基于异靛蓝衍生物的电子传输型聚合物及有机场效应晶体管的制作方法
本发明涉及半导体材料技术领域,更具体地说,涉及一种电子传输型聚合物半导体材料和有机场效应晶体管。
背景技术:
由于聚合物半导体材料可采用溶液旋涂、印刷、打印等加工手段制备低成本、大面积的有机场效应晶体管等电子器件,近年来备受关注。有机场效应晶体管的活性层部分是有机半导体层。有机半导体层按照传输载流子种类的不同可以分为p型材料(传输空穴),n型材料(传输电子)和双极传输材料(即能传输空穴也能传输电子)。就目前的研究情况而言,p型材料的研究较多且发展很快,聚合物材料的迁移率已经超过10cm2/V·s,达到多晶硅的水平。相比而言,n型聚合物材料则发展缓慢,器件的迁移率明显低于p型材料,且空气稳定性不好。而互补性反相器和逻辑电路的发展离不开高性能的n型传输材料。因此,发展空气稳定性好的n型(传输电子)聚合物材料成为当前研究的重点。目前,空气稳定且迁移率超过1cm2/V·s的n型聚合物材料主要是基于萘二酰亚胺衍生物(J.Am.Chem.Soc.2016,138,3679)和基于引入内酰胺的对苯撑乙烯类化合物(中国专利CN201410004856.6;Adv.Mater.2016,28,7213)构建的。
异靛蓝类聚合物作为场效应晶体管材料近年来进行了很多研究。这些研究都只是针对基于异靛蓝类材料的空穴或双极传输性能,然而对其电子传输性能的研究明显滞后。例如,基于异靛蓝的p型聚合物的空穴迁移率可以达到3.6cm2/V·s(Adv.Mater.2012,24,6457);基于卤素取代异靛蓝的双极传输聚合物的空穴和电子迁移率在1cm2/V·s左右(J.Am.Chem.Soc.2012,134,20025;Chem.Sci.,2013,4,2447);基于氮杂异靛蓝的聚合物表现出高的空穴传输能力,迁移率达到8cm2/V·s(Chem.Mater.2016,28,2209)。相比之下,基于异靛蓝的聚合物的电子迁移率仅在0.22cm2/V·s(Chem.Commun.,2014,50,2180)。
目前,高迁移率共轭聚合物主要采用传统的Stille过渡金属催化的偶联缩聚反应制备,不仅需要高纯度的含锡聚合单体,合成过程复杂繁琐,而且同时生成有机锡化合物等有害副产物。碳氢直接芳基化反应是近年来发展起来的一类新型偶联反应,具有原子经济性好、绿色环保等优点,但受限于C-H键的反应活性较低以及反应选择性较差的问题,一直难以应用到高迁移率共轭聚合物的合成中。因此,采用高效的碳氢直接芳基化聚合反应也是本领域研究人员关注的热点。
因此,本发明提供一种基于异靛蓝衍生物的电子传输型聚合物及聚合物半导体材料,该聚合物半导体材料具有高的电子迁移率,从而满足在有机场效应晶体管中的应用。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种聚合物及聚合物半导体材料,该聚合物具有高的电子迁移率;本发明还提供基于异靛蓝衍生物的电子传输型聚合物及有机场效应晶体管。
具体技术方案如下:
本发明提供一种式(I)所示的基于异靛蓝衍生物的电子传输型聚合物:
其中,X为C-F或N,R为C26~C40的支化链烷基,n为聚合度,为自然数5-50,Ar为第二聚合芳香单元,为式(II)和式(III)所示结构中的一种,
具体的,所述含异靛蓝衍生物的聚合物为式(IV)和(V)所示结构中的一种。
更具体的,所述含异靛蓝衍生物的聚合物根据X为C-F或N及Ar的不同,所述含异靛蓝衍生物的聚合物为式(IV-1)、(IV-2)、(V-1)或(V-2)所示结构中的一种。
优选的,所述含异靛蓝衍生物的聚合物为式(VI)、(VII)、(VIII)和(IX)所示结构中的一种。
其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等,且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
优选的,R1和R2所含碳原子数目为相等。
本发明还提供一种制备上述方案所述的聚合物的制备方法:
在手套箱中,向耐压管中加入6,6′-二溴代异靛蓝衍生物(式(A)或式(B))、(E)-1,2-双(3,4-二氟噻吩-2-基)乙烯(式(C),简称为四氟代二噻吩乙烯)或3,3′,4,4′-四氟-2,2′-联噻吩(式(D),简称为四氟代联噻吩)、Herrmann催化剂、P(o-MeOPh)3、特戊酸,碳酸铯和甲苯,封闭反应管,在加热搅拌条件下进行反应,得到式(I)所示的聚合物。所得式(I)所示的聚合物经过如下纯化处理:将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮和正己烷抽提。将聚合物溶于邻二氯苯中再次在甲醇中沉降。
