括与香烟烟雾暴露相关联的c>a颠换的增加频率(ding,l.等somaticmutationsaffectkeypathwaysinlungadenocarcinoma.nature455,1069–1075(2008))、以及与12种癌症类型相关联的c>t转换和c>g颠换的增加频率(kandoth,cyriac等“mutationallandscapeandsignificanceacross12majorcancertypes.”nature502.7471(2013):333-339)。
如本文所用,术语“来源组织”是指样品中的核酸的组织来源,其中“组织”用于描述同一类型的细胞的组或群体。一些组织可具有多种细胞类型,例如肝细胞、肺泡细胞或血细胞,而其他组织可来源于不同的生物体,例如母体和胎儿,或来源于健康相对于疾病组织。同样,“参考组织”可用于确定甲基化特异性组织模式或水平。例如,来自多个受试者的参考组织可用于确定测试样品的组织组分。在一些实施方案中,不同来源的dna分子是母体来源的dna分子和胎儿来源的dna分子。在一些实施方案中,不同来源的dna分子是第一组织来源的dna分子和第二组织来源或白细胞来源的dna分子。在一些实施方案中,确定来源组织可指示疾病(例如,癌症)的存在,或者可用于确定来自特定组织的dna(例如,母体样品中的胎儿cfdna)的相对量或绝对量。因此,本文所述的组合物和方法可用于区分并识别dna的组织来源。鉴于组织的未知来源,dna可使用标准方法来提取,本文所述的组合物和方法可用于生成组织特异性数据,并且所述数据可适配最可能的组织参考仓,如在实施例中进一步描述的。
如本文所用,术语“多核苷酸”、“核酸”和“核酸分子”可互换使用,并且是指dna分子(例如,cdna或基因组dna)、rna分子(例如,mrna)、dna-rna杂交体以及使用核苷酸类似物生成的dna或rna的类似物。核酸分子可以是核苷酸、寡核苷酸、双链dna、单链dna、多链dna、互补dna、基因组dna、非编码dna、信使rna(mrna)、微小rna(mirna)、核仁小rna(snorna)、核糖体rna(rrna)、转运rna(trna)、小干扰rna(sirna)、不均一核rna(hnrna)或小发夹rna(shrna)。在某些实施方案中,所述方法可在核酸样品(诸如dna或rna,例如基因组dna)上进行。在一些实施方案中,核酸分子可以是细胞-游离dna(cfdna)。细胞-游离dna被认为是由细胞坏死或凋亡引起,其中基因组细胞dna被消化并且成为片段化的胞外dna。凋亡来源的细胞-游离dna可来自非宿主(例如,移植的器官或组织)、胎儿(例如,来自产生细胞-游离胎儿dna的胎盘)、或患病组织(例如,来自产生循环肿瘤dna的肿瘤)。细胞-游离dna可在一系列样品中检测,所述样品包括但不限于血液、血浆和尿液。在一些实施方案中,核酸分子与外来体缔合,所述外来体是从各种不同细胞(包括癌细胞)释放的微泡。在一些实施方案中,本文所述的组合物和方法可能够基于其甲基化状态区分坏死和凋亡来源的cfdna。核酸样品可以本领域普通技术人员已知的任何方式分离(例如,通过离心)。
如本文所用,术语“样品”是指通常来源自生物流体、细胞、组织、器官或生物体的样品,其包含核酸或核酸混合物,所述核酸或核酸混合物包含至少一个待筛查例如癌症或非整倍性的核酸序列。在一些实施方案中,样品是血液样品,诸如全血样品、血清样品或血浆样品。在一些实施方案中,样品包含至少一个怀疑其基因组已发生变异的核酸序列。此类样品包括但不限于痰/口腔液、羊膜液、血液、血液级分或细针活检样品(例如手术活检、核芯针活检、细针活检等)、尿液、粪便、腹膜液、胸膜液、脑脊液、胃肠液、细胞系、包埋在石蜡中的组织、新鲜冷冻组织等。虽然样品通常取自人类受试者(例如,患者),但是所述测定可用于在来自任何哺乳动物的样品中检测疾病或病状、或检测疾病或病状的状态、或确定受试者是否具有对于疾病或病状的易感性,所述哺乳动物包括但不限于狗、猫、马、山羊、绵羊、牛、猪等。所述样品可在从生物来源获得时直接使用,或者在预处理以改变样品的特征之后使用。例如,这种预处理可包括由血液制备血浆、稀释粘性流体等。预处理的方法还可涉及但不限于亚硫酸氢盐转化、过滤、沉淀、稀释、蒸馏、混合、离心、冷冻、冻干、浓缩、扩增、核酸片段化、干扰组件的失活、试剂的添加、溶胞等。如果关于样品采用此类预处理方法,则此类预处理方法通常使得目标核酸优选以与未处理的测试样品(例如,即未经受任何此类预处理方法的样品)中的浓度成比例的浓度保留在测试样品中。根据所使用的样品的类型,可使用处理方法来进行另外的处理和/或纯化步骤以获得所需纯度或大小的核酸片段,所述处理方法包括但不限于超声处理、雾化、凝胶纯化、pcr纯化系统、核酸酶裂解、特定大小捕获或排阻、靶向捕获或这些方法的组合。任选地,可在进一步分析之前从样品中分离细胞-游离dna。在一些实施方案中,样品来自其疾病或病状将由本发明的系统和方法来确定的受试者,也称为“测试样品”。
如本文所用,术语“mip”是指分子倒置探针(也称为环状捕获探针)。如本文所用,术语“引物”、“探针”或“捕获探针”在其选择性地结合到核酸分子的能力的情况下也可以是指mip。分子倒置探针是核酸分子,其含有两个靶向性多核苷酸臂、一个或多个独特分子标签(也称为独特分子标识符(umid))和多核苷酸接头(例如,通用主链接头)。多核苷酸接头的范围可以是10、20、30、40、50、60、70、80、90、100、125、150、175、200、225、250、275、300、400、500、1000、1500、2000个或更多个碱基。参见例如图5。在一些实施方案中,mip可包含多于一个独特分子标签,诸如两个独特分子标签、三个独特分子标签或更多个。在一些实施方案中,每个mip中的多核苷酸臂位于mip的5'和3'端,而独特分子标签和多核苷酸接头位于中间。例如,在一些实施方案中,所述mip依次包含以下组件:第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂。在一些实施方案中,mip中的多核苷酸接头(或主链接头)在本发明的方法中使用的所有mip中是通用的。在一些实施方案中,所述mip可不包含任何独特分子标签。
在mip中,多核苷酸臂被设计成在基因组核酸样品中的靶序列(或位点)的上游和下游杂交。这些多核苷酸臂与在基因组核酸样品中的侧接靶序列的一个或多个重复序列(本文中被称为“空位序列”或“独特空位序列”)基本上互补。在一些实施方案中,空位序列的长度是1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、40、50、60、70、80、90、100、100、125、150、175、200、225、250、275、300、400、500、1000、1500、2000个碱基或更大。当询问细胞-游离dna时,空位序列的长度通常小于150或200个碱基。在一些实施方案中,靶向性多核苷酸臂包含连接序列和延伸序列。mip可包含与dna样品中的多个重复序列基本上互补的靶向性多核苷酸臂。例如,mip可与dna样品(例如,包含人类基因组的样品)中的数十个、数百个、数千个、数十万个、或数百万个目标靶序列杂交。在一些实施方案中,mip靶向例如大于1,000、大于10,000、大于20,000、大于30,000、大于40,000、大于50,000、大于60,000、大于70,000、大于80,000、大于90,000、大于100,000、大于200,000、大于300,000、大于400,000、大于500,000、大于600,000、大于700,000、大于800,000、大于900,000、和/或大于1,000,000个目标靶序列。在一些实施方案中,“基本上互补”是指两个臂中的0个错配或仅一个臂中的最多1个错配(例如,当靶向性多核苷酸臂与核酸中的分别侧接目标位点的第一区域和第二区域杂交时)。在一些实施方案中,“基本上互补”是指在两个臂中最多有少量错配,诸如1、2、3、3、5、6、7或8个错配。
术语“靶序列”、“目标序列”和“目标靶序列”可互换使用来是指由本发明的引物或探针结合或捕获的序列并且包含一个或多个cpg位点。除其他考虑之外,目标靶序列被选择来包含至少一个cpg位点;然而,并非每一个由本发明的引物或探针捕获的核酸序列均包含cpg位点。在一些实施方案中,由本发明的引物或探针捕获的核酸序列中的30%或更多、31%或更多、32%或更多、33%或更多、34%或更多、35%或更多、36%或更多、37%或更多、38%或更多、39%或更多、40%或更多、41%或更多、42%或更多、43%或更多、44%或更多、45%或更多、46%或更多、47%或更多、48%或更多、49%或更多、50%或更多、51%或更多、52%或更多、53%或更多、54%或更多、55%或更多、56%或更多、57%或更多、58%或更多、59%或更多、60%或更多、61%或更多、62%或更多、63%或更多、64%或更多、65%或更多、66%或更多、67%或更多、68%或更多、69%或更多、70%或更多包含一个或多个cpg位点。这些目标靶序列可包括靶向性多核苷酸臂与其杂交的重复序列以及重复序列侧接的空位序列。在某些实施方案中,重复序列在与靶向性多核苷酸臂杂交时具有0、1、2、3、4或更多个错配。在一些实施方案中,相同的重复序列存在于目标靶序列中,在这种情况下,靶多核苷酸臂是相同的。在其他实施方案中,两个不同的重复序列(例如,重复a和重复b)存在于目标靶序列中,在这种情况下,靶多核苷酸臂不是相同的。在具体实施方案中,重复序列在与靶向性多核苷酸臂杂交时具有0或1个错配。在一些实施方案中,mip结合到alu重复序列。在一些实施方案中,mip不结合基因组中的长散布核苷酸元件(line)。
在一些实施方案中,独特分子标签是随机生成的短核苷酸序列。在某些实施方案中,独特分子标签设计成不与位于基因组核酸片段上或基因组核酸样品中的任何序列或位点杂交。在某些实施方案中,独特分子标签是可被并入或连接到核酸(例如,多核苷酸)的具有合适的可检测标记的任何标签,其允许检测和/或识别包含或连接于所述标签的核酸。在某些实施方案中,具有足够长度的独特分子标签以确保每个mip包含分子标签的独特组合的浓度被引入,从而使得每个捕获事件不同。通过追踪个体捕获事件,能够识别重复并且减少捕获偏差。虽然本文所述的发明已实现几乎一致的捕获效率,因为使用相同的捕获探针来询问整个基因组上的许多位点,但是将捕获偏差考虑在内(即,针对捕获效率的差异进行归一化)的能力进一步提高所述测定的定量方面,例如当进行cnv分析时。在一些实施方案中,在测序方法期间(例如通过聚合酶)将标签并入或连接到核酸。标签的非限制性实例包括核酸标签、核酸索引或条形码、放射性标记(例如,同位素)、金属标记、荧光标记、化学发光标记、磷光标记、荧光淬灭剂、染料、蛋白质(例如,酶、抗体或其部分、接头、结合对的成员)等或其组合。在一些实施方案中,标签(例如,核酸索引或条形码)是核苷酸或核苷酸类似物的独特的、已知的和/或可识别的序列。在一些实施方案中,标签或umid通过在生物信息学分析期间允许识别独特分子来帮助减少或去除扩增误差和测序误差。在一些实施方案中,标签是四个、五个、或六个或更多个连续核苷酸。包含寡核苷酸的独特分子标识符描述于美国专利申请号11/186,636,其作为us20070020640a1公开,并且在mip的情况下寡核苷酸独特分子标识符的用途描述于美国专利申请号12/027,039,其作为us20080269068a1公开。多种荧光团可用于各种不同的激发和发射光谱。任何合适类型和/或数量的荧光团均可用作标签。在一些实施方案中,1个或多个、2个或更多个、3个或更多个、4个或更多个、5个或更多个、6个或更多个、7个或更多个、8个或更多个、9个或更多个、10个或更多个、20个或更多个、30个或更多个、50个或更多个、100个或更多个、500个或更多个、1000个或更多个、10,000个或更多个、100,000个或更多个、10^6个或更多个、10^7个或更多个、10^8个或更多个、10^9个或更多个、10^10个或更多个、10^11个或更多个、10^12个或更多个不同的标签在本文所述的方法(例如,核酸检测和/或测序方法)中使用。在一些实施方案中,一种或两种类型的标签(例如,荧光标记)与文库中的每种核酸连接。在一些实施方案中,染色体特异性标签用于使染色体计数更快或更容易。标签的检测和/或定量可通过合适的方法、机器或设备来进行,所述合适的方法、机器或设备的非限制性实例包括流式细胞术、定量聚合酶链式反应(qpcr)、凝胶电泳、发光计、荧光计、分光光度计、合适的基因芯片或微阵列分析、蛋白质印迹、质谱、色谱、细胞荧光分析、荧光显微镜、合适的荧光或数字成像方法、共聚焦激光扫描显微镜、激光扫描细胞术、亲和色谱、手动分批模式分离、电场悬浮、合适的核酸测序方法和/或核酸测序装置等及其组合。在特定的实施方案中,标签适合与微阵列分析一起使用。
将mip引入核酸(例如,核酸片段)以进行对位于核酸样品(例如,基因组dna)上的靶序列或位点的捕获。在一些实施方案中,例如,如果基因组dna存在于样品中,则片段化可帮助分子倒置探针捕获靶核酸。如本文更详细描述的,在捕获目标靶序列(例如,基因座)之后,所捕获的靶标可进一步经受酶促空位填充和连接步骤,使得靶序列的拷贝被并入圆环中,所述圆环在本文中被称为复制子。通过延长杂交和空位填充孵育期,可改进mip对核酸片段上的靶序列的捕获效率。(参见例如,turnereh,等,natmethods.2009年4月6:1-2.)。
