一种喜树碱‑阿霉素前药及其制备方法和应用与流程
本发明涉及生物医药技术、纳米医药及药物控制释放技术领域,具体地说是一种还原降解释药的喜树碱-阿霉素前药,及其制备方法和应用。
背景技术:
癌症是全球第二大死亡原因。根据世界卫生组织统计,2015年癌症造成880万人死亡。全球接近六分之一的人死亡是由于癌症,其中约70%的癌症死亡发生在低收入和中等收入国家。目前,化疗是癌症的主要治疗方法之一。然而,大多数化疗药物,比如喜树碱(camptothecin),紫杉醇(paclitaxel)等,在水中溶解度有限,而且有着不良副作用。因此,对化疗药物的改性以及设计有效的药物投递系统一直是药物控制释放领域的研究焦点。纳米药物已被广泛研究用于癌症治疗。纳米颗粒的使用不仅可以改善这些药物的水分散性,而且还可以增强它们的药代动力学和生物体内分布,改善治疗效果和减少副作用。
喜树碱(camptothecin,CPT)最早是从喜树中提取出来的生物碱,它是DNA拓扑异构酶I(TOPO I)抑制剂,在临床前研究中具有显著的抗肿瘤活性。但由于低溶解度和严重不良副作用,在临床试验中失败。目前,喜树碱类似物伊立替康(Irinotecan)和拓扑替康(Topotecan)已被美国食品和药物管理局(FDA)批准用于治疗结肠癌。伊立替康在水中能够水解出7-乙基-10-羟基喜树碱(SN-38),后者比伊立替康本身高1000倍的抗肿瘤活性。但伊立替康在人体内仅能够水解少量的7-乙基-10-羟基喜树碱,故其对癌细胞的杀伤力也明显受损。阿霉素(Doxorubicin,DOX)是DNA拓扑异构酶II(TOPO II)抑制剂,具有很强的抗肿瘤活性,但是有多重副作用,包括恶心、呕吐和心律不齐。由于阿霉素具有一定的亲水性,导致阿霉素会迅速的从纳米颗粒释放出来,导致其抗肿瘤效果受损。
由于喜树碱和阿霉素均是拓扑异构酶抑制剂,因而具有协同的抗癌活性。在体外细胞实验中,两种药物经常一起给药,发现具有很强的协同作用。但是在临床试验中,喜树碱和阿霉素副作用也一起增强,因此大多数在临床二期实验前就被终止。阿霉素和喜树碱也可以一起连在同一个高分子聚合物上,如Journal of Controlled Release,Volume 210,28July 2015,Pages 198–207。但是他们很难从高分子聚合物上释放下来,并且释放的速度也不会一致,导致疗效不是最好的。而且高分子聚合物载药量底,合成复杂,因而有必要发明新的方法能够 简单有效的一起投递喜树碱和阿霉素。
技术实现要素:
本发明的首要目的是提供一种可在肿瘤细胞选择性释放喜树碱和阿霉素的前药,具有显著的癌细胞抑制效果,同时不良副作用低。
本发明的另一目的是提供制备所述的前药的方法,合成过程简单易行,载药量高。
本发明的再一目的是提供所述的前药在制备癌症治疗药物中的应用。
本发明的上述目的通过以下技术方案实现:
首先,提供一种同时载有喜树碱类和阿霉素的前药,它是喜树碱或其衍生物与阿霉素通过连接基团形成的喜树碱类-阿霉素前药,其结构式如式(I)
其中,
R1为喜树碱或其衍生物基团;
R2为或者中的一种;
R3为或者中的一种;
X为S或者-CH2-中的一种;
n1和n2为重复单元数,为0-10的整数;优选0-5的整数;更优选0-2的整数。
本发明优选的方案中,所述的喜树碱或其衍生物基团来自式(II)~式(III)所示结构中的一种:
式(II)为喜树碱的化学结构式,式(III)为7-乙基-10-羟基喜树碱化学结构式。
本发明进一步优选的方案中,所述的式(I)中R1为喜树碱基团,R2为R3为X为S或者-CH2-,n1=2,n2=2。
本发明最优选的方案中,所述的喜树碱类-阿霉素前药结构如下式(VI)所示。
本发明还提供一种制备所述喜树碱类-阿霉素前药的方法,是以喜树碱或其衍生物为原料,在有机溶剂中,通过与带有相应基团的原料反应,制成喜树碱或其衍生物前药中间体,最后所述中间体再和阿霉素反应制备得到所述的喜树碱类-阿霉素前药。