其中,R为C26~C40的支化链烷基
优选的,所述6,6′-二溴代异靛蓝衍生物为式(A-1)和(B-1)所示结构中的一种,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
优选的,所述具有式(A-1)或式(B-1)结构的化合物与具有式(C)或式(D)结构的化合物的摩尔比为1:1。
优选的,所述Herrmann催化剂与具有式(A-1)或式(B-1)结构的化合物的摩尔比为(0.01~0.04):1。
优选的,所述P(o-MeOPh)3与Herrmann催化剂的摩尔比为(1~3):1。
优选的,所述特戊酸与具有式(A-1)或式(B-1)结构的化合物的摩尔比为(0.5~1.5):1。
优选的,所述特戊酸与具有式(A-1)或式(B-1)结构的化合物的摩尔比为(1~4):1。
优选的,所述甲苯的体积使得具有式(A-1)或式(B-1)结构的化合物的浓度为0.01摩尔每升。
优选的,所述加热搅拌条件为120℃和12小时。
本发明还提供一种上述技术方案所述的聚合物构成的聚合物半导体材料。
本发明还提供一种有机场效应晶体管,电荷传输层为上述技术方案所述的聚合物半导体材料,电子迁移率最高达到5.0cm2/V·s。
本发明提供一种具有式(I)结构的聚合物及聚合物半导体材料,本发明优选吸电子能力强的氟代异靛蓝(见式(A))或氮杂异靛蓝(见式(B))为第一聚合单体,选取平面性好、最高被占据轨道(HOMO)能级低的四氟代二噻吩乙烯(见式(C))或四氟代联噻吩(见式(D))为第二聚合单体。多个氟原子的引入使得本发明提供的聚合物具有如下优势,(1)低的最高未占据轨道(LUMO)和HOMO能级,导致在有机场效应晶体管器件中电子能够有效注入并能够在空气中稳定传输;同时空穴难以注入到活性层中,从而使得本发明所提供的聚合物具有空气稳定的n型(电子)传输特性。(2)好的共轭骨架平面性和强的分子间相互作用,从而保证了使用该聚合物半导体材料的有机场效应晶体管具有高的电子迁移率,最高达到5.0cm2/V·s。(3)适合采用高效低毒的直接芳基化聚合反应制备本发明提供的n型聚合物,从而避免了使用目前被广泛采用的较为复杂和高毒性的Stille聚合反应,进一步提高了聚合物材料的应用价值。
附图说明
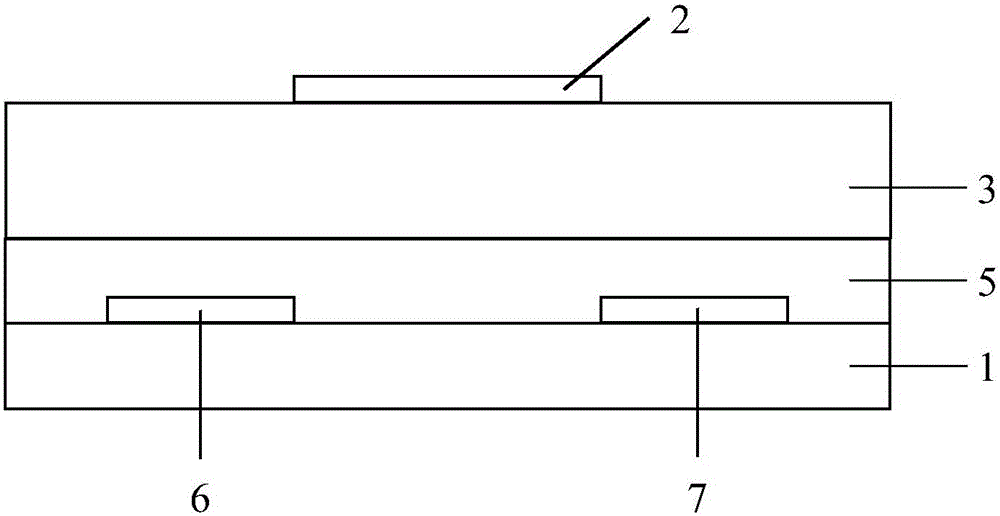
图1为本发明提供的底栅顶接触型(BGTC)有机场效应晶体管的结构示意图;
图2为本发明提供的顶栅底接触型(TGBC)有机场效应晶体管的结构示意图;
图3为本发明实施例10制备的P1作为电荷传输层制备的BGTC结构的有机场效应晶体管的输出特性曲线图;
图4为本发明实施例10制备的P1作为电荷传输层制备的BGTC结构的有机场效应晶体管的转移特性曲线图;
图5为本发明实施例10制备的P1作为电荷传输层制备的TGBC结构的有机场效应晶体管的输出特性曲线图;
图6为本发明实施例10制备的P1作为电荷传输层制备的TGBC结构的有机场效应晶体管的转移特性曲线图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明公开了一种式(I)所示的聚合物:
其中,X为C-F或N,R为C26~C40的支化链烷基,n为聚合度,为自然数5-50,Ar为第二聚合芳香单元,为式(II)和式(III)所示结构中的一种,
具体的,所述含异靛蓝衍生物的聚合物为式(IV)和(V)所示结构中的一种。
更具体的,所述含异靛蓝衍生物的聚合物为式(IV-1)、(IV-2)(V-1)和(V-2)所示结构中的一种。
(S-1)下面对本发明提供的具有式(I)中X为C-F结构的聚合物的制备方法进行说明。
步骤(S-1-1):1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝:
按照如上反应方程式,根据文献(J.Am.Chem.Soc.2012,134,20025)提供的方法制备,将6-溴-7-氟靛红和6-溴-7-氟吲哚-2-酮和冰醋酸混合。用高纯氩气置换气体后,加入浓盐酸,加热到130℃反应12小时,得到深红色固体为6,6′-二溴-7,7′-二氟异靛蓝。在氩气保护下,向溶有6,6′-二溴-7,7′-二氟异靛蓝和氢氧化钾(KOH)粉末的干燥二甲基亚砜(DMSO)溶液中,滴加溶有碘代烷烃(R-I)的四氢呋喃(THF)溶液。室温(R.T.)搅拌1天后,倒入大量水中淬灭。纯化得深红色固体1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝(式(A)所示结构)。