mip技术可用于检测或扩增复杂混合物中的特定核酸序列。使用mip技术的优点之一在于其高度复用的能力,所述mip技术允许在含有数千个mip的单个反应中捕获数千个靶序列。mip技术的各个方面描述于,例如,hardenbol等,“multiplexedgenotypingwithsequence-taggedmolecularinversionprobes,”naturebiotechnology,21(6):673-678(2003);hardenbol等,“highlymultiplexedmolecularinversionprobegenotyping:over10,000targetedsnpsgenotypedinasingletubeassay,”genomeresearch,15:269-275(2005);burmester等,“dmetmicroarraytechnologyforpharmacogenomics-basedpersonalizedmedicine,”methodsinmolecularbiology,632:99-124(2010);sissung等,“clinicalpharmacologyandpharmacogeneticsinagenomicsera:thedmetplatform,”pharmacogenomics,11(1):89-103(2010);deeken,“theaffymetrixdmetplatformandpharmacogeneticsindrugdevelopment,”currentopinioninmoleculartherapeutics,11(3):260-268(2009);wang等,“highqualitycopynumberandgenotypedatafromffpesamplesusingmolecularinversionprobe(mip)microarrays,”bmcmedicalgenomics,2:8(2009);wang等,“analysisofmolecularinversionprobeperformanceforallelecopynumberdetermination,”genomebiology,8(11):r246(2007);ji等,“molecularinversionprobeanalysisofgenecopyalternationsrevealsdistinctcategoriesofcolorectalcarcinoma,”cancerresearch,66(16):7910-7919(2006);以及wang等,“allelequantificationusingmolecularinversionprobes(mip),”nucleicacidsresearch,33(21):e183(2005),其每一个出于所有目的特此以引用的方式整体并入。还参见美国专利号6,858,412;5,817,921;6,558,928;7,320,860;7,351,528;5,866,337;6,027,889以及6,852,487,所述专利各自出于所有目的特此以引用的方式整体并入。
先前mip技术已成功应用于其他研究领域,包括癌症中的生物标志物的新型识别和细分。参见例如,brewster等,“copynumberimbalancesbetweenscreen-andsymptom-detectedbreastcancersandimpactondisease-freesurvival,”cancerpreventionresearch,4(10):1609-1616(2011);geiersbach等,“unknownpartnerforusp6andunusualss18rearrangementdetectedbyfluorescenceinsituhybridizationinasolidaneurysmalbonecyst,”cancergenetics,204(4):195-202(2011);schiffman等,“oncogenicbrafmutationwithcdkn2ainactivationischaracteristicofasubsetofpediatricmalignantastrocytomas,”cancerresearch,70(2):512-519(2010);schiffman等,“molecularinversionprobesrevealpatternsof9p21deletionandcopynumberaberrationsinchildhoodleukemia,”cancergeneticsandcytogenetics,193(1):9-18(2009);press等,“ovariancarcinomaswithgeneticandepigeneticbrca1losshavedistinctmolecularabnormalities,”bmccancer,8:17(2008);以及deeken等,“apharmacogeneticstudyofdocetaxelandthalidomideinpatientswithcastration-resistantprostatecancerusingthedmetgenotypingplatform,”pharmacogenomics,10(3):191-199(2009),其每一个出于所有目的特此以引用的方式整体并入。
mip技术还已被应用于识别新的药物相关的生物标志物。参见例如,caldwell等,“cyp4f2geneticvariantaltersrequiredwarfarindose,”blood,111(8):4106-4112(2008);以及mcdonald等,“cyp4f2isavitamink1oxidase:anexplanationforalteredwarfarindoseincarriersofthev433mvariant,”molecularpharmacology,75:1337-1346(2009),其每一个出于所有目的特此以引用的方式整体并入。其他mip应用包括药物开发和安全性研究。参见例如,mega等,“cytochromep-450polymorphismsandresponsetoclopidogrel,”newenglandjournalofmedicine,360(4):354-362(2009);dumaual等,“comprehensiveassessmentofmetabolicenzymeandtransportergenesusingtheaffymetrixtargetedgenotypingsystem,”pharmacogenomics,8(3):293-305(2007);以及daly等,“multiplexassayforcomprehensivegenotypingofgenesinvolvedindrugmetabolism,excretion,andtransport,”clinicalchemistry,53(7):1222-1230(2007),其每一个出于所有目的特此以引用的方式整体并入。mip技术的另外的应用包括基因型和表型数据库化。参见例如,man等,“geneticvariationinmetabolizingenzymeandtransportergenes:comprehensiveassessmentin3majoreastasiansubpopulationswithcomparisontocaucasiansandafricans,”journalofclinicalpharmacology,50(8):929-940(2010),其出于所有目的特此以引用的方式整体并入。
如本文所用,术语“捕获(capture)”或“捕获(capturing)”是指引物或探针(例如,分子倒置探针)与对应的靶向性位点之间的结合或杂交反应。
如本文所用,术语“灵敏度”是指测定(例如,方法、测试)的性能的统计学量度,其通过将真阳性的数量除以真阳性和假阴性的总和来计算。
如本文所用,术语“特异性”是指测定(例如,方法、测试)的性能的统计学量度,其通过将真阴性的数量除以真阴性和假阳性的总和来计算。
如本文所用,术语“复制子”是指通过捕获反应或扩增反应生成的核酸。在一些实施方案中,扩增子是单链核酸分子。在一些实施方案中,扩增子是单链环状核酸分子。在一些实施方案中,扩增子是双链核酸分子。例如,mip捕获靶序列或位点或者与靶序列或位点杂交。在捕获反应或杂交之后,引入连接/延伸混合物来延伸并连接两个靶向性多核苷酸臂之间的空位区域以形成单链环状核苷酸分子,即mip复制子。复制子中的空位填充序列可被认为是“插入物”或“插入物序列”。可通过聚合酶链式反应(pcr)来扩增mip复制子以产生多个mip扩增子,其为双链核苷酸分子。可以由多个第一目标靶序列(例如,含有已知或怀疑的cpg位点的序列)和多个第二目标靶序列(例如,分布在整个基因组中的靶序列)来产生mip复制子和扩增子。
如本文所用,术语“测序”在广义上使用,并且可以是指本领域已知的允许识别核酸的至少一部分(包括但不限于延伸产物或载体插入片段的至少一部分)中的至少一些连续核苷酸的顺序的任何技术。测序还可以是指允许检测核酸序列中的核苷酸碱基之间的差异的技术。示例性测序技术包括靶向测序、单分子实时测序、基于电子显微镜的测序、晶体管介导的测序、直接测序、随机鸟枪测序、桑格双脱氧终止测序、靶向测序、外显子测序、全基因组测序、杂交测序(例如,在诸如微阵列的阵列中)、焦磷酸测序、毛细管电泳、凝胶电泳、双重测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模平行签名测序、乳液pcr、低变性温度共扩增pcr(co-amplificationatlowerdenaturationtemperature-pcr)(cold-pcr)、多重pcr、通过可逆染料终止子的测序、双端测序(paired-endsequencing)、近期测序、核酸外切酶测序、边连接边测序(sequencingbyligation)、短读取测序、单分子测序、边合成边测序(sequencing-by-synthesis)、实时测序、反向终止子测序、离子半导体测序、纳米球测序、纳米孔测序、454测序、solexa基因组分析仪测序、miseq(illumina)、hiseq2000(illumina)、hiseq2500(illumina)、illumina基因组分析仪(illumina)、iontorrentpgmtm(lifetechnologies)、miniontm(oxfordnanoporetechnologies)、实时smrttm技术(pacificbiosciences)、探针-锚定分子连接(cpaltm)(completegenomics/bgi)、
本文所述的方法和组合物可替代地采用微阵列技术来定量mip产物。“微阵列”或“阵列”是指具有表面的固相支持物,所述表面优选但非排他地为平面或基本上平坦的表面,其携带含有核酸的位点阵列使得所述阵列的每个位点包含基本上相同或相同的寡核苷酸或多核苷酸的拷贝,并且在空间上限定并且不与阵列的其他成员位点重叠;也就是说,所述位点在空间上是离散的。阵列或微阵列还可包含具有表面的非平面可询问结构,诸如珠粒或孔。阵列的寡核苷酸或多核苷酸可与固体支持物共价结合,或者可非共价结合。常规微阵列技术综述于例如schena,编,microarrays:apracticalapproach,irlpress,oxford(2000)。“阵列分析”、“通过阵列分析”或“通过微阵列分析”是指使用微阵列对一种或多种生物分子的分析例如像序列分析。在一些实施方案中,每个样品单独与单个微阵列杂交。在其他实施方案中,通过将多个微阵列物理连接到单个多微阵列板上可增强处理通量从而实现便利的高通量处理。在某些实施方案中,可制造例如来自affymetrix公司(santaclara,calif.,usa)的定制dna微阵列以特异性定量mip测定的产物。
本领域普通技术人员应理解,本文所述的组合物和方法可如适用于正在讨论的应用进行适配和修改,并且本文所述的组合物和方法可用于其他合适的应用中,并且此类其他添加和修改将不脱离本发明的范围。
本发明将根据以下实验细节得到更好的理解。然而,本领域的技术人员将易于理解,所论述的特定方法和结果仅仅是说明性的,在随后的实施方案中将对本发明进行更充分地描述。
用于检测甲基化状态的方法
用于检测甲基化状态的现有方法采用全基因组测序技术,所述技术固有地需要大量的输入dna和大量的读取来实现所需甲基化位点的覆盖。本发明的实施方案提供对于现有甲基化检测方法的问题的解决方案。这些实施方案用捕获方法替代了先前的文库制备,所述捕获方法使用包含与重复序列杂交的靶向性多核苷酸臂的少量寡核苷酸mip,所述臂是连接到高性能通用主链结构的臂。这些mip被设计成侧接并且合并在整个人类基因组上的独特对齐序列,但是富含与甲基化有关的靶标(即,含有cpg位点的靶标)。cpg位点是位于整个基因组上的位点,其中甲基化在所述位点的胞嘧啶核苷酸处发生。通过进行磺化、水解脱氨和脱磺(例如,亚硫酸氢盐转化或简单地脱氨),将非甲基化的胞嘧啶转化为尿嘧啶。相比之下,甲基化的胞嘧啶被保护而不受此反应影响,并且因此保留胞嘧啶。因此,通过进行亚硫酸氢盐转化或保护甲基化的胞嘧啶的可替代程序并且随后对cpg位点进行测序,本文所述的实施方案提供一种检测cpg位点是否甲基化的方法。设想的选择捕获分子的方法实现在所需的区域中选择独特序列以用于定量,并且不依赖于在合宜的重复序列扩增中存在一些独特序列。
在优化的捕获方法中使用重复序列允许在文库制备期间在针对单分子动力学产生带条形码的靶标时在很少或没有相似序列的干扰的情况下密集平铺靶区域。在一些实施方案中,所述方法比先前的方法更具有经济效益。具体地,这些方法通过使用仍能够调查全基因组指数的少量捕获试剂(引物或探针)来提供节约。此外,捕获试剂不仅可提供关于甲基化状态的信息,并且更通常地还提供关于靶位点的序列的信息。此信息可用于确定例如拷贝数变异或突变图谱。此信息还可用于检测染色体异常,例如非整倍性诸如三体性,或组织特异性甲基化评分和模式以及疾病或病状特异性突变图谱或模式,例如血液中组织特异性循环肿瘤dna(ctdna)的存在。
在一些实施方案中,所述方法还提供一种在易于多重使用的测定中具有低读取计数的快速分析。例如,在所述方法内可使用多层独特分子标签和/或条形码来识别特定的引物种类以及去卷积多重数据以将信号追溯到个体样品。例如,第一mip群体可用于获得甲基化状态(以及任选地序列信息),而第二mip群体提供序列信息。此外,所述方法可用于超低覆盖的应用,诸如在100%胎儿样品诸如受孕产物或非胎儿诊断性样品中检测三体性。样品可进行混合(例如,胎儿相对于母体或患病相对于未患病)或不混合(例如,怀疑患有疾病或病状的个体),在这种情况下,“覆盖”或读深可能相当低,因为信号会很强。在一些实施方案中,所述方法与全基因组测序、全外显子组测序和靶向测序相比也是快的。本文所述的方法还提供需要与全基因组亚硫酸氢盐测序相比相对少量的输入dna的优点,所述全基因组亚硫酸氢盐测序在严苛的亚硫酸氢盐转化过程期间具有输入dna损失的问题,而本文所述的方法允许在转化步骤之后捕获亚硫酸氢盐转化的dna,从而保护输入dna并且减少偏差。更具体地,大部分文库制备试剂盒需要双链输入dna用于衔接子连接步骤。因为亚硫酸氢盐转化使dna变性,所以亚硫酸氢盐转化步骤需要在衔接子连接步骤之后但是在pcr之前进行。严苛的亚硫酸氢盐转化可损害一些连接的分子,并且因此使其不可用。另外,使用常规方法,连接衔接子需要进行甲基化,否则胞嘧啶将被转化,这增加另外的成本。