本发明所述的制备喜树碱类-阿霉素前药的方法中,制备R1为喜树碱或7-乙基-10-羟基喜树碱的前药的方法,具体包括以下步骤:
1)在有机溶剂中,在酰化催化剂存在下使用三光气活化喜树碱或其衍生物内酯环上的羟基,再加入过量的2,2'-二硫代二乙醇或1,6-己二醇反应,得到中间体1;所述的喜树碱或其衍生物是喜树碱或7-乙基-10-羟基喜树碱;
2)在有机溶剂中,使用氯甲酸-4-硝基苯酯与步骤1)得到的中间体醇的羟基进行酯交换反应,得到中间体2;
3)将步骤2)得到的中间体2和阿霉素在有机溶剂中混合,并加入有机碱搅拌反应,通过氨酯交换反应生成的酰胺键将中间体2和阿霉素连接,得到本发明所述的部分喜树碱类-阿霉素前药。
本发明所述的制备方法中,步骤1)所述的有机溶剂可选自二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环或二甲基甲酰胺中的任意一种;优选二氯甲烷。
本发明所述的制备方法中,步骤1)所述的酰化催化剂可选自4-(二甲基氨基)吡啶、三乙胺或N,N-二异丙基乙胺中的任意一种;优选4-(二甲基氨基)吡啶。
本发明所述的制备方法中,步骤2)所述的有机溶剂可选自二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环或二甲基甲酰胺中的任意一种;优选二氯甲烷。
本发明所述的制备方法中,步骤3)所述的有机溶剂可选自DMF或二甲基亚砜中的任意一种;优选DMF。
本发明所述的制备方法中,步骤3)所述的有机碱可选自三乙胺或N,N-二异丙基乙胺中的任意一种或两种的混合物;优选三乙胺。
本发明所述制备方法优选的一种实施方式中,制备了式(IV)所示前药,其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的2,2'-二硫代二乙醇反应,制备得到中间体1;然后使用氯甲酸-4-硝基苯酯在有机溶剂中将中间体1通过酯交换反应转化为中间体2;最后,将中间体2和阿霉素一起在DMF中混合,并加入三乙胺一起搅拌,制备得到式(IV)所示前药:CPT-ss-DOX。
本发明制备方法优选的另一种实施方式中,制备了以下式(V)所示的前药:
其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的1,6-己二醇反应,制备得到中间体1;然后使用氯甲酸-4-硝基苯酯在有机溶剂中将中间体1通过酯交换反应转化为中间体2;最后,将中间体2和阿霉素一起在DMF中混合,并加入三乙胺一起搅拌,制备得到式(V)所示前药:CPT-cc-DOX。
本发明的前药可以直接单独用作癌症治疗药物,也可以进一步制成其他药物学上可接受的剂型用于癌症治疗。因此,本发明还提供所述喜树碱类-阿霉素前药在药物学上可接受的制剂。
本发明优选的方案中,所述的制剂是纳米颗粒制剂,所述的纳米颗粒外部为赋型剂和/或药用载体,所述的赋型剂和/或药用载体内部包裹本有发明所述的喜树碱类-阿霉素前药。
所述的赋型剂和药用载体包括但不限于吐温系列乳化剂(如吐温80、吐温20等)、聚氧乙烯蓖麻油(Cremophor EL)、脂质体(liposome)或者两亲性高分子中的任意一种或两种以上的混合物。本发明优选两亲性高分子。
所述的两亲性高分子选自聚乙二醇-b-聚乳酸(polyethylene glycol-b-polylactide,PEG-b-PLA)、聚乙二醇-b-聚己内酯(polyethylene glycol-b-polycaprolactone,PEG-b-PCL)或聚乙二醇-b-聚磷酯(polyethylene glycol-b-polyphosphoester,PEG-b-PPE)中的任意一种。
本发明还提供所述的喜树碱类-阿霉素前药在制备所述癌症治疗药物中的应用。
本发明优选的应用方案中,使用两亲性高分子对所述的喜树碱类-阿霉素前药进行物理包裹,制备得到纳米颗粒。
本发明的一种优选的实施方式中,使用聚乙二醇-b-聚乳酸作为药用载体对所述的喜树碱类-阿霉素前药进行物理包裹;