其中,烷基R为C26~C40的支化链烷基,
优选的,1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝为式(A)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
步骤(S-1-2):四氟二噻吩乙烯制备:根据文献(Adv.Mater.2015,27,6753)提供的方法制备。
按照如上反应方程式,用干冰-丙酮浴冷却,氩气保护下,将正丁基锂的正己烷溶液逐滴加入到溶有2-溴-5-三甲基硅基-3,4-二氟噻吩的干燥乙醚中。在-78℃反应1小时后,向反应液中一次性加入无水N,N-二甲基甲酰胺(DMF)。纯化得清亮无色液体2-醛基-5-三甲基硅基-3,4-二氟噻吩。在氩气保护下,在-20℃,将四氯化钛(TiCl4)缓慢加入到干燥的THF中,反应15分钟。一次性加入锌粉,体系升温并回流1小时。再次降温至-20℃,向反应液中加入2-醛基-5-三甲基硅基-3,4-二氟噻吩以及吡啶,升温回流过夜。次日用冰水淬灭,乙醚萃取。乙醇重结晶得到针状无色晶体(E)-1,2-二(5-三甲基硅基-3,4-二氟噻吩基)乙烯。在冰水浴冷却下,向溶有(E)-1,2-二(5-三甲基硅基-3,4-二氟噻吩基)乙烯的二氯甲烷溶液中,逐滴加入三氟乙酸。室温反应1小时后,加水淬灭。纯化后得到白色晶体四氟二噻吩乙烯(式(C)所示结构)。
步骤(S-1-3):四氟联噻吩制备:
按照如上反应方程式,在干冰-丙酮浴中冷却,氩气保护下,将正丁基锂的正己烷溶液加入到溶有化合物2-溴-5-三甲基硅基-3,4-二氟噻吩的干燥乙醚中。反应半小时后,向反应液中逐滴加入三丁基氯化锡。体系逐渐恢复至室温,搅拌过夜。经处理后制得2-三丁基锡-5-三甲基硅基-3,4-二氟噻吩。在氩气保护下,将化合物2-溴-5-三甲基硅基-3,4-二氟噻吩和化合物2-三丁基锡-5-三甲基硅基-3,4-二氟噻吩以及Pd(PPh3)4溶于干燥的甲苯和DMF中,80℃加热搅拌过夜。次日冷至室温,倒入KF水溶液淬灭。纯化得5,5′-二(三甲基硅)-3,3′,4,4′-四氟-2,2′-联噻吩。将化合物5,5′-二(三甲基硅)-3,3′,4,4′-四氟-2,2′-联噻吩与n-Bu4NF·3H2O反应即可得到四氟联噻吩(式(D)所示结构)。
步骤(S-1-4)具有式(I)结构中X为C-F,Ar为式(II)的聚合物的制备,即符合式(IV-1)的聚合物:
按照如上反应方程式,在手套箱中,向耐压管中,加入化合物1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝(式(A))、四氟二噻吩乙烯(式(C))、Herrmann催化剂、配体P(o-MeOPh)3、特戊酸和碳酸铯。氩气气氛下加入甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物,为具有式(I)结构中X为C-F,Ar为式(II)的聚合物。
其中,烷基R为C26~C40的支化链烷基,n为聚合度,为自然数5-50。
优选的,1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝为式(A-1)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
步骤(S-1-5)具有式(I)结构中X为C-F,Ar为式(III)的聚合物的制备,即符合式(IV-2):同步骤(S-1-4),将四氟二噻吩乙烯(式(C))替换为四氟联噻吩(式(D))即可。
优选的,1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝为式(A-1)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
(S-2)下面对本发明提供的具有式(I)结构中X为N的聚合物的制备方法进行说明。
步骤(S-2-1):N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝的制备