在一些实施方案中,所述方法涉及遗传分析领域。通常,这些方法可用作检测并定量甲基化状态的快速且经济的手段。因为甲基化状态可通过测序来确定,所以通过本文所述的方法获得的序列信息允许检测突变并且在从完整染色体和染色体的臂延伸到微观缺失和重复、亚微观缺失和缺失以及甚至单核苷酸特征(包括单核苷酸多态性、缺失和插入)的范围内检测遗传特征的缺失和重复。在某些实施方案中,这些方法可用于检测亚染色体遗传损伤,例如微缺失。此外,所述方法可用于确定突变或与疾病或病状相关的其他序列元件(例如,通过检测一个snp或多个snp)。因为所述方法在单个测定中提供不同类型的信息,所以它们比当前的方法更简单、更高效并且更便宜。在某些实施方案中,所述方法还提供最大可能性估算(k),这实现增加的准确性和探针捕获效率的估算并且减少对于在拷贝数变异(cnv)检测期间的外来测序的需要。这可能由于探针统一性而产生低变异系数(cv),因为使用少量的探针(例如,一个、两个或更多个)。这些捕获探针允许另外探针的组合而没有干扰或交叉测定反应。将来自若干探针及其独特读取的信息组合起来极大地减少所述系统的误差。事实上,靶向探针增加可极大地增强测定实用性,同时减少成本。
与靶向测序相比,一些实施方案提供的方法具有特定优点。在某些实施方案中,本文所述的方法使用捕获点处两个序列元件的同时识别,并且两个臂被限制接近。相比之下,典型的靶向测序方法将允许聚合酶在单个位点处起始。由典型测序创建的产物上运行(runon-product)低效率地产生,但也可能产生具有第二引物的内部或“脱靶引发”。一些实施方案的核酸的固有“双重识别”增加了严格性,即延续到通过mip结构中的分子标识符元件进行定量的效应。可将独特分子标签置于mip主链中的一个位点处,但是在使用分子标识符的标准靶向测序中,在两个引物中使用随机序列。另外,所述方法允许较低的试剂成本,因为与靶向测序所需的数百或数千个多重pcr引物相比,可用非常少的mip实现在整个基因组上的覆盖。尽管如此,所述方法仍然享有靶向测序显示优于鸟枪法的大部分(如果不是所有的话)经济和性能优点。
总之,一些实施方案的方法和核酸提供优于先前所述的遗传方法的明显优点。例如,全基因组测序和大规模平行签名测序通常需要基因组的大量非信息部分的昂贵分析;而本发明的方法可使用一部分基因组产生类似的答案,从而减少了测定成本和时间。其他方法依赖选择性测定基因组的信息部分。虽然某些方面具有一些相似性,但是在一些实施方案中,所述方法使用识别重复引物结合位点的新型综合方法,本发明的方法允许更大的测定设计参数(序列不可知–例如但不限于重复线性元件)、更多候选引物(例如,因为列举了所有可能的引物)、对于临床实用性而言足够特异和敏感的简单更低成本的测定以及更大的多重能力。
本文所述的组合物和方法可用于通过血浆dna的序列分析组装甲基化组。根据母体血浆确定胎盘或胎儿甲基化组的能力提供一种确定、检测并监测与怀孕相关病状(诸如先兆子痫、宫内生长受限、早产以及其他病状)相关联的异常甲基化特征的非侵入性方法。例如,检测疾病特异性异常甲基化特征允许筛查、诊断并监测此类怀孕相关病状。测量母体血浆甲基化水平允许筛查、诊断并监测此类怀孕相关病状。除关于怀孕相关病状的调查的直接应用以外,所述方法可应用于目的在于血浆dna分析的医学的其他领域。例如,癌症的甲基化组可根据癌症患者的血浆dna确定。如本文所述,根据血浆进行的癌症甲基化组分析潜在地是根据血浆(例如,检测众所周知的癌症相关体细胞突变)进行癌症基因组分析的协同技术。
对于早期癌症检测,确定甲基化状态可用于筛查癌症。当血浆样品的甲基化测试比率显示与健康对照(参考比率)相比的异常水平时,可怀疑出现癌症。使用本文所述的组合物和方法,可进行癌症类型和癌症的来源组织的进一步确认和评估。本文所述的组合物和方法还允许检测肿瘤相关拷贝数畸变、染色体易位和在整个基因组上的单核苷酸变体(突变图谱)。在一些实施方案中,放射和成像调查(例如,计算机断层扫描、磁共振成像、正电子发射断层扫描)或内镜检查(例如,上胃肠道内镜检查或结肠镜检查)可用于基于血浆甲基化评分进一步调查怀疑患有癌症的个体。
对于癌症筛查或检测,确定血浆(或其他生物学)样品的甲基化评分可与用于癌症筛查或检测的其他形式结合使用,诸如前列腺特异性抗原测量(例如,用于前列腺癌)、癌胚抗原(例如,用于结肠直肠癌、胃癌、胰腺癌、肺癌、乳腺癌、甲状腺髓样癌)、甲胎蛋白(例如,用于肝癌或生殖细胞肿瘤)、ca125(例如,用于卵巢和乳腺癌)、以及ca19-9(例如,用于胰腺癌)。
另外,可对其他组织进行测序以获得细胞甲基化组。例如,可分析肝脏组织以确定对于肝脏特异性的甲基化模式,所述甲基化模式可用于识别肝脏病变。也可进行分析的其他组织包括脑细胞、骨、肺、心脏、肌肉和肾等。各种组织的甲基化特征可例如由于发育、衰老、疾病过程(例如,炎症或肝硬化或自身免疫过程(诸如在全身性红斑狼疮中))或治疗(例如,使用去甲基化剂诸如5-氮杂胞苷和5-氮杂脱氧胞苷进行的治疗)而随时间变化。dna甲基化的动态性质使得此类分析对于监测生理和病理过程是潜在地有价值的。例如,如果检测个体的血浆甲基化组与当所述个体健康时获得的基线值相比的变化,则可检测贡献血浆dna的器官中的疾病过程。
另外,移植器官的甲基化组可根据器官移植接受者的血浆dna确定。因为血浆dna通常被视为细胞死亡的标志物,从移植器官释放的dna的血浆水平的增加可用作来自所述器官的增加的细胞死亡的标志物,诸如排斥事件或涉及所述器官的其他病理过程(例如,感染或脓肿)。在抗排斥疗法成功实施的事件中,由移植器官释放的dna的血浆水平预期将降低。
所述方法的示例性应用包括甲基化相关疾病和病状的检测、诊断、预后、复发、最小的残余风险评估。例如,应用可包括确定受试者是否具有对于与核酸的甲基化状态相关联的疾病或病状的易感性;诊断受试者的疾病或病状的方法,所述疾病或病状与核酸的甲基化状态相关联;检测受试者的疾病或病状的状态的方法,所述疾病或病状与核酸的甲基化状态相关联。具体的疾病和病状包括例如癌症。癌细胞的标志是它们比非癌细胞更快速地分裂。因此,癌细胞和非癌细胞将具有不同的甲基化模式。本文所述的实施方案和方法提供对于肿瘤活检或循环肿瘤dna中的甲基化、cnv和突变图谱状态的测定。具体地,所述实施方案和方法可用于提供发展癌症的诊断、预后、分期和/或可能性,所述癌症例如像前列腺癌、结肠直肠癌、肺癌、乳腺癌、肝癌、或膀胱癌。某些实施方案提供关于癌症、或告知治疗决定、或评估最小残余风险和复发的诊断、或分期或预后信息。已知受甲基化影响或已知影响甲基化的病状包括但不限于衰老、饮食、生活方式、种族、发育、躁郁症、多发性硬化症、糖尿病、精神分裂症、癌症、神经退行性疾病、炎症、病灶、感染、免疫应答、暴露于:毒品、酒精、烟草、杀虫剂、重金属、放射、uv其他环境因素。在一些实施方案中,本文提供的方法可提供循环的细胞-游离胎儿dna的甲基化状态和序列信息,例如作为非侵入性产前测试。使用本文所述的方法进行的非侵入性产前测试可用于例如确定先兆子痫或早产分娩的风险。使用本文所述的方法进行的另外的测试包括非整倍性的儿科诊断、受孕产物或过早流产的风险的测试、非侵入性产前测试(定性且定量的遗传测试,诸如孟德尔疾病、插入/缺失和染色体不均衡)、测试着床前遗传学、肿瘤表征、产后测试(包括细胞遗传学)和诱变剂效应监测。
所述方法的另一种示例性应用包括一种区分来源于受试者和一个或多个另外个体的核酸种类的方法,所述受试者和所述一个或多个另外个体具有核酸的不同甲基化状态。例如,受试者可以是妊娠女性,并且一个或多个另外个体可以是未出生胎儿。在这些实施方案中,血液样品可以是母体血浆或母体血清。可替代地,受试者可以是组织移植物接受者,并且一个或多个另外个体可以是组织移植物供体。
所述方法的另一种示例性应用包括一种区分来源于受试者的不同组织的核酸种类的方法。例如,本发明的方法可用于在目标组织(例如,胎儿、肿瘤或疾病来源的组织)与其他组织(例如,母体、非肿瘤或无疾病来源)之间进行区分。在一些实施方案中,受试者可以是妊娠女性。在这些实施方案中,血液样品可以是母体血浆或母体血清。可替代地,受试者可以是组织移植物接受者或癌症患者。
所述方法的另一种示例性应用包括一种确定受试者或受试者的组的年龄或“生物年龄”或“甲基化年龄”的方法。更具体地,已知个体的遗传物质随时间变化,并且本文所述的方法通过在单个测定中确定数千个或数十万个cpg位点的甲基化状态来实现来自个体的遗传物质的基于甲基化的年龄确定。这对于法医学目的和并且对于年龄相关病变诸如阿尔茨海默氏病两者均具有实用性。所述方法还可用于确定特定组织的年龄。如在实施例中进一步描述的,所述方法可用于确定特定组织(诸如结肠直肠组织)的“生物年龄”或胎儿的胎龄。
在一些实施方案中,捕获引物和探针(例如,mip)还具有与不是相同分子的一部分的常规pcr引物对相比结合稳定性增加的益处。在某些实施方案中,确切的靶向性臂序列对pcr引物而言稍短,并且因此在pcr环境中将具有非常低的解链温度。然而,在mip构型中,引物将通过协作以稳定相互作用来增强结合特异性。如果一个臂具有高结合效率,则即使相对臂具有较低的效率,捕获也会增强。所述对的加成长度改进捕获的“开/关”平衡,因为较低效率的臂在mip中通常比其作为游离pcr引物更接近其靶标。
在一些实施方案中,提供一种用于确定受试者是否具有对于与核酸的甲基化状态相关联的疾病或病状的易感性的方法。在一些实施方案中,本发明提供一种用于诊断受试者的疾病或病状的方法,所述疾病或病状与核酸的甲基化状态相关联。在一些实施方案中,本发明提供一种用于检测受试者的疾病或病状的状态的方法,所述疾病或病状与核酸的甲基化状态相关联。在某些实施方案中,这些方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于步骤g)中的所述比较来确定受试者是否易感所述疾病或病状、或诊断所述受试者中的所述疾病或病状、或检测所述受试者中的所述疾病或病状的状态。
在一些实施方案中,提供一种区分来源于受试者和一个或多个另外个体的核酸种类的方法,所述受试者和所述一个或多个另外个体具有核酸的不同甲基化状态,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品,所述血液样品包含来源于所述受试者和所述一个或多个另外个体的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有所述核酸的不同甲基化状态的个体分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来区分来源于所述受试者和所述一个或多个另外个体的核酸种类。
在一些实施方案中,提供一种区分来源于受试者的第一组织和一个或多个另外组织的核酸种类的方法,所述第一组织和所述一个或多个另外组织具有核酸的不同甲基化状态,所述方法包括:
a)获得从来自所述受试者的无细胞体液样品分离的核酸样品,所述无细胞体液样品包含来源于所述第一组织和所述一个或多个另外组织的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从具有所述核酸的不同甲基化状态的参考组织分离的参考核酸样品计算的多个参考比率进行比较;
h)基于在步骤g)中的所述比较来区分来源于所述第一组织和所述一个或多个另外组织的核酸种类。
在一些实施方案中,提供一种确定受试者中的核酸的甲基化状态并且检测拷贝数变异的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的所述目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量,以便确定核酸的甲基化状态;以及
f)确定在步骤d)测序的所述独特mip扩增子的数量;至少部分地基于独特mip扩增子序列的所述数量确定读取密度;以及通过将所述读取密度与基于从参考受试者分离的参考核酸样品计算的多个参考读取密度进行比较来检测拷贝数变异。
在另一个实施方案中,靶向性分子标签用于去除重复并且从而改进分析。在某些实施方案中,针对限定区域确定独特mip扩增子序列的数量以确定读取密度。
在一些实施方案中,提供一种确定受试者的甲基化年龄的方法,所述方法包括:
a)获得从来自所述受试者的血液样品分离的核酸样品;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来确定所述受试者的甲基化年龄。
在一些实施方案中,提供一种确定受试者的组织的甲基化年龄的方法,所述方法包括:
a)获得从来自所述受试者的无细胞体液样品分离的核酸样品,所述无细胞体液样品包含来源于所述组织的核酸;
b)进行所述核酸样品的亚硫酸氢盐转化;
c)通过使用分子倒置探针(mip)的一个或多个群体在步骤b)中获得的所述核酸样品中捕获多个目标靶序列以产生多个复制子,
其中所述mip群体中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接所述多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)对由步骤c)中获得的所述复制子扩增的多个mip扩增子进行测序;
e)确定在步骤d)测序的所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
f)基于在步骤e)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
g)将所述测试比率与基于从参考组织分离的参考核酸样品计算的多个参考比率进行比较;以及
h)基于在步骤g)中的所述比较来确定所述组织的甲基化年龄。
在一些实施方案中,所述方法是一种确定受试者是否具有对于与核酸的甲基化状态相关联的疾病或病状的易感性的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入用于多重测序的包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来确定所述受试者是否易感所述疾病或病状。
在一些实施方案中,所述方法是一种诊断受试者的疾病或病状的方法,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来诊断所述受试者中的所述疾病或病状。