按照如上反应方程式,根据文献(Chem.Mater.2016,28,2209)提供的方法合成,氢化钠先与6-溴-1H-吡咯并[2,3-b]吡啶反应,在与碘代烷烃(R-I)在室温搅拌过夜。次日,加水淬灭反应,纯化得到无色油状液体6-溴-1-烷基-1H-吡咯并[2,3-b]吡啶。向装有氯铬酸吡啶盐(PCC)的无水1,2-二氯乙烷和乙腈的悬浊液中滴加6-溴-1-烷基-1H-吡咯[2,3-b]并吡啶的1,2-二氯乙烷溶液,接着加入催化剂量的AlCl3,反应体系加热回流。纯化得到黄色固体6-溴-1-烷基-1H-吡咯[2,3-b]并吡啶-2,3-二酮。在0℃下,向6-溴-1-烷基-1H-吡咯[2,3-b]并吡啶-2,3-二酮的无水甲苯溶液中滴加P(NMe2)3。滴加结束后,缓慢升到室温并继续搅拌1小时。纯化得到深红色固体N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝(式(B)所示结构)。
其中,烷基R为C26~C40的支化链烷基,
优选的,N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝为式(B-1)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
步骤(S-2-2)具有式(I)结构中X为N,Ar为式(II)的聚合物的制备,即符合式(V-1):同步骤(S-1-4),将1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝(式(A))替换为N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝(式(B))即可。
优选的,N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝为式(B-1)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
步骤(S-2-3)具有式(I)结构中X为N,Ar为式(III)的聚合物的制备,即符合式(V-2):同步骤(S-1-5),将1,1′-双烷基-6,6′-二溴-7,7′-二氟异靛蓝(式(A))替换为N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝(式(B))即可。
优选的,N,N-双烷基-6,6′-二溴-7,7′-二氮杂异靛蓝为式(B-1)所示结构,其中,m=0-5的自然数,R1和R2为直烷基链,碳原子数目可以相等或不相等。且满足R1和R2所含碳原子数目之和加上m的数值与式(I)中R的碳原子数目相一致,在26~40之间。
更优选的,R1和R2所含碳原子数目为相等。
此外,本发明还提供一种由上述技术方案所述的聚合物构成的聚合物半导体材料。
与现有技术相比,本发明提供一种具有式(I)结构的聚合物及聚合物半导体材料,本发明优选吸电子能力强的氟代异靛蓝(见式(A))或氮杂异靛蓝(见式(B))为第一聚合单体,选取平面性好、最高被占据轨道(HOMO)能级低的四氟代二噻吩乙烯(见式(C))和四氟代联噻吩(见式(D))为第二聚合单体。多个氟原子的引入使得本发明提供的聚合物具有如下优势,(1)低的最高未占据轨道(LUMO)和HOMO能级,导致在有机场效应晶体管器件中电子能够有效注入并能够在空气中稳定传输;同时空穴难以注入到活性层中,从而使得本发明所提供的聚合物具有空气稳定的n型(电子)传输特性。(2)好的共轭骨架平面性和强的分子间相互作用,从而保证了使用该聚合物半导体材料的有机场效应晶体管具有高的电子迁移率,最高达到5.0cm2/V·s。(3)适合采用高效低毒的直接芳基化聚合反应制备本发明提供的n型聚合物,从而避免了使用目前被广泛采用的较为复杂和高毒性的Stille聚合反应,进一步提高了聚合物材料的使用价值。
本发明还提供一种有机场效应晶体管,结构示意图如图1或图2所示,图1为底栅顶接触型(BGTC)器件结构,图2为顶栅底接触型(TGBC)器件结构。在图1和图2中:1为基板,2为栅极,3为介电层,4为修饰层,5为电荷传输层,由本发明所诉的聚合物半导体材料构成;6代表金属源电极;7代表金属漏电极。
按照本发明,在BGTC器件结构(图1)中以重掺杂的n型硅片为基板1和栅极2,其上有300nm厚的二氧化硅作为介电层3,电容为10nF/cm2。二氧化硅介电层3可以选择性地进行修饰以形成一薄层修饰层4,以改变介电层3与电荷传输层间的界面性质,从而提高有机场效应晶体管的性能;本发明中所用的修饰试剂优选为含硅类化合物,可与介电层3上的自由羟基发生化学反应,从而形成一薄层修饰层4;所述含硅类化合物优选包括十八烷基三氯硅烷(OTS-C18)、辛基三氯硅烷(OTS-C8)、六甲基二硅胺烷(HMDS)、苄基三氯硅烷(BTS)、苯基三氯硅烷或含氟烷基三氯硅烷,具体试剂和修饰方法可参考应用物理杂志上的文献(J.Appl.Phys.,2004,96,6431-6438);本发明制备的聚合物半导体材料通过溶液法沉积形成电荷传输层5,电荷传输层厚度优选为30~100nm,更优选为30~60nm,电荷传输层5的厚度优选用台阶仪测定,40nm厚的金作为源金属电极6和漏金属电极7或TGBC器件结构中的栅极2,优选经掩模板热蒸发沉积,制备的器件的导电沟道宽长比优选为30。