在一些实施方案中,所述方法是一种检测受试者的疾病或病状的状态的方法,所述疾病或病状与核酸的甲基化状态相关联,所述方法包括:
a)从来自所述受试者的血液样品获得基因组dna样品;
b)进行所述基因组dna样品的亚硫酸氢盐转化;
c)将所述亚硫酸氢盐转化的基因组dna样品加入到多孔板的每个孔中,其中所述多孔板的每个孔包含探针混合物,其中所述探针混合物包含分子倒置探针(mip)群体和缓冲液;
其中所述mip群体中的每个mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
其中每个所述mip中的所述第一靶向性多核苷酸臂与第一重复区域基本上互补,并且每个所述mip中的所述第二靶向性多核苷酸臂与第二重复区域基本上互补,另外其中所述第一重复区域和所述第二重复区域分别侧接多个目标靶序列中的每个序列;
其中每个所述mip中呈组合的所述第一独特靶向性分子标签和所述第二独特靶向性分子标签在每个所述mip中是不同的;
其中所述多个目标靶序列中的每个目标靶序列具有一个或多个cpg位点;
其中每个cpg位点在所述目标靶序列内具有对应的已知位置;
d)将所述亚硫酸氢盐转化的基因组dna样品与所述mip的所述探针混合物一起孵育以捕获所述多个目标靶序列;
e)将延伸/连接混合物加入所述mip和所述多个目标靶序列的d)的所述样品中以形成多个mip扩增子,其中所述延伸/连接混合物包含聚合酶、多种dntp、连接酶和缓冲液;
f)将核酸外切酶混合物加入所述靶向性mip扩增子和对照mip扩增子中以去除过量的探针或过量的基因组dna;
g)将索引pcr混合物加入f)的所述样品以向所述多个扩增子加入包含独特分子标签的一对测序衔接子;
h)使用大规模平行测序方法以确定所述mip扩增子内的每个对应cpg位点处的胞嘧啶核苷酸的出现的数量;
i)基于在步骤h)确定的胞嘧啶核苷酸的出现的所述数量的第一总和与在所述多个目标靶序列中的cpg位点的所述已知数量的第二总和来确定测试比率;
j)将所述测试比率与基于从具有与所述核酸的甲基化状态相关联的所述疾病或病状的参考受试者分离的参考核酸样品计算的多个参考比率进行比较;以及
k)基于在步骤j)中的所述比较来检测所述受试者中的所述疾病或病状的状态。
在某些可替代的实施方案中,本发明的mip可不包含任何独特分子标签。根据本公开的方法,可能使用不包含独特分子标签的mip来确定甲基化评分、拷贝数变异、突变图谱等。
在可替代的实施方案中,本文所述的任一种方法的亚硫酸氢盐转化可由另一种类型的脱氨反应替代。
以上方法还可用于检测胎儿或非胎儿受试者的非整倍性。在某些实施方案中,作为检测非整倍性的替代方案,所述方法可用于检测和定量染色体臂中遗传特征的缺失和重复,以及微观缺失和重复、亚微观缺失和缺失以及单核苷酸特征(包括单核苷酸多态性、缺失和插入)。
在一些实施方案中,本发明的方法使用单个种类的mip。在可替代的实施方案中,所述方法可使用2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50或更多种类的mip。例如,多个种类的mip可用于检测单个样品中的不同疾病或病状(例如,癌症、怀孕相关病状诸如先兆子痫或早产分娩、或染色体异常,诸如非整倍性)。在某些实施方案中,单个mip可用于检测单个样品中的不同疾病或病状(例如,癌症、怀孕相关病状诸如先兆子痫或早产分娩、或染色体异常,诸如非整倍性)。
技术人员将理解,第一靶向性多核苷酸臂和第二靶向性多核苷酸臂的长度可适当地改变,以提供靶向性多核苷酸与核酸样品之间的有效杂交。例如,第一靶向性多核苷酸臂和/或第二靶向性多核苷酸臂可在14与30个碱基之间,例如18-21个碱基。在某些实施方案中,第一靶向性多核苷酸臂和/或第二靶向性多核苷酸臂的长度是14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个碱基。在某些实施方案中,靶向性多核苷酸臂具有在45℃与80℃之间(例如,45℃、46℃、47℃、48℃、49℃、50℃、51℃、52℃、53℃、54℃、55℃、56℃、57℃、58℃、59℃、60℃、61℃、62℃、63℃、64℃或65℃)的解链温度(tm)和/或在10%与80%之间(例如,大约10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%或80%)的gc含量。在某些实施方案中,靶向性多核苷酸臂具有在45℃与55℃之间的tm和/或在15%与25%之间的gc含量。在某些实施方案中,靶向性多核苷酸臂具有在45℃与66℃之间的tm和/或在10%与40%之间的gc含量。在一些实施方案中,第一靶向性多核苷酸臂的序列是cactacactccaacctaa(seqidno:4)或ttctcctacctcaacctc(seqidno:5)。在一些实施方案中,第二靶向性多核苷酸臂的序列是caaaaaactaaaacaaaa(seqidno:6)或ccaaactaaaatacaata(seqidno:7)。在一些实施方案中,靶向性多核苷酸臂靶向例如大于1,000、大于10,000、大于20,000、大于30,000、大于40,000、大于50,000、大于60,000、大于70,000、大于80,000、大于90,000、大于100,000、大于200,000、大于300,000、大于400,000、大于500,000、大于600,000、大于700,000、大于800,000、大于900,000、和/或大于1,000,000个目标序列。在一些实施方案中,mip不结合基因组中的长散布核苷酸元件(line)。
独特分子标签提供一种确定给定扩增子的捕获事件数量的方式。mip可包含一个或多个独特分子标签,例如1、2、3、4或5个独特分子标签。在某些实施方案中,第一独特分子标签和/或第二独特分子标签的长度在4与15个碱基之间,例如4、5、6、7、8、9、10、11、12、13、14或15个碱基。
多核苷酸接头桥接两个靶向性多核苷酸臂之间的空位。在一些实施方案中,多核苷酸接头直接位于第一独特分子标签与第二独特分子标签之间。在某些实施方案中,多核苷酸接头与受试者的任何基因组区域基本上不互补。在某些实施方案中,多核苷酸接头具有在20与1,000个碱基之间(例如,20、25、30、35、40、45、50、55、60或65个碱基)的长度和/或在45℃与85℃之间(例如,45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃或85℃)的解链温度和/或在10%与80%之间(例如,大约10%、15%、20%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、或80%)的gc含量。在某些实施方案中,多核苷酸接头包含至少一个扩增引物结合位点,例如正向扩增引物结合位点。在一些实施方案中,所述接头包含反向扩增引物结合位点。在一些实施方案中,反向引物结合位点在使用正向扩增引物进行pcr的第一延伸之后生成。例如,正向扩增引物的序列可包含ccgtaatcgggaagctgaag(seqidno:1)的核苷酸序列并且/或者反向扩增引物的序列可包含gcacgatccgacggtagtgt(seqidno:2)的核苷酸序列。因此,多核苷酸接头的核苷酸序列可包含cttcagcttcccgattacgggcacgatccgacggtagtgt(seqidno:3)的核苷酸序列。
在某些实施方案中,mip包含cactacactccaacctaa(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)caaaaaactaaaacaaaa(seqidno:8)的核苷酸序列,其中(n1-10)表示第一独特分子标签并且(n11-20)表示第二独特分子标签。在一些实施方案中,mip包含ttctcctacctcaacctc(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)ccaaactaaaatacaata(seqidno:9)的核苷酸序列,其中(n1-10)表示第一独特分子标签并且(n11-20)表示第二独特分子标签。在一些实施方案中,mip包含5’磷酸根以促进连接。
在一些实施方案中,mip群体具有在10fm与100nm之间的浓度,例如0.5nm。在某些实施方案中,mip的浓度将随着被靶向的序列的数量而变化,例如,如通过将目标靶序列的数量乘以反应中基因组当量的数量所计算(“总靶标数量”)。在特定的实施方案中,mip分子数量与总靶标数量的近似比率是1:50、1:100、1:150、1:200、1:250、1:300、1:350、1:400、1:450、1:500、1:550、1:600、1:650、1:700、1:750、1:800、1:850、1:900、1:950或1:1,000。在某些实施方案中,每个mip复制子和/或扩增子是单链环状核酸分子。
在一些实施方案中,通过以下产生mip复制子:i)第一靶向性多核苷酸臂和第二靶向性多核苷酸臂分别与核酸样品中的第一区域和第二区域杂交,其中所述第一区域和所述第二区域侧接目标靶序列;以及ii)杂交之后,使用连接/延伸混合物来延伸并连接两个靶向性多核苷酸臂之间的空位区域以形成单链环状核酸分子。在某些实施方案中,通过例如通过pcr扩增mip复制子来产生mip扩增子。
在一些实施方案中,测序步骤包括下一代测序方法,例如大规模平行测序方法或短读取测序方法。在一些实施方案中,测序可通过本领域已知的任何方法进行,例如靶向测序、单分子实时测序、基于电子显微镜的测序、晶体管介导的测序、直接测序、随机鸟枪测序、桑格双脱氧终止测序、靶向测序、外显子测序、全基因组测序、杂交测序、焦磷酸测序、毛细管电泳、凝胶电泳、双重测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模平行签名测序、乳液pcr、低变性温度共扩增pcr(cold-pcr)、多重pcr、通过可逆染料终止子的测序、双端测序、近期测序、核酸外切酶测序、边连接边测序、短读取测序、单分子测序、边合成边测序、实时测序、反向终止子测序、纳米孔测序、454测序、solexa基因组分析仪测序、
可在各种实施方案中使用的测序技术包括例如
一些实施方案包括在测序(例如,如上所述的测序步骤d))之前的扩增mip扩增子的pcr反应以实现测序。此pcr反应可以是索引pcr反应。在某些实施方案中,索引pcr反应向mip扩增子中的每一个引入以下组件:包含独特样品条形码的一对索引引物和一对测序衔接子。在特定的实施方案中,带条形码的靶向性mip扩增子在5’至3’方向上依次包含以下组件:
第一测序衔接子-第一测序引物结合位点-第一独特靶向性分子标签-第一靶向性多核苷酸臂-捕获的核酸-第二靶向性多核苷酸臂-第二独特靶向性分子标签-第二测序引物结合位点-独特样品条形码-第二测序衔接子。
在一些实施方案中,目标靶序列位于单条染色体上。在可替代的实施方案中,目标靶序列位于多条染色体上。在特定的实施方案中,目标靶序列在其中甲基化状态与疾病或病状相关的特定位点处选择。因为可使用单个mip序列来靶向整个基因组上的目标靶序列,所以在某些实施方案中,本发明的方法提供能够一次检测多于一个染色体的甲基化状态的益处。通过选择提供在整个基因组上的足够覆盖的mip,目标序列的甲基化状态可充当基因组的甲基化状态的代表。此外,因为mip提供序列信息以及甲基化状态,所以mip可用于检测与甲基化状态和/或染色体或其他序列异常相关联的1、2、3、4、5、6、7、8、9、10种或更多种病状。
在一些实施方案中,提供的方法包括一种从用于检测受试者的甲基化的多个候选mip选择分子倒置探针(mip)方法,所述方法包括:
a)接收所述多个候选mip的核酸序列,其中所述多个候选mip中的每个所述mip依次包含以下组件:
第一靶向性多核苷酸臂-第一独特分子标签-多核苷酸接头-第二独特分子标签-第二靶向性多核苷酸臂;
b)对于所述多个候选mip中的每个相应的mip,
i)计算预测在所述结合臂序列上没有错配的情况下由所述相应的mip捕获的独特cpg位点的第一数量(a);
ii)计算预测在所述结合臂序列上有一个错配的情况下由所述相应的mip捕获的独特cpg位点的第二数量(c);
iii)计算预测在所述结合臂序列上没有错配的情况下在整个基因组上由所述相应的mip捕获的独特位点的第三数量(e);
iv)计算预测在所述结合臂序列上有一个错配的情况下在整个所述基因组上由所述相应的mip捕获的独特位点的第四数量(g);
v)计算预测在所述结合臂序列上没有错配的情况下在整个所述基因组上由所述相应的mip捕获的非独特位点的第五数量(f);
vi)计算预测在所述结合臂序列上有一个错配的情况下在整个所述基因组上由所述相应的mip捕获的非独特位点的第六数量(h);
vii)计算存在于所述第一靶向性多核苷酸臂上的cpg位点的第七数量(i);
viii)计算存在于所述第二靶向性多核苷酸臂上的cpg位点的第八数量(j);
ix)至少部分地基于所述第一、第二、第三、第四、第五、第六、第七和第八数量来计算所述相应的mip的性能度量;
c)至少部分地基于针对所述多个候选mip中的每个mip在步骤b)ix)中计算的所述性能度量来选择mip。
例如,在以上方法中,选择步骤c)的所述mip使得所述第七数量(i)和所述第八数量(j)的总和小于所述候选mip的剩余集合的对应总和。在某些实施方案中,第一总和是所述第一数量(a)和所述第二数量(c)的总和,第二总和是所述第三数量(e)、所述第四数量(g)、所述第五数量(f)、和所述第六数量(h)的总和;并且选择步骤c)的所述mip使得所述第一总和与所述第二总和之间的比率小于所述候选mip的剩余集合的比率。在某些实施方案中,第三总和是所述第三数量(e)和所述第四数量(g)的总和;第四总和是所述第三数量(e)、所述第四数量(g)、所述第五数量(f)、和所述第六数量(h)的总和;并且选择步骤c)的所述mip使得所述第三总和与所述第四总和之间的比率小于所述候选mip的剩余集合的比率。在某些实施方案中,步骤c)的所述mip基于所述结合臂序列上的一个错配位点的平均捕获系数(k1)和零个错配位点的平均捕获系数(k0)的比率(ke)来选择:
并且其中所述比率(ke)通过实验来估算。在某些实施方案中,步骤b)的所述性能度量包括对应于所述第一数量(a)和所述第二数量(c)的加权总和的因数。在某些实施方案中,所述加权总和对应于a+ke×c。在某些实施方案中,步骤b)的所述性能度量包括对应于所述第三数量(e)和所述第四数量(g)的加权总和的因数。在某些实施方案中,所述加权总和对应于e+ke×g。在某些实施方案中,选择步骤c)的所述mip使得第一加权总和a+ke×c与第二加权总和e+ke×g之间的乘积大于所述候选mip的剩余集合的乘积。
在一些实施方案中,提供一种核酸分子,其包含cactacactccaacctaa(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)caaaaaactaaaacaaaa(seqidno:8)的核苷酸序列,其中(n1-10)表示第一独特分子标签并且(n11-20)表示第二独特分子标签。在一些实施方案中,提供一种核酸分子,其包含ttctcctacctcaacctc(n1-10)cttcagcttcccgattacgggcacgatccgacggtagtgt(n11-20)ccaaactaaaatacaata(seqidno:9)的核苷酸序列,其中(n1-10)表示第一独特分子标签并且(n11-20)表示第二独特分子标签。本发明的另外的mip分子包括以下各项:
mrop208-f,
tcctacctcaacctccta(6n)bb(6n)ccaaactaaaatacaata(seqidno:11),
/5phos/tcctacctcaacctcctannnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnccaaactaaaatacaata(seqidno:12)
mrop208-r,
cactacactccaacctaa(6n)bb(6n)caaaaaactaaaacaaaa(seqidno:13),
/5phos/cactacactccaacctaannnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnncaaaaaactaaaacaaaa(seqidno:14)
mrop206-f,
ttctcctacctcaacctc(6n)bb(6n)ccaaactaaaatacaata(seqidno:15,
/5phos/ttctcctacctcaacctcnnnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnccaaactaaaatacaata(seqidno:16)
mrop206-r,
cactacactccaacctaa(6n)bb(6n)aaaactaaaacaaaaaaa(seqidno:17),
/5phos/cactacactccaacctaannnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnaaaactaaaacaaaaaaa(seqidno:18)
如本文所用,“bb”指代主链序列。技术人员将理解,任何通用主链序列可在此应用。
如上所述,在具体实施方案中,a)所述第一独特分子标签的长度在4与15个碱基之间;并且/或者b)所述第二独特分子标签的长度在4与15个碱基之间。
用于识别mip的方法
图1是用于进行本文所述的过程中的任一种(包括过程200、300和400)的计算设备100的框图。如本文所用的,术语“处理器”或“计算设备”是指被配置有进行本文所述的计算机化技术中的一种或多种的硬件、固件和软件的一个或多个计算机、微处理器、逻辑设备、服务器或其他设备。处理器和处理设备还可包括用于存储当前正在处理的输入、输出和数据的一个或多个存储设备。计算设备100可包括“用户接口”,其可包括但不限于一个或多个输入设备(例如,小键盘、触摸屏、轨迹球、语音识别系统等)和/或一个或多个输出设备(例如,视觉显示器、扬声器、触觉显示器、打印设备等)的任何合适的组合。计算设备100可包括但不限于配置有进行本文所述的计算机化技术中的一个或多个的硬件、固件和软件的一个或多个设备的任何合适的组合。本文所述的组件中的每一个可在一个或多个计算设备100上实施。在某些方面,这些系统的多个组件可被包括在一个计算设备100内。在某些实施方案中,组件和存储设备可在若干计算设备100上实施。
计算设备100包括至少一个通信接口单元108、输入/输出控制器110、系统存储器以及一个或多个数据存储设备。系统存储器包括至少一个随机存取存储器(ram102)和至少一个只读存储器(rom104)。所有这些元件均与中央处理单元(cpu106)通信以促进计算设备100的操作。计算设备100可以许多不同的方式来配置。例如,计算设备100可以是常规的独立计算机,或者可替代地,计算设备100的功能可分布在整个多个计算机系统和体系结构中。在图1中,计算设备100通过网络或局部网络被链接到其他服务器或系统。
计算设备100可被配置成分布式体系结构,其中数据库和处理器被容纳在分开的单元或位置中。一些单元进行主要处理功能,并且至少含有通用控制器或处理器和系统存储器。在分布式体系结构实施方案中,这些单元中的每一个可通过通信接口单元108附接到通信集线器或端口(未示出),其充当与其他服务器、客户端或用户计算机和其他相关设备的主要通信链路。通信集线器或端口本身可具有最小的处理能力,其主要充当通信路由器。多种通信协议可以是系统的一部分,所述通信协议包括但不限于:ethernet、sap、sastm、atp、bluetoothtm、gsm和tcp/ip。
cpu106包括处理器(诸如一个或多个常规微处理器)以及用于从cpu106卸载工作负载的一个或多个辅助协处理器(诸如数学协处理器)。cpu106与通信接口单元108和输入/输出控制器110通信,cpu106通过通信接口单元108和输入/输出控制器110与其他设备(诸如其他服务器、用户终端或设备)通信。通信接口单元108和输入/输出控制器110可包括用于与例如其他处理器、服务器或客户终端同时通信的多个通信信道。
cpu106也与数据存储设备通信。数据存储设备可包括磁性、光学或半导体存储器的适当组合,并且可包括例如ram102、rom104、闪存驱动器、光盘(诸如压缩盘或硬盘)或驱动器。例如,cpu106和数据存储设备各自可完全位于单个计算机或其他计算设备内;或通过通信介质彼此连接,所述通信介质诸如usb端口、串行端口电缆、同轴电缆、以太网电缆、电话线、射频收发器或者其他类似的无线或有线介质或者上述的组合。例如,cpu106可通过通信接口单元108连接到数据存储设备。cpu106可被配置来进行一种或多种特定的处理功能。
数据存储设备可存储例如(i)计算设备100的操作系统112;(ii)一个或多个应用程序114(例如,计算机程序代码或计算机程序产品),其适于根据在此所述的系统和方法,并且特别是根据关于cpu106详细描述的过程来引导cpu106;或(iii)数据库116,其适于存储可用于存储程序所需信息的信息。
操作系统112和应用程序114可例如以压缩格式、未编译格式和加密格式被存储,并且可包括计算机程序代码。程序的指令可从除了数据存储设备之外的计算机可读介质(诸如从rom104或从ram102)读取到处理器的主存储器中。虽然程序中的指令序列的进行使得cpu106进行本文所述的过程步骤,但是可使用硬连线电路代替用于本发明的过程的实施方案的软件指令或与所述软件指令组合。因此,描述的系统和方法并不限于硬件和软件的任何特定组合。
可提供合适的计算机程序代码用于进行如本文所述的一种或多种功能。程序还可包括程序元件,诸如操作系统112、数据库管理系统和“设备驱动程序”,所述设备驱动程序允许处理器通过输入/输出控制器110与计算机外围设备(例如,视频显示器、键盘、计算机鼠标等)交互。
如本文所用,术语“计算机可读介质”是指提供或参与向计算设备100的处理器(或本文所述的设备的任何其他处理器)提供指令以供进行的任何非暂时性介质。此种介质可采取许多形式,其包括但不限于非易失性介质和易失性介质。非易失性介质包括例如光盘、磁盘或光磁盘或者集成电路存储器(诸如闪存)。易失性介质包括通常构成主存储器的动态随机存取存储器(dram)。计算机可读介质的常见形式包括例如软盘、软磁盘、硬盘、磁带、任何其他磁介质、cd-rom、dvd、任何其他光学介质、穿孔卡、纸带、任何其他具有孔样式的物理介质、ram、prom、eprom或eeprom(电子可擦除可编程只读存储器)、flash-eeprom、任何其他存储芯片或盒式磁带或者计算机可从中读取的任何其他非暂时性介质。
各种形式的计算机可读介质可涉及将一个或多个指令的一个或多个序列携带至cpu106(或本文所述的设备的任何其他处理器)以供进行。例如,最初可将指令载于远程计算机的磁盘上(未示出)。远程计算机可将指令载入它的动态存储器中并使用调制解调器通过以太网连接、电缆线或甚至电话线发送指令。计算设备100本地的通信设备(例如,服务器)可接收在相应的通信线路上的数据,并将数据放置在处理器的系统总线上。系统总线将数据携带至主存储器,处理器从所述主存储器检索并进行指令。主存储器接收的指令可任选地在处理器进行之前或之后存储在存储器中。另外,指令可通过通信端口作为电信号、电磁信号或光信号被接收,这些信号是携带各种类型的信息的无线通信或数据流的示例性形式。
图2是根据说明性实施方案的用于设计和选择探针(例如,mip)的过程200的流程图。过程200包括以下步骤:确定约束的集合(步骤202);使用所述约束的集合来识别引物(例如,靶多核苷酸臂)(步骤204);进行优化技术以使mip的延伸臂和连接臂上的cpg位点的数量最小化、使捕获的cpg位点的总数量最大化并且使独特可映射的位点的数量最大化(步骤206);以及基于优化技术选择探针(步骤208)。如本文所用,“引物”可以是指捕获探针(诸如分子倒置探针)的杂交部分。例如,“引物”可以是指mip的一个或多个靶向性多核苷酸臂。
在步骤202,确定约束的集合。所述约束的集合可例如通过cpu106使用其上实施的软件或应用程序来确定。在一些实施方案中,cpu106也可使用软件或应用程序来进行过程200的后续步骤中的任何一个或多个。例如,cpu106可使用软件和应用程序来基于所确定的约束在给定的参考基因组(例如,hg19)中找到丰富的引物对,并且自动地为基因组文件创建基于后缀阵列的索引。
在一些实施方案中,所述约束的集合可替代地被称为算法标志。例如,约束(或算法标志)可包括左侧引物和右侧引物(或分别连接臂和延伸臂)的长度、引物对的最小频率、引物之间的最大距离(例如,扩增子长度)、引物的最小和/或最大总频率、以百分比表示的每个引物的最小gc含量、以百分比表示的非相同扩增子的最小量、基因组中引物的分布或其任何合适的组合。在说明性实施方案中,可在设计引物对时使用以下约束的集合:
·左侧引物或连接臂的长度:18、19、20、21个碱基
·右侧引物或延伸臂的长度:18、19、20、21个碱基
·引物对的频率:100,250,500,2500,5000,10,000,100,000,500,000,1,000,000
·扩增子长度:50-150个碱基对,例如小于85个碱基对
·每个引物的最小gc含量:10%,15%,20%,30%,40%
·扩增子独特性(独特目标靶序列的百分比):大于约40%
·基因组中引物的分布:迭代运行,其中每个存储块(bucket)大小(bs)在1%至50%的范围内,并且存储块填充(bf)在1至bs-1的范围内,其中存储块大小(bs)是指基因组长的bs%,并且每个存储块必须含有所有命中的bf%。
应理解,以上约束的集合在本质上仅是说明性的,并且在探针选择过程中可使用其他约束的集合。
在步骤204,使用在步骤202确定的约束的集合来识别引物的集合。具体地,对于每个引物设计,可提供以下参数的任何组合:左侧引物序列(例如,以及它们在基因组的正链和负链上的出现数量)、右侧引物序列(例如,以及它们在基因组的正链和负链上的出现数量)、包括独特对和非独特对两者的所述对的频率(例如,左侧引物序列和右侧引物序列配对在一起,其中扩增子长度受到约束的限制)、独特存在的扩增子的频率和百分比以及来自独特对和非独特对的扩增子序列。在一些实施方案中,每个引物对可能能够扩增基因组上的多个区域(例如,多于数百个、多于数千个、多于数万个、多于数十万个或多于数百万个)。
在一些实施方案中,预测的引物对被转化以靶向亚硫酸氢盐转化的基因组。所生成的引物对可识别或预测扩增子位点,而不允许在亚硫酸氢盐转化的基因组上在左侧引物序列或右侧引物序列(即,左臂或右臂)中发生任何错配。可替代地,为了识别或预测另外的扩增子位点,可允许少量错配,诸如允许:
左臂中1个错配和右臂中0个错配
左臂中0个错配和右臂中1个错配
左臂中1个错配和右臂中1个错配
左臂中2个错配和右臂中0个错配,或
左臂中0个错配和右臂中2个错配。
在一些实施方案中,上述扩增子预测方案提供亚硫酸氢盐转化的基因组中预测的扩增子的基因组坐标。然而,在一些实施方案中,同样提供预测的扩增子的基因组坐标对于识别扩增子位点而不允许发生任何错配的方案可能是计算密集的。在这种情况下,所述方案可以分为两个部分。在第一部分中,识别扩增子位点而不允许发生任何错配,并且不提供所识别的扩增子位点的基因组坐标。在第二部分中,识别包括少量错配(例如,上面列举的错配的集合)的扩增子位点,并且提供这些扩增子位点的基因组坐标以及无错配扩增子位点的基因组坐标。将所述方案分解成这两个模块部分可节省计算复杂度。然而,通常,应理解,所述两个部分可组合以提供无错配扩增子位点、错配扩增子位点以及它们在单个函数中的基因组坐标的集合。
在一些实施方案中,可(例如,通过过滤操作)去除在步骤204识别的扩增子位点中的一个或多个。例如,在一些实施方案中,去除含有cg二核苷酸的臂序列。通过过滤操作的那些引物(在下文中被称为“候选引物”)的扩增子位点应靶向参考基因组的多个区域(例如,通常为2500或更多个)。另外,在一些实施方案中,候选引物的左臂和右臂序列两者均具有40℃至高60℃范围内的解链温度(tm),如通过dna结合稳定性的最近邻模型计算的,其中根据核酸序列来合计经验稳定性参数。参见例如,santalucia和hicks2004。
在去除(或过滤)操作之后,剩余的扩增子位点将被进一步处理,以便为每个候选引物生成参数值的集合。在一些实施方案中,将计算cpg的数量和已通过过滤操作的扩增子位点的总数量的比例。对于每个候选引物,可将富集信息(例如,计算的比例)、相关联的扩增子位点信息以及任何其他参数值保存在数据库(诸如数据库116)中。
在步骤206,进行优化技术以识别具有最佳预测性能的引物。优化技术涉及评价每个候选引物的目标函数。具体地,可能需要使用目标函数,所述目标函数使mip的延伸臂和连接臂上的cpg位点的数量最小化、使捕获的cpg位点的总数量最大化、使独特可映射的位点的数量最大化、或其任何合适的组合。