按照本发明,在TGBC器件结构(图2)中以重掺杂的n型硅片为基板1,栅极2为金属电极。绝缘性聚合物为介电层3,所述绝缘性聚合物优选包括聚甲基丙烯酸甲酯(PMMA)、聚乙烯基苯酚(PVP)、聚乙烯醇(PVA)、聚苯乙烯(PS)、聚氯乙烯(PVA)、聚酰亚胺(PI)、苯并环丁烯树脂(BCB,美国陶氏(Dow)化学公司产品)或无定型氟树脂(Cytop,日本旭硝子(Asahi)公司产品),优选为PMMA、PS、BCB或Cytop。其余各部分同上所述。
由于本发明提供的聚合物半导体材料具有良好的溶解性,因此,本发明制备的聚合物半导体材料优选通过溶液法加工成膜,作为电荷传输层5,制备方法包括:旋涂成膜:将本发明提供的聚合物半导体材料溶解在氯苯和邻二氯苯中,优选为邻二氯苯,浓度优选为1~5毫克/毫升,优选为2毫克/毫升,经四氟乙烯滤膜过滤,滴涂在置于涂膜仪上的已制备好的上述基板上,旋涂速度优选为700~1500rpm,更优选为1000rpm,旋涂时间优选为30~60秒,更优选为60秒。薄膜制备过程在大气气氛下进行,电荷传输层5的厚度优选为30~100纳米,更优选为30~60纳米。
本发明提供的聚合物半导体材料通过溶液法加工成为电荷传输层5后,为了提高电荷传输层5的有序度,对上述该层进行热退火处理,可以在单一温度下退火或在不同温度下进行梯度退火,热退火温度范围在100~300℃。更优选为150~250℃。利用上述后处理方法得到的薄膜可以利用原子力显微镜(AFM)和X-射线衍射仪进行薄膜形貌的表征。
为了进一步说明本发明的技术方案,下面结合实施例对本发明实施方案进行描述,但不以任何方式限制本发明的范围,特别是本发明涉及到的烷基R(式(I)中)的范围。
实施例1至实施例9为聚合单体的制备方法
实施例1:1,1′-双(2-十四烷基十六烷基)-6,6′-二溴-7,7′-二氟异靛蓝的合成
在氩气保护下,向溶有6,6′-二溴-7,7′-二氟异靛蓝(912mg,2.00mmol)和KOH粉末(448mg,8.00mmol)的50mL干燥DMSO中,滴加溶有15-(3-碘代甲基)二十九烷(2.74g,5.00mmol)的50mL干燥THF。室温搅拌1天后,倒入大量水中淬灭。过滤得红色固体,用氯仿溶解,饱和食盐水洗涤,无水硫酸镁干燥后过滤,旋干。粗产物经硅胶柱色谱(淋洗剂为石油醚(PE):CH2Cl2=5:1)纯化得深红色固体2.16g,产率80%。结构表征数据如下:1H NMR(400MHz,CDCl3)δ8.87(d,J=8.6Hz,2H),7.20(dd,J=8.7,6.4Hz 2H),3.92(t,J=7.2Hz,4H),1.40-1.14(m,90H),0.89(t,J=6.9Hz,12H)。
实施例2:1,1′-双(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氟异靛蓝的合成
在氩气保护下,向溶有6,6′-二溴-7,7′-二氟异靛蓝(912mg,2.00mmol)和KOH粉末(448mg,8.00mmol)的50mL干燥DMSO中,滴加溶有15-(3-碘代丙基)二十九烷(2.88g,5.00mmol)的50mL干燥THF。室温搅拌1天后,倒入大量水中淬灭。过滤得红色固体,用氯仿溶解,饱和食盐水洗涤,无水硫酸镁干燥后过滤,旋干。粗产物经硅胶柱色谱(淋洗剂为石油醚(PE):CH2Cl2=5:1)纯化得深红色固体2.16g,产率80%。结构表征数据如下:1H NMR(400MHz,CDCl3)δ8.89(d,J=8.6Hz,2H),7.19(dd,J=8.7,6.4Hz 2H),3.90(t,J=7.2Hz,4H),1.67(m,4H),1.38-1.13(m,110H),0.88(t,J=6.9Hz,12H)。
实施例3:1,1′-双(4-十六烷基二十三烷基)-6,6′-二溴-7,7′-二氟异靛蓝的合成
在氩气保护下,向溶有6,6′-二溴-7,7′-二氟异靛蓝(912mg,2.00mmol)和KOH粉末(448mg,8.00mmol)的50mL干燥DMSO中,滴加溶有15-(3-碘代己基)三十三烷(3.37g,5.00mmol)的50mL干燥THF。室温搅拌1天后,倒入大量水中淬灭。过滤得红色固体,用氯仿溶解,饱和食盐水洗涤,无水硫酸镁干燥后过滤,旋干。粗产物经硅胶柱色谱(淋洗剂为石油醚(PE):CH2Cl2=5:1)纯化得深红色固体2.16g,产率80%。结构表征数据如下:1H NMR(400MHz,CDCl3)δ8.89(d,J=8.6Hz,2H),7.20(dd,J=8.7,6.4Hz 2H),3.88(t,J=7.2Hz,4H),1.65(m,4H),1.36-1.11(m,142H),0.87(t,J=6.9Hz,12H)。
实施例4:6-溴-1-(4-十四烷基十八烷基)-1H-吡咯并[2,3-b]吡啶的制备:
在0℃下,向氢化钠(分散在煤油中重量比为60%,235mg,5.88mmol)的DMF(10mL)悬浊液中加入6-溴-1H-吡咯并[2,3-b]吡啶(1.00g,5.08mmol)。接着在室温搅拌15分钟,然后向反应体系加入15-(3-碘代丙基)二十九烷(3.22g,5.58mmol),在室温搅拌过夜。次日,加水淬灭反应,用乙酸乙酯萃取,无水硫酸镁干燥有机相后过滤,旋干。粗产物经硅胶柱色谱(淋洗剂为PE)纯化得到无色油状液体(2.62g,产率80.0%)1H NMR(400MHz,CDCl3):δ7.75(d,J=8.1Hz,1H),7.23(d,J=3.6Hz,1H),7.20(d,J=8.1Hz,1H),6.45(d,J=3.6Hz,1H),4.13(d,J=7.2Hz,2H),1.67(m,2H),1.38-1.13(m,55H),0.88(t,J=6.9Hz,6H).13C NMR(100MHz,CDCl3):δ147.86,134.70,131.11,129.08,119.48,119.32,99.99,48.82,39.22,32.37,31.80,30.33,30.10,30.06,29.98,29.80,26.67,23.13,14.32.
实施例5:6-溴-1-(4-十四烷基十八烷基)-1H-吡咯并[2,3-b]吡啶-2,3-二酮的制备
向装有氯铬酸吡啶盐(PCC)(2.00g,9.28mmol)和硅胶粉(2.0g)的无水1,2-二氯乙烷(10mL)和乙腈(15mL)的悬浊液中滴加6-溴-1-(4-十四烷基十八烷基)-1H-吡咯并[2,3-b]吡啶(2.50g,3.49mmol)的1,2-二氯乙烷(5mL),加料过程中保证良好的搅拌。加入催化剂量的AlCl3(8mg),反应体系加热回流3小时。结束反应后将溶剂蒸除,剩余物进行硅胶柱色谱(淋洗剂为PE:甲苯)纯化,得到黄色固体(1.53g,产率65.1%)。结构表征数据如下:1H NMR(400MHz,CDCl3):δ7.64(d,J=7.7Hz,1H),7.27(d,J=7.7Hz,1H),3.69(d,J=7.1Hz,2H),1.97(m,2H),1.23-1.17(m,55H),0.88(t,J=6.5Hz,6H).13C NMR(100MHz,CDCl3):δ181.45,164.96,158.93,150.28,134.32,123.62,110.77,44.07,36.57,32.37,31.82,30.35,30.10,29.80,26.57,23.13,14.32.
实施例6:N,N-双(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氮杂异靛蓝的制备
在0℃下,向6-溴-1-(4-十四烷基十八烷基)-1H-吡咯并[2,3-b]吡啶-2,3-二酮(0.811g,1.2mmol)的无水甲苯(5mL)中滴加P(NMe2)3(0.196g,1.2mmol)。滴加结束后,缓慢升到室温并继续搅拌1小时。用水淬灭反应,有机相用无水硫酸镁干燥。去除溶剂后,进行硅胶柱色谱(淋洗剂为PE:甲苯)纯化,得到深红色固体(0.475g,产率30.0%)结构表征数据如下:1H NMR(400MHz,CDCl3)δ9.28(d,J=8.3Hz,2H),7.18(d,J=8.3Hz,2H),3.74(d,J=7.0Hz,4H),1.67(m,4H),1.38-1.13(m,110H),0.88(t,J=6.9Hz,12H).13C NMR(100MHz,CDCl3)δ168.15,158.73,144.02,139.34,131.71,122.11,114.91,44.14,36.54,32.38,31.90,30.39,30.14,30.12,30.06,29.81,26.62,23.14,14.33.