在一些实施方案中,每个候选mip的目标函数可基于以下矩阵来建立:
表1:来自预测位点的预测cpg计数
表2:整个基因组上的预测位点计数
表3:每个臂上的cpg计数的数量
在以上探针矩阵中,参考亚硫酸氢盐转化的基因组,标记为“0个错配”的行指示在两臂中具有完美匹配的mip,并且标记为“1个错配”的行指示在其一个臂中容许最多1个错配的引物。若干直观的目标函数可易于从这些探针矩阵推导出来。例如,使i+j最小化(例如,i和j的总和是0)的目标函数将确保探针性能不是个体的甲基化状态的函数(例如,因为在臂序列结合位点中存在cpg位点)。在第二个实例中,使(a+c)/(e+f+g+h)最大化的目标函数可产生特异性地靶向cpg位点的读取。作为第三个实例,使(e+g)/(e+f+g+h)最大化的目标函数选择在亚硫酸氢盐转化的基因组上具有比非独特捕获位点显著更多的独特捕获位点的引物。为了进一步说明这个概念,以下详细解释三个示例性目标函数。
a.延伸臂和连接臂上的cpg位点的总数量(p1)
每个候选引物或探针的示例性目标函数可定义为探针的延伸臂和连接臂上的cpg位点的总数量:
p1=i+j(1)
b.可用cpg位点的总数量(p2)
每个候选引物或探针的另一个示例性目标函数可定义为捕获的可用cpg位点的总数量:
p2=g(a,b,c...h;k0,k1)(2)
其中k0是0个错配位点的平均捕获系数,并且k1是1个错配位点的平均捕获系数。更具体地:
p2=a+kec(3)
其中ke可从实验数据估算,并且:
c.独特可映射的读取的总数量(p3)
每个候选引物或探针的另一个示例性目标函数可定义为整个亚硫酸氢盐转化的基因组上独特可映射的读取的总数量:
p3=g(a,b,c...h,k0,k1)(5)
其中k0和k1如上定义。更具体地,p3可定义为:
p3=e+keg(6)
其中ke定义为方程(4)。
d.综合探针性能函数
评估每个候选引物或探针的目标函数的综合方式是首先去除其中p1不等于零的任何候选引物或探针。换言之,可仅考虑其中p1=0的候选引物或探针,使得延伸臂或连接臂上不存在cpg位点。在第二步骤中,性能函数可对应于:
p=p2×p3(7)
合并方程(3)和(6),方程(7)可写成:
p=((a+kec)×(e+keg))/((e+f)+ke(g+h))(8)
注意,如上文关于方程(4)所述,可使用实验数据来估算ke的值。更具体地:
在步骤208,基于在步骤206进行的优化技术从候选引物集合中选择引物。例如,所选择的引物可对应于具有最佳预测性能的引物,即,具有p1=0并且使如关于步骤206所描述的目标函数最大化的引物。
预期过程200的步骤或描述可与本公开的任何其他实施方案一起使用。另外,关于图2描述的步骤和描述可以交替的顺序或并行地进行以进一步实现本公开的目标。例如,这些步骤中的每一个可以任何顺序或并行地或基本上同时进行,以减少滞后或增加系统或方法的速度。此外,应注意,可使用计算设备100,并且更具体地,使用计算设备100的cpu106来实施过程200。
图3是根据说明性实施方案的用于预测测试受试者的疾病状态的过程300的流程图。过程300包括以下步骤:接收测试受试者的测序数据(步骤302);计算测试受试者的甲基化比率(步骤304);接收参考受试者的集合的甲基化比率(步骤306);以及基于测试受试者的甲基化比率与参考受试者的甲基化比率的比较预测测试受试者的疾病状态(步骤308)。
甲基化评分根据涵盖cpg位点的测序读取中含有的信息来计算。每次cpg位点被读取覆盖,检索的信息在以下式中被认为是一个计数(甲基化的或非甲基化的)。如果单个读取涵盖多个cpg位点,则所述单个读取可生成多个计数。像bismark甲基化提取器(bismarkmethylationextractor)的程序如下计算甲基化比率:
在步骤302,接收测试受试者的测序数据。具体地,测试受试者可具有未知的疾病状态或对于特定疾病状态的易感性。所接收的测序数据通过从测试受试者获得核酸样品、使用亚硫酸氢盐转化来处理样品并且使用引物群体(诸如分子倒置探针(mip))捕获核酸样品中的位点的集合来获得。如关于图5详细描述的,每个mip依次包括第一靶向性多核苷酸臂、第一独特靶向性分子标签、多核苷酸接头、第二独特靶向性分子标签和第二靶向性多核苷酸臂。第一靶向性多核苷酸臂和第二靶向性多核苷酸臂在整个群体中的mip上是相同的,而第一独特靶向性分子标签和第二独特靶向性分子标签在整个群体中的mip上是不同的。mip扩增子来自于位点的捕获,并且对扩增子进行测序以获得测序数据。
在步骤304,通过评价靶区域内的甲基化胞嘧啶核苷酸的数量与已知的cpg位点的总数量之间的比率来计算测试受试者的甲基化比率。如关于图6详细描述的,亚硫酸氢盐转化的过程将非甲基化的胞嘧啶核苷酸转化为尿嘧啶核苷酸(其随后在pcr期间转化为胸腺嘧啶核苷酸),并且对于甲基化的胞嘧啶核苷酸不具有影响。因此,在样品使用亚硫酸氢盐转化进行处理之后,在cpg位点处剩余胞嘧啶核苷酸的存在指示这些胞嘧啶核苷酸是甲基化的。甲基化比率提供甲基化的胞嘧啶核苷酸与cpg位点的总数量相比的比例量度。
在步骤306,接收参考受试者的集合的甲基化比率的集合。具体地,参考受试者可对应于表现出已知疾病状态或对于患有疾病的已知易感性的一组人。以与关于步骤304所述的相同的方式来计算参考受试者的甲基化比率,但是是对于每个参考受试者。
在步骤308,将测试受试者的甲基化比率(在步骤304计算)与参考受试者的甲基化比率(在步骤306获得)进行比较,并且基于此比较预测测试受试者的特定疾病的疾病状态或易感性。具体地,可使用统计测试来将测试甲基化比率与参考甲基化比率的群体进行比较,并且确定测试甲基化比率是否属于与相同疾病状态或易感性相关联的任何参考甲基化比率的聚类。
图4是根据说明性实施方案的用于预测测试受试者的疾病状态的过程400的流程图。在一个实例中,过程400可用于实施关于图3示出并描述的过程300的步骤304和308。如关于图3所描述的,甲基化比率可用于预测具有未知的疾病状态或对于疾病的易感性的测试受试者的疾病状态。
过程400包括以下步骤:接收从使用亚硫酸氢盐转化处理的样品记录的测序数据(步骤402);过滤测序读取以去除已知假象(步骤406);将读取与亚硫酸氢盐转化的人类基因组对齐(步骤408);将cpg位点迭代参数k初始化至1(步骤412);以及确定胞嘧啶核苷酸是否在第k个cpg位点处存在(步骤414)。当所有k个cpg位点均已考虑时,过程400还包括以下步骤:计算在步骤414确定的胞嘧啶核苷酸的数量的总和s(步骤420);计算测试样品的甲基化比率s/k(步骤422,其中k对应于cpg位点的总数量);以及通过将测试样品的甲基化比率与参考甲基化比率的集合进行比较来选择测试样品的疾病状态(步骤424)。
在步骤402,接收从测试样品记录的数据。测试受试者具有未知的疾病状态。所述样品可以是从测试受试者分离并使用亚硫酸氢盐转化处理的核酸样品。所述数据可包括从核酸样品获得的测序数据。在一个实例中,测序数据通过使用mip群体扩增核酸样品中位点的集合以产生mip扩增子的集合来获得。然后可对mip扩增子进行测序以获得在步骤402接收的测序数据。
在步骤406,将测试样品的测序读取进行过滤以去除已知假象。在一个实例中,可处理在步骤402接收的数据以去除探针与探针相互作用的影响。在一些实施方案中,将所有mip的连接和延伸靶向性臂与双端序列读取匹配。未能与mip的两个臂匹配的读取被判定为无效并丢弃。在一些具体实施中,允许每个臂中最多一个碱基对错配,但是可丢弃具有更多错配的任何读取。剩余的有效读取的臂序列被去除,并且来自连接和延伸末端两者的分子标签也可从所述读取中去除。
在步骤408,将所得修整的读取与人类基因组对齐。在一些实施方案中,可使用对齐工具将读取与参考人类基因组对齐。具体地,可评估对齐评分用于表示特定读取与参考的对齐程度如何。具有高于阈值的对齐评分的读取在本文中可被被称为主要对齐,并且被保留。相比之下,具有低于阈值的对齐评分的读取在本文中可被称为次要对齐,并且被丢弃。任何沿着参考基因组与多个位置对齐的读取在本文中可被称为多重对齐,并且被丢弃。
在步骤412,将cpg位点迭代参数k初始化至一。cpg位点的数量和位置是已知的。
在步骤414,检查第k个cpg位点以确定胞嘧啶核苷酸是否存在。如关于图6详细描述的,亚硫酸氢盐转化的过程将非甲基化的胞嘧啶核苷酸转化为尿嘧啶核苷酸(其随后在pcr期间转化为胸腺嘧啶核苷酸),但是对于甲基化的胞嘧啶核苷酸不具有影响。因此,在样品使用亚硫酸氢盐转化进行处理之后,在cpg位点处剩余胞嘧啶核苷酸的存在指示这些胞嘧啶核苷酸是甲基化的。在步骤414检查第k个cpg位点之后,cpg位点迭代参数k在步骤418递增,直至已考虑所有k个cpg位点。当已考虑所有k个cpg位点时,过程400进行到步骤420以计算测试样品的胞嘧啶核苷酸的总和s。
在步骤422,计算测试样品的甲基化比率s/k。甲基化比率对应于通过k归一化的在k个cpg位点处存在的胞嘧啶核苷酸的总数量,并且提供甲基化的胞嘧啶核苷酸与cpg位点的总数量相比的比例量度。
在步骤424,将测试样品的甲基化比率与参考甲基化比率的集合(已从具有已知疾病状态的参考受试者进行计算)进行比较,并且进行统计测试来为测试受试者选择预测的疾病状态。
图4中步骤的顺序仅出于说明性目标而示出,并且不是限制性的。
因为甲基化变化并非随机分布在基因组中,所以在一些实施方案中,甲基化比率可通过过滤掉或分离接近于基因组的关键元件的靶标来计算甲基化比率。例如,为了增加癌症样品中甲基化不足的检测的灵敏度,接近cpg岛的靶标可被过滤掉,因为它们往往变得过甲基化。在第二种情况下,甲基化比率可使用基因间区域中含有的靶标来计算,因为已知它们显示较高水平的甲基化不足。
在一些实施方案中,测试样品的甲基化不足的水平可通过将其甲基化密度与对照样品的集合(5、10、50、100、500、1000、10,000个或更多个对照样品)进行比较来确定。甲基化密度定义为对于限定的区域或对于限定的仓大小(1,000、10,000、100,000、1,000,000、10,000,000个或更多个碱基)在cpg环境中的甲基化的c的平均百分比。对于每个仓,如下计算z评分并且确定高于限定阈值的z甲基化的百分比。
z甲基化=md测试–md对照(12)
mdsd-对照
其中:
md测试是测试样品的限定仓的甲基化密度;
md对照是对照样品的集合的限定仓的甲基化密度的平均值;并且
mdsd-对照是对照样品的集合的甲基化密度的标准偏差。
另外,cnv(包括cna)可通过将甲基化密度用读取密度替代以相同方式计算。
还可进行比较分析来检测测试样品组相对于对照组中的差异甲基化cpg位点。methylkit是用于dna甲基化分析的r程序包(altunaakalin,matthiaskormaksson,shengli,francinee.garrett-bakelman,mariae.figueroa,arimelnick,christophere.mason.(2012).“methylkit:acomprehensiverpackagefortheanalysisofgenome-widednamethylationprofiles.”genomebiology,13:r87.)methylkit可用于进行样品关联和聚类以及差异甲基化分析。可识别在测试组与对照组之间具有差异甲基化的cpg位点。一些cpg位点可能仅在测试样品的子集中显示差异甲基化状态。因此,识别cpg位点与限定“重量”的组合可能更适于生成在未知样品属于测试组的情况下允许评价的算法。
本领域普通技术人员应理解,本文所述的组合物和方法可如适用于正在讨论的应用进行适配和修改,并且本文所述的组合物和方法可用于其他合适的应用中,并且此类其他添加和修改将不脱离本发明的范围。
以上提及的实施方案将根据以下实验细节得到更好的理解。然而,本领域的技术人员将易于理解,所讨论的具体方法和结果本身仅是说明性的。
实施例
实施例1:用于捕获目标靶序列的mip设计和方法
探针设计
针对关于任何重复区域的基因组中的半冗余位点创建单个靶向捕获探针。设计另外的准则来靶向短(~160个bp)循环核酸并且具有>150,000个位点(具有完全引物匹配或1个错配)的所述重复位点的数量/亚硫酸氢盐转化的人类基因组。探针臂解链温度在42℃与50℃之间。
探针构建
合成大小在80-105个bp之间的范围内(取决于重复靶向性序列的长度)的单个寡核苷酸,诸如在图5和图11a中所见的寡核苷酸
dna制备
根据下游用途,dna可从各种来源提取,包括来自全血的基因组dna、片段化的血浆dna或从福尔马林固定的石蜡包埋(ffpe)组织提取的dna。
在提取之后,dna通过诸如以下的标准工作流进行亚硫酸氢盐转化:输入dna→使dna变性→孵育→亚硫酸氢盐转化→孵育→基于柱的磺化-水解脱氨-脱磺→洗脱至所需浓度。因为探针被设计成使得其仅对于亚硫酸氢盐转化的基因组是可扩增的,所以它允许捕获在整个基因组上已被亚硫酸氢盐转化的代表性位点。
位点捕获反应
将捕获探针(在经验确定的浓度下)与从各种来源提取的10-20ng亚硫酸氢盐处理的dna混合。将混合物在热循环仪中在已针对探针与模板的退火(98℃持续5min→从98℃降落至50℃持续24min→50℃持续120min)进行优化的温度下孵育。在该孵育期间,探针分子在与探针序列互补的特定染色体位置处与dna模板退火。最容易预测的位点是具有与探针臂完全互补的序列的那些位点(不变位点),但是也以稍微更低的效率靶向在任一个臂中具有一个或多个变异的位点。每个反应的探针的最佳量取决于四个主要考虑因素:1)用作模板的基因组的数量,其可在样品之间变化,2)由特定探针靶向的位点的总体数量,3)不变位点与变异位点的比率,以及4)所需的基因组cpg位点的多样性。
杂交程序完成之后,加入酶和试剂的5μl混合物,并且将所述混合物在50℃下孵育1hr,然后在72℃下孵育20min,然后保持在4℃下。在该步骤期间,dna聚合酶(优选地不具有链置换特性的聚合酶)填充空位,并且使用在连接臂的5’处的磷酸根通过dna连接酶使探针共价环化(图6)。仅与模板退火并延伸的探针在该步骤期间被环化,并且线性探针因为引物结合位点之间的空位而不能扩增。这些线性探针随后在之后的步骤中被消化。
核酸外切酶消化
加入核酸外切酶的混合物。这通过核酸外切酶消化的方式去除剩余的探针和基因组dna。然后对核酸外切酶进行加热灭活。
捕获位点索引和扩增
将10μl清理和捕获的探针混合物(即,复制子)加入到含有热稳定聚合酶、dntp、pcr缓冲液以及与探针主链互补的通用引物的50μl反应混合物(图7和图11b)。每个样品使用具有不同测序条形码序列的引物进行扩增,从而允许对汇集的样品文库进行多重测序。对反应进行以经验确定数量的pcr循环,这可通过存在由电泳可观察到干净的扩增子条带来确认。将pcr产物使用ampure珠粒进行纯化并且使用qubit荧光计进行定量。
对捕获的位点进行测序
接下来,将纯化的pcr产物汇集到文库中。使用单端或双端测序对文库进行测序,使用75-100个循环以便确定位点特异性空位的完整序列。如果使用单端测序,则读取将由连接臂、随后的分子标签以及在延伸/连接步骤期间填充的独特空位序列组成。不必要测序到延伸臂中,因为所述序列从探针中已知。例如,使用大规模平行测序来识别该区域中的两种甲基化模式(甲基化不足或过甲基化)、受到的dna损伤的类型和量(例如,突变图谱)、以及待针对大染色体异常或基因组不稳定性测定的位点的计数。将来自测序仪的读取与电脑转化的基因组对齐以确定观察到c核苷酸而非预期的t的位置(亚硫酸氢盐转化产生u核苷酸,其通过测序方法读出为t核苷酸)。甲基化比率计算为在cpg位点中观察到的c的数量除以目标靶序列中的cpg二核苷酸的总数量。这确定靶区域中非转化的(即,甲基化的)胞嘧啶核苷酸的比率。然后报告样品的平均甲基化比率。
去除pcr重复
以下描述的是在位点特异性甲基化分析之前识别并去除pcr重复的重要性的实例。使用独特分子标识符允许表征每个捕获事件。更具体地,这些标识符用于对由相同捕获事件产生的读取进行分仓、去除重复并且报告单个共有读取。
从promega获得从十二个个体的血液提取的人类基因组dna(gdna)的混合物(目录号g3041)。根据销售商的手册,使用ez-96dnamethylation-gold对一、五、十或二十纳克的血液gdna和两百纳克的鲑精dna载体进行亚硫酸氢盐转化。如本文所述进行mip捕获。如在本发明实施例中所述还进行随后的pcr,不同的是加入40ul的核酸外切酶反应物而非10ul,并且在100ul而非50ul的五个体积中组装pcr。将文库汇集并且使用双端试剂v2化学在hiseq2500上进行测序。根据图13所述的路线并且在整个说明书中对测序数据进行处理。所述路线包括在bismark对齐之后压缩独特分子标识符以去除pcr重复的步骤(1312)。将bam文件输入到bismark甲基化提取器中,并且将bismark覆盖文件上传到seqmonk中以用于进一步分析(1314)。
为了确定在低输入gdna下测定的稳健性,来自不同输入gdna文库的cpg位点处的甲基化的差异通过将其与20ng文库对照进行比较来确定。首先,将具有低于20x覆盖的cpg位点过滤掉并且确定高于20x覆盖的位点的数量(参见以下表4和表5)。
表4:在没有pcr重复去除的情况下在样品之间观察到的差异甲基化
表5:在没有pcr重复去除的情况下在样品之间观察到的差异甲基化
表4示出在没有pcr重复去除的情况下的数据,并且表5示出在重复去除之后的数据。当通过消除20ng输入样品的pcr重复仅失去4.5%的cpg位点时(上表295,235个位点→下表282,059个位点),在1ng样品中过滤掉87.5%的位点(119,691个位点→14,865个位点),从而示出在低输入下pcr重复的增加。
接下来,在20ng对照样品和每个对应输入文库两者中识别具有高于20x覆盖的cpg位点。共有位点的数量在表4和表5的第3列中报告。过滤掉在20ng文库对照和对应的文库(20、10、5或1ng)两者中至少在20x覆盖下缺少的cpg位点。最后,在第4-9列中报告在对照20ng文库与20、10、5或1ng之间显示高于100%、80%、60%、50%、40%、30%的甲基化差异的cpg位点的数量。在20ng输入下,在去除或不去除pcr重复的情况下在两个数据集合中,当与20ng文库对照相比时,很少位点显示高于30%差异的差异甲基化。在10ng下,pcr重复去除使测定的再现性稍微提高约2倍。在5ng和1ng输入下,去除pcr重复成为分析的关键元素。通过去除重复使在20ng对照文库的情况下具有不一致甲基化状态的cpg位点的数量降低(上表相对于下表)。然而,消除pcr重复还极大地减少在低输入dna文库中具有高于20x覆盖的cpg位点的数量。
总之,检测低输入dna中的差异甲基化可通过在分析之前去除pcr重复来改进,这可减少假阳性的数量。
突变图谱
如上所述,可对使用本文所述的组合物和方法生成的pcr产物进行测序,并且序列信息可告知甲基化状态、cnv状态并且还提供基因组的突变图谱。所有这三种量度可以是不同疾病和病状(包括癌症)的标志。
癌症是由对于细胞dna的损伤或改变导致的失调细胞生长的疾病。随着细胞远离调控内稳态的状态发展,它需要破坏关键控制途径(诸如细胞周期调控、细胞死亡和能量代谢)的dna改变。
最近认识到的癌症的标志是基因组稳定性和dna修复过程的失调。在具有称为“突变图谱”的有差别的不稳定性模式的不同癌症的情况下,基因组稳定性的失调可通过多种途径发生。例如,一个个体的结肠直肠肿瘤可具有与来自其他个体的结肠直肠肿瘤不同的突变图谱。该图谱包括所有单核苷酸取代或变异、小插入和缺失以及较大非整倍性和染色体重排的总和。已提出突变图谱的该模式来响应于免疫疗法区分治疗有效性。参见rizvi等“mutationallandscapedeterminessensitivitytopd-1blockadeinnon–smallcelllungcancer”science;2015年4月3日:第348卷,第6230期,第124-128页。
检测并分类肿瘤的突变图谱的能力具有临床价值,尤其对于改进的预后而言。本文所述的组合物和方法尤其可用于确定突变图谱。例如,在捕获目标dna并对其进行测序之后,可使用标准或定制方法将dna与基因组对齐,并且继续应用一般变体识别器。在应用变体识别器和过滤变体的另外方法之后,可将转换、颠换、缺失和插入的数量分仓到相应的类别中,并且计算所述数量/分析的dna的兆碱基。因为dna损伤的位置分散在整个基因组上,所以不需要关注于预先确定的靶向位置。相反,像本文所述的测定整个基因组上的许多重复区域的技术允许在单个测定中阐明突变图谱。
实施例2:生物信息学工作流
必须处理原始测序数据,以便其用于检测甲基化状态。首先,对序列读取进行过滤以去除已知假象,诸如探针与探针的相互作用、主链序列或衔接子序列。然后将mip的连接臂和延伸臂(即,第一靶向性多核苷酸臂和第二靶向性多核苷酸臂)与序列读取匹配,从而允许每个臂中最多一个碱基对错配。不符合该准则的读取被视为无效并被丢弃。同时,将来自连接末端和延伸末端两者的分子标签分开保存,用于在之后的步骤中对捕获事件计数-虽然在一些实施方案中,将标签保存在一起。通过babraham生物信息学使用bismark亚硫酸氢盐映射器将修整的读取与亚硫酸氢盐转化的人类基因组(hg19)对齐。检查独特对齐的读取(以sam/bam格式的文件)以对具有独特探针空位序列的每个靶向位点的独特分子标签进行计数。这些计数是在下一代测序(ngs)平台(例如,illuminahiseq2500流动池(flowcell))中进行测序的探针-与-靶标的杂交事件的初始数量。可替代地或另外,可使用大规模平行测序或边合成边测序。独特对齐读取(以bam格式的文件)最终运行通过bismark甲基化提取器以确定样品的甲基化状态。对于该步骤,仅使用空位序列(即,连接和延伸臂序列不包括在计算中)。将重复甲基化比率(或评分)报告为整个样品中cpg环境中的甲基化c的比率。例如,cpg中的甲基化c的数量可除以cpg中的甲基化c加上cpg中的非甲基化c的总和。
根据疾病或病状筛查显示异常高水平的技术变异或群体基线变异的靶标或区域,以得出比可通过捕获和测序的其他随机方法获得的更低的变异系数。
实施例3:癌细胞系中甲基化状态的确定
使用以上所述的方法,处理总计12个样品(lncap和pc3癌细胞系、5个健康对照女性样品和5个健康男性样品)。将甲基化比率平均到1mb仓中用于可视化,以得到甲基化密度。针对每个仓对5个女性样品的甲基化密度进行平均。相似地,针对每个仓对5个男性样品的甲基化密度进行平均。图8描绘获得的数据的图形表示;两个内轨表示平均的男性样品与相应的癌细胞系之间每个1mb仓中独特捕获事件的比率,它可用于分析样品的拷贝数变异。另外,关于在整个基因组上其甲基化状态,在癌细胞系dna与非癌血液来源的dna样品之间存在明显的分离。此外,血液来源的dna存在于由本文所述的组合物和方法询问的稳定范围的总体甲基化中。
然后通过查看样品的甲基化指数,在样品之间检测到差异,如图9的上部曲线所示,其中y轴表示甲基化评分,并且x轴表示在如下所述的降低百分比下混合到血液人类基因组dna中的lncapgdna的百分比量。最左侧样品对应于来源自健康个体的血液。随着x轴朝向右侧增加,增加百分比的癌细胞系dna(lncap)从2.5%至50%加入到从健康个体的血液分离的dna。图9的下部曲线描绘来自图9的上部曲线的健康样品和2.5%样品的放大版本。图9所示的结果指示甲基化百分比的变化可至少在2.5%下区分,因为2.5%的重复甲基化评分显著低于健康个体的那些重复甲基化评分。在此可检测至少2.5%或3%的混合物。因此,本文所述的mip可用于确定在整个基因组上或在所需间隔下的甲基化并且呈现为评分(指数、比率、百分比、密度等),其对于临床医生可以是可用于比较健康组织和患病组织,如在描绘癌性样本相对于非癌性样本、侵略性地生长的癌组织相对于非侵略性地生长的癌组织的情况下,或帮助诊断在血浆dna中的其中可整合各种类型的非正常组织的疾病,包括预测早产、先兆子痫,和作为用于从血样样品检测早期癌症的筛查方法。
基于这些数据,可使用mip从血液抽取物检测例如结肠直肠癌的存在。来自肿瘤的片段化小dna存在于血液的血浆组分中,并且可分离该dna。在从血液分离该dna之后,可通过本文所述的方法询问所述dna。可使用在询问各种非癌性个体之后获得的信息计算群体来源的基线。
在进一步对具有例如结肠直肠癌的各种个体进行测序之后,可建立第二疾病状态评分。在随后测序后,将能够确定个体样品与疾病状态或非患病状态的最佳拟合。这些方法针对癌症(例如,结肠直肠癌)对于此目的是理想的,因为所述评分可针对癌症的多种现象得出并且同时进行计算。具体地,甲基化状态作为重复甲基化评分给出,这对应于甲基化比率。通常,可使用染色体非整倍性评分或另一种评分。癌症的标志包括基因组不稳定性(例如,拷贝数变异)和整体甲基化不足,尤其是在与可能含有情况特异性过甲基化的非重复区域相反的重复区域中。使用本文所述的方法是有利的,因为它们减少靶向全基因组的噪声,从而产生由mip靶向的特定重复内的较低样品间变异,同时使用较少的读取并且降低成本。
实施例4:结肠直肠样品中的基因组不稳定分析
该实施例描述本发明的组合物和方法测量从结肠或直肠分离的腺瘤和腺癌的甲基化状态的用途。相同的组合物和方法还用于识别癌样品中甲基化不足的普通cpg位点。最后,从相同的数据集合检测拷贝数改变。
材料
使用以下mip捕获探针和pcr引物:
捕获探针:
:/5phos/ttctcctacctcaacctcnnnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnccaaactaaaatacaata(seqidno:16)(整合dna技术(idt),4nmolultramertmdna寡聚物,标准脱盐)5’磷酸化(idt)
pcr_正向_引物:
aatgatacggcgaccaccgagatctacacatacgagatccgtaatcgggaagctgaag(seqidno:19)(idt,250nmol,标准脱盐)
pcr_反向_索引_引物:
caagcagaagacggcatacgagatnnnnnnnnacacgcacgatccgacggtagtgt(seqidno:20),其中n可以是a、t、c或g,对于每个索引这是不同组合。(idt,100nmol,标准脱盐)
方法
从来自十四个腺瘤和四个腺癌以及其对应的正常相邻组织的新鲜冷冻组织提取人类基因组dna(hgdna)。使用来自qiagen的allprepdna/rna/蛋白质微型试剂盒根据销售商的手册提取hgdna。使用nanodroptm分光光度计定量提取的hgdna,并且根据销售商的手册使用ez-96dnamethylation-gold将10ng样品和200ng鲑精dna载体用于亚硫酸氢盐转化。
将亚硫酸氢盐转化的dna加入到第一靶捕获反应(在图11a、图11b中所述)。使用特别设计的分子倒置探针(mip)来捕获来自如本文所述的亚硫酸氢盐转化的基因组的cpg位点中富含的重复元件。首先,将mip与其在亚硫酸氢盐dna上的靶标退火。将反应物在包含最终浓度为1xampligase缓冲液和1pmol探针的20ul最终体积中组装。运行以下循环程序:98℃持续5分钟,降落(每30秒下降1℃以达到50℃),50℃持续120分钟。
在捕获之后,立即通过高保真dna聚合酶将退火探针在其3’端延伸(参见图11a)。当新合成的dna与mip的连接臂会合时延伸停止,因为dna聚合酶缺乏链置换活性。将新3’端使用在该位置处的磷酸根修饰的能量连接到探针的5’端,从而创建单链环状分子(或复制子)。将延伸/连接反应物在40ul的最终体积中组装,包括整个捕获反应物和最终浓度的1xampligase缓冲液、150nm的dntp、1mmnad+、375mm的甜菜碱、10个单位的ampligase以及1个单位的
在延伸/连接步骤之后,立即通过核酸外切酶消化未连接的探针和gdna。进行该步骤以避免在随后的pcr扩增反应中形成潜在的不需要的产物。将核酸外切酶反应物在50ul的最终体积中组装,包含40ul的延伸连接反应物和最终浓度的1xnebuffer1以及40个单位的核酸外切酶i和200个单位的核酸外切酶iii。将该反应物在37℃下孵育55分钟。然后将核酸外切酶酶在90℃下灭活40分钟并且保持在4℃或-20℃下,直至处理到pcr反应为止。
将pcr反应物在50μl的最终体积中组装,使用10μl的核酸外切酶反应物以及最终浓度的1x
在570nm的最终浓度下使用快速运行模式和针对读取1和2以及针对索引读取的定制引物在hiseq2500上对文库进行测序。针对读取1和读取2进行106个循环的双端读取以及8个循环的索引读取。(参见图12)。读取1定义为从连接臂朝向延伸臂测序的读取。索引在读取1之后读取。在双端转向之后,读取2从延伸臂的反向元件朝向连接臂的反向元件进行测序(图12)。
使用以下步骤进行如在图13中概括的分析。使用casava(illumina)进行解复用(从每个多重索引文库检索测序数据)和fastq文件生成(1302)。使用trimmomatic(bolger,a.m.,lohse,m.和usadel,b.(2014)(1304).trimmomatic:aflexibletrimmerforilluminasequencedata.bioinformatics)修整illumina测序衔接子以及mip主链(对于mip结构,参见图11a)。过滤掉在连接臂或延伸臂上具有多于一个错配的读取(对于探针结构,参见图11a)(1306)。在读取1上,使用定制路线修整掉延伸臂序列。在读取2上,使用定制路线修整掉连接臂序列(1308)。使用bismark三字母对齐器(threeletteraligner)(felixkrueger,babrahaminstitute)进行对齐,其中bowtie2选项生长sam文件,然后使用samtools生成bam文件(1310)(lih.*,handsakerb.*,wysokera.,fennellt.,ruanj.,homern.,marthg.,abecasisg.,durbinr.and1000genomeprojectdataprocessingsubgroup(2009)thesequencealignment/map(sam)formatandsamtools.bioinformatics,25,2078-9)。bowtie2输出文件含有独特对齐的读取。如果对齐具有一个独特最佳的对齐评分,则读取(或读取对)独特地对齐。换言之,丢弃具有多个最佳对齐评分的读取。bismark最小对齐评分通过选项定义:--评分-最小l,0,-0.4,其中l是读取长度(修整之后~75个碱基)。最小对齐评分=0+-0.4*75=-30,其对应于5个错配(每个-6)或多至-30的总和的错配和插入缺失的组合(插入缺失评分:1个碱基:-8,2个碱基-11,3个碱基-14,4个碱基-17,5个碱基-20,6个碱基-23…)bowtie2:(langmeadb,salzbergs.fastgapped-readalignmentwithbowtie2.naturemethods.2012,9:357-359)。
在以下选项的情况下使用bismark甲基化提取器:-p,--无_重叠,--忽略18,--忽略_r218,--综合性,–bedgraph,--胞嘧啶_报告,其中–p:双端分析,--无_重叠:即使测序两次,读取1和2覆盖的胞嘧啶仅报告一次,--忽略18:从分析排除来自读取1中的mip的序列,--忽略_r218:从分析排除来自读取2中的mip的序列,--综合性:显示合并全部四个可能的链特异性甲基化信息的在cpg、chg和chh环境下的甲基化状态。最后,也使用–bedgraph和--胞嘧啶_报告选项来生成覆盖文件(cov)。覆盖输出具有以下特征:<染色体><起始位置><结束位置><甲基化百分比><甲基化计数><非甲基化计数>并且使用基于1的染色体坐标并且仅报告cpg环境。将bam和cov文件输入到seqmonk(simonandrews,babrahaminstitute)中以用于甲基化状态的可视化并且用于进一步的统计分析。
结果
在cpg环境处的甲基化百分比如下通过bismark甲基化提取器计算并报告:cpg处的%甲基化=100*cpg处的甲基化c/(cpg处的甲基化c+cpg处的非甲基化c)。表1示出肿瘤和对应的正常相邻样品的cpg处的甲基化%。
表6
通过比较肿瘤样品和正常样品的甲基化密度来确定肿瘤样品的甲基化不足的水平。甲基化密度定义为对于限定的一兆碱基仓cpg环境中的甲基化c的平均百分比。为了进行该分析,将从bismark甲基化提取器获得的覆盖文件输入到seqmonk中。通过在最少25个不同计数的情况下对在每兆碱基仓处的甲基化状态进行平均来确定甲基化密度。每次cpg位点被读取覆盖,检索的信息被认为是一个计数。如果单个读取涵盖多个cpg,则所述单个读取可生成多个计数。平均而言,每个读取覆盖2个cpg位点。过滤掉y染色体上的仓并且分析总计2852个仓。对于每个仓,如下计算zmeth:
其中zd肿瘤是肿瘤样品的一兆碱基的仓中的甲基化密度;
md正常是来自所有正常样品的一兆碱基的仓的甲基化密度的平均值(n=18);并且
mdsd=来自所有正常样品的甲基化密度的标准偏差(n=18)。
针对有效的2852个仓计算zmeth。如果对应的zmeth低于-5,则仓被认为甲基化不足。表2描绘具有显著甲基化不足的仓的百分比。
表7
还可使用圆环图来使样品的甲基化不足状态可视化。在图14中,针对所有兆碱基报告甲基化状态。左侧的圆环图(正常)示出整个基因组上的高水平的甲基化,其中大部分仓(点)在红色区域中。右侧的图(肿瘤)显示显著量的仓(点),其中较低的甲基化在灰色和绿色区中。十八个肿瘤样品中,有十三个肿瘤样品示出一定水平的甲基化不足。再次使用seqmonk,识别在那些肿瘤样品中通常甲基化不足的位点。首先,过滤掉具有小于30x的覆盖的cpg位点。使用卡方检验p<0.01分析总计89,998个cpg。识别到总计120个位点。有趣的是,在具有明显整体甲基化不足的全部十三个样品中,基因trps1上游的两个cpg位点通常甲基化不足,但是在没有甲基化不足的五个样品(样品726、548、236、245、708)中并非如此(表8)。
表8:具有显著整体甲基化的十三个样品中的常见的甲基化不足位点
通过比较来自肿瘤和正常样品的读取密度(rd)来确定腺瘤和腺癌样品中存在的拷贝数改变。读取密度在此定义为存在于一兆碱基的仓中的读取的总数量。首先,针对读取的总数量将读取归一化到具有最高读取总数量的样品。从人类基因组hg19(3,137,161,264个碱基)创建总计3114个仓。从分析去除具有小于50个读取的仓。保留总计2874个仓。去除的仓大部分对应于由‘n’标记的hg19组件中的未知碱基(7.6%)(ucsc)。应注意,从分析去除的仓的比例(2874/3114*100=7.7%)匹配hg19基因组的未知碱基,从而表明测定靶位点良好地分布在整个可用的hg19基因组中。也过滤掉染色体y上的仓,因为样品中的一些是女性并且不具有映射到染色体y的读取。使用总计2867个仓来计算在肿瘤与正常相邻样品之间的每个限定仓处的读取的log2比率并对其进行作图(图15,上图)。
可替代地,可使用来自biodiscovery的像nexus8.0的专用软件来基于读取深度计算拷贝数变异。图15(下图)示出在腺癌样品中的一个(样品781)中的cna。在chr3q、chr10p、chr13q、chr20q处存在增益,并且在chr5q、chr17p和chr18q中可见损失。nexus8.0软件详细示出样品781中的cna事件以及定位在cna事件处的识别的癌症相关基因(参见表4)。
表9:cna事件和相关联的癌症相关基因的详细描述
可将基因组不稳定性报告为具有显著cna(增益或损失)的仓的百分比。通过首先确定如上所述的肿瘤和正常样品的每兆碱基仓处的读取密度来计算该基因组不稳定性指数。对于每个仓,如下计算zcna:
其中rd肿瘤是限定肿瘤样品的一兆碱基的仓中的读取密度。
rd正常是所有正常样品的一兆碱基的仓中的读取密度的平均值(n=18)
rdsd=来自所有正常样品的读取密度的标准偏差(n=18)
计算总计2867个不同zcna。判断小于-3且大于3的zcna显著不同于正常样品。具有显著cna的仓的百分比在表5中报告。
表10:具有显著cna的仓的百分比
还可在肿瘤与正常样品之间评估特定碱基处的不同甲基化状态。例如,将覆盖文件输入到seqmonk中并且针对正常和肿瘤样品分析具有至少30x的覆盖的cpg位点的甲基化状态。对于样品781,总计191,490个cpg位点满足此准则。在正常与肿瘤样品之间,总计811个cpg位点表现出至少45%的显著差异。另外,所述811个位点在肿瘤样品中比在正常样品中较少甲基化。在肿瘤样品中没有位点比正常样品甲基化多δ45%。有趣的是,在肿瘤样品中表现出甲基化不足的位点的大部分与具有拷贝数增益的位点共定位。针对具有至少一百个分析的cpg位点的每条染色体臂,对在正常与肿瘤之间具有δ45%的cpg位点的百分比进行作图。参见图16。
实施例5:确定来源组织
已示出血浆中的细胞-游离dna(cfdna)的分析可用于不同的诊断目的,包括但不限于非侵入性产前测试和癌症检测;然而,目前证明还难以确定混合物中的不同种类的cfdna的来源或位置(例如,以区分来自造血细胞的血浆cfdna相对于来自肝细胞的血浆cfdna),从而使得cfdna测定的临床用途在一定程度上受限。
用于确定cfdna的来源组织的现有方法包括使用组织特异性rna表达模式或组织特异性甲基化模式。例如,winstonkoh等示出血浆中的细胞-游离rna的rna表达模式可能与某些组织类型相关(参见koh等pnas2014111(20)7361-7366)。然而,rna众所周知是不稳定的,所以当通过rnaseq或rt-qpcr测量时,已证明它对于临床使用是不可靠的。使用组织特异性甲基化模式来确定来源组织先前依赖于全基因组亚硫酸氢盐测序。(参见sun等pnas2015112(40)。已显示来源组织可根据基因组中在特定基因座处的cfdna的甲基化模式来确定。然而,为了从全基因组亚硫酸氢盐测序得到所需的信号,需要进行深度测序,这是耗时且昂贵的。
实验和结果
使用本文所述的组合物和方法,可确定不同组织对于包括来自不同组织类型的细胞-游离dna的混合物的生物样品的贡献,借此可分析dna混合物的甲基化模式(例如,基因组中在重复位点处的甲基化水平)并且确定各种组织类型对于dna混合物的分数补偿。在一些实施方案中,可确定对于dna混合物潜在地做出贡献的组织类型(候选组织)的甲基化模式。然后,确定目标dna混合物的甲基化模式。例如,可计算在各个位点处的甲基化水平。因为dna混合物由来自候选组织的dna构成,所以可通过将dna混合物的甲基化模式与候选组织类型进行比较来确定dna混合物的组成。
在一些实施方案中,相对于参考值的dna中的特定组织类型的贡献百分比的分离值(例如,减去的差值或比率)可指示疾病状态。参考值可对应于健康个体中确定的贡献百分比,并且大于阈值的分离值可确定疾病状态,因为患病组织比健康组织释放更多细胞-游离dna分子。
在概念验证研究中,发明人能够在来自肝脏与来自全血的dna之间进行区分。提取全血来源dna的四个样品,每个具有三个重复,并且提取肝脏来源dna的四个样品,每个具有三个重复。使用本文所述的组合物和方法确定样品中的每个的甲基化状态。具体地,使用以下mip:/5phos/ttctcctacctcaacctcnnnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnccaaactaaaatacaata(seqidno:16)
分析和结果
一般而言,确定来源组织的分析的途径是相似的,与生物学或测量无关。参见图17。首先,从具有组织特异性信号的测定生成参考文库。接下来,使用去卷积算法来解释未知样品并且提供未知样品的百分比估算。
对于在此所述的概念验证研究,通过所述测定测量的所有cpg位点的聚类分析或主组分分析显示不同dna种类之间的不同甲基化模型。如图18所示,血液来源dna(红色)与肝脏dna(蓝色)之间的分离在种类之间比在一个种类内大。
为了验证本文所述的组合物和方法是否可区分另外的组织类型,从不同个体获得来自不同组织来源的dna。参见图19,其从左至由按顺序示出:肝脏(6)、肾(2)、子宫(3)、卵巢(3)、胎盘(1)、胰腺(3)、结肠(3)、胃(3)、肺(3)、膀胱(3)。使用本文先前所述的组合物和方法以及标准对齐和数据处理包,发明人能够在不同组织类型之间进行区分。参见图19中提供的树状图,其示出来自分组在一起的相同组织来源的所有样品。使用以下mip:/5phos/ttctcctacctcaacctcnnnnnnnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnnnnnccaaactaaaatacaata(seqidno:21)。
实施例6:甲基化年龄的确定
已知甲基化状态随时间变化,其中特定组织根据一定范围的因素在不同速率下变得过甲基化或甲基化不足,所述因素包括暴露于环境因素或疾病的存在。因此,如本文所述确定甲基化状态可提供“生物年龄”的量度,这可提供年龄相关病变的存在的早期指示。例如,使用本文所述的组合物和方法连同实施例4所述的组织样品,显示总体甲基化随受试者的年龄降低。参见图20。使用基于小波的滑动窗口分析来计算甲基化的区域综述,并且检查与受试者的年龄的相关的系数。针对每个基因组区域,计算区域系数与样品年龄之间的相关以及相关联的p值。使用benjamini-hochberg方法针对多个假设校正p值,并且示出校正之后的显著相关。
如通过整体甲基化指数(gmi)预测的胎龄
使用本文所述的组合物和方法,在怀孕期间在五个时间点上14个妇女将血液采集到
/5phos/ttctcctacctcaacctcnnnnnncttcagcttcccgattacgggcacgatccgacggtagtgtnnnnnnccaaactaaaatacaata(seqidno:16),以及
本文所述的方法,在五个胎龄(ga)时间点中的每个处针对每个样品确定整体甲基化指数(gmi)。如图21所示,gmi以线性方式随ga降低,直至出生为止。随着胎盘dna的丰度作为总血浆dna的百分比增加,预期甲基化不足的dna增加。
实施例7:5’羟甲基化的检测
5’羟甲基胞嘧啶(5hmc)来源于5’甲基胞嘧啶(5mc)的氧化。5mc向5hmc的转化是活性去甲基化过程的中间步骤。在细胞中,该反应通过十-十一种易位酶家族(tet)催化。5’羟甲基胞嘧啶水平经常在癌症中失调,并且可能促进肿瘤发展和进程。
亚硫酸氢盐转化在5hmc与5mc之间不进行区分。两种修饰均防止胞嘧啶向尿嘧啶转化。为了辨别5mc与5hmc,使用本文所述的组合物和方法,从样品提取gdna并且分到2个反应中:1)常规亚硫酸氢盐转化和2)gdna的变性,接着使用高钌酸钾将5hmc氧化为5-甲酰胞嘧啶,接着使用亚硫酸氢盐将5-甲酰胞嘧啶转化为尿嘧啶。在如本文所述的mip捕获和测序之后,通过比较来自反应1和2的数据来检测5hmc的位点:在反应1中,发现5hmc和5mc两者均为胞嘧啶,而发现非甲基化胞嘧啶为胸腺嘧啶。在反应2中,发现5mc为胞嘧啶,但是发现5hmc和非甲基化胞嘧啶为胸腺嘧啶。羟甲基化状态以及羟甲基化密度可如实施例4中所述计算。
- 还没有人留言评论。精彩留言会获得点赞!