实施例7:2-三丁基锡-5-三甲基硅基-3,4-二氟噻吩的制备
在干冰-丙酮浴中冷却,氩气保护下,将正丁基锂的正己烷溶液(2.5M,4.0mL,9.9mmol)逐滴加入到溶有化合物2-溴-5-三甲基硅基-3,4-二氟噻吩(2.55g,9.39mmol)的30mL干燥乙醚中。-78℃反应半小时后,向反应液中逐滴加入三丁基氯化锡(2.8mL,10.3mmol)。体系逐渐恢复至室温,搅拌过夜。加水淬灭反应,用轻石油醚萃取,有机相用饱和食盐水洗,无水硫酸镁干燥后过滤,蒸除溶剂后得淡黄色液体4.6g,直接用于下一步。
实施例10:5,5′-二(三甲基硅)-3,3′,4,4′-四氟-2,2′-联噻吩
在氩气保护下,将化合物2-溴-5-三甲基硅基-3,4-二氟噻吩(272mg,1.00mmol)和化合物2-三丁基锡-5-三甲基硅基-3,4-二氟噻吩(579mg,1.20mmol)以及Pd(PPh3)4(23mg,0.020mmol)溶于1mL干燥甲苯和1mL干燥DMF中,80℃加热搅拌过夜。次日冷至室温,倒入KF水溶液淬灭。乙醚萃取,有机相用饱和食盐水洗,无水硫酸镁干燥后过滤,蒸除溶剂。粗产物经硅胶柱色谱(淋洗剂为石油醚)纯化得白色固体(306mg,产率80%)。结构表征数据如下:1H NMR(400MHz,CDCl3)δ0.36(s,18H);19F NMR(376MHz,CDCl3)δ-128.54(dd,J=12.5,2.1Hz,2F),-134.45(dd,J=12.5,2.1Hz,2F);13C NMR(100MHz,CDCl3)δ150.0(dd,J=253,20.0Hz),142.7(dd,J=265,22.0Hz),115.8(d,J=47.3Hz),115.3,0.9;GC/MS m/z(%):382(100)。
实施例9:3,3′,4,4′-四氟-2,2′-联噻吩
在冰水浴冷却下,向溶有化合物5,5′-二(三甲基硅)-3,3′,4,4′-四氟-2,2′-联噻吩(200mg,0.520mmol)的8mL THF中,加入n-Bu4NF·3H2O(32.8mg,0.100mmol),室温下反应3小时。加水淬灭后,乙醚萃取,有机相用饱和食盐水洗涤,无水硫酸镁干燥后过滤,蒸除溶剂。粗产物经硅胶柱色谱(淋洗剂为石油醚)纯化得白色固体112mg,产率90%。1H NMR(400MHz,CDCl3)δ6.76(d,J=2.3Hz,2H);19F NMR(376MHz,CDCl3)δ-134.97(dd,J=11.0,2.3Hz,2F),-136.38(dd,J=11.0,2.3Hz,2F);13C NMR(100MHz,CDCl3)δ150.0(dd,J=260,17.5Hz),142.0(dd,J=267,20.9Hz),110.8(m),102.6(m);GC/MS m/z(%):238(100)。
实施例10至实施例16为符合式(I)所述聚合物的制备方法
实施例10:聚合物P1的制备
在30mL耐压管中,加入化合物1,1′-(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氟异靛蓝(135mg,0.100mmol)、四氟二噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜132mg,产率91%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=57.9kDa,元素分析(C90H136N2F6O2S2(%)):计算值:C 74.23,H9.41,N 1.92,S 4.40;测量值:C 74.02,H 9.21,N 1.87,S 4.61。
实施例11:聚合物P2的制备
在30mL耐压管中,加入化合物1,1′-(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氟异靛蓝(135mg,0.100mmol)、四氟联噻吩(24.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜135mg,产率94%。结构表征数据如下:GPC:Mn=88.0kDa,元素分析(C88H134N2F6O2S2(%)):计算值:C 73.90,H 9.44,N 1.96,S 4.48;测量值:C 73.71,H 9.29,N 1.88,S 4.65。
实施例12:聚合物P3的制备
在30mL耐压管中,加入化合物1,1′-(2-十四烷基十六烷基)-6,6′-二溴-7,7′-二氟异靛蓝(118mg,0.100mmol)、四氟二噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜132mg,产率91%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=52.9kDa,元素分析(C78H114N2F6O2S2(%)):计算值:C 72.63,H 8.91,N 2.17,S 4.97;测量值:C 72.82,H 9.01,N 1.97,S 4.71。
实施例13:聚合物P4的制备
在30mL耐压管中,加入化合物N,N-双(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氮杂异靛蓝(132mg,0.100mmol)、四氟二噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜129mg,产率92%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=50.9kDa,元素分析(C88H136N4F4O2S2(%)):计算值:C 74.32,H 9.64,N 3.94,S 4.51;测量值:C 74.08,H 9.31,N 3.87,S 4.61。
实施例14:聚合物P5的制备
在30mL耐压管中,加入化合物N,N-双(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氮杂异靛蓝(132mg,0.100mmol)、四氟联噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜127mg,产率90%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=60.9kDa,元素分析(C86H134N4F4O2S2(%)):计算值:C 73.98,H 9.67,N 4.01,S 4.59;测量值:C 73.87,H 9.51,N 3.89,S 4.65。
实施例15:聚合物P6的制备
在30mL耐压管中,加入化合物1,1′-(4-十六烷基二十三烷基)-6,6′-二溴-7,7′-二氟异靛蓝(155mg,0.100mmol)、四氟二噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(1.9mg 2.0mol%)、配体P(o-MeOPh)3(1.4mg,4.0mol%)、特戊酸(10.2mg,0.100mmol)和碳酸铯(98mg,0.30mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜132mg,产率91%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=60.9kDa,元素分析(C104H166N2F6O2S2(%)):计算值:C 75.53,H 10.11,N 1.69,S 3.88;测量值:C 75.82,H 9.81,N 1.77,S 4.01。
实施例16:聚合物P7的制备
在30mL耐压管中,加入化合物1,1′-(4-十四烷基十八烷基)-6,6′-二溴-7,7′-二氟异靛蓝(135mg,0.100mmol)、四氟二噻吩乙烯(27.0mg,0.100mmol)、Herrmann’s催化剂(0.8mg 1.0mol%)、配体P(o-MeOPh)3(0.7mg,2.0mol%)、特戊酸(20.4mg,0.200mmol)和碳酸铯(130mg,0.40mmol)。惰性气氛下加入10mL甲苯,封闭反应管,120℃搅拌加热12小时。冷至室温后,将聚合物在甲醇中沉降,过滤,将收集到的聚合物在索氏提取器中依次用丙酮、正己烷抽提洗涤。将聚合物溶于邻二氯苯再次在甲醇中沉降,过滤后得到亮黑色聚合物膜132mg,产率91%。结构表征数据如下:凝胶渗透色谱(GPC):Mn=42.9kDa,元素分析(C90H136N2F6O2S2(%)):计算值:C 74.23,H 9.41,N 1.92,S 4.40;测量值:C 74.12,H 9.11,N 1.77,S 4.81。
实施例17~23
以重掺杂的n型硅片为基底和栅极,其上覆盖有300nm厚的二氧化硅介电层,二氧化硅介电层用BCB修饰(厚度40nm),复合介电层的电容为8nF/cm2,分别采用实施例10~16中制备的共轭聚合物作为半导体材料,分别配成邻二氯苯溶液,浓度均为2毫克/毫升,转速均为1000rpm,旋转时间均为60秒,薄膜厚度均在30-60纳米之间;然后进行热退火处理,退火温度及时间见列表1;最后沉积厚度为50纳米的金(Au)以形成薄膜晶体管器件的源/漏电极,退火温度、时间以及薄膜晶体管器件的载流子迁移率列于表1中:
表1实施例17~23制备的薄膜晶体管的性能测定结果
实施例24~27
以重掺杂的n型硅片为基底,沉积厚度为40纳米的金(Au)以形成薄膜晶体管器件的源/漏电极,分别采用实施例10,11,13,14中制备的共轭聚合物作为半导体材料,分别配成邻二氯苯溶液,浓度均为2毫克/毫升,转速均为1000rpm,旋转时间均为60秒,薄膜厚度均在30-60纳米之间。然后进行热退火处理,退火温度及时间见列表2;接着旋涂PMMA作为介电层,厚度为500nm;最后沉积厚度为40纳米的金(Au)以形成薄膜晶体管器件的栅电极,退火温度、时间以及薄膜晶体管器件的载流子迁移率性质列于表2中:
表2实施例24~27制备的薄膜晶体管的性能测定结果
如图3、图4所示,为本发明实施例10制备的共轭聚合物半导体材料P1作为电荷传输层制备的BGTC型有机场效应晶体管的输出特性曲线和转移特性曲线,二氧化硅介电层用BCB修饰,薄膜退火温度为200℃,溶剂为邻二氯苯,迁移率达到0.36cm2/V·s。
如图5、图6所示,为本发明实施例10制备的共轭聚合物半导体材料P1作为电荷传输层制备的TGBC型有机薄膜晶体管的输出特性曲线和转移特性曲线,绝缘性聚合物为PMMA,薄膜退火温度为200℃,溶剂为邻二氯苯,迁移率达到5.0cm2/V·s。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- 一种新型罗汉果醇衍生物单体的制作方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 一种新型罗汉果醇衍生物单体的制作方法
- 通过预先占位调控邻位碳硼烷衍生物结构的方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗菌药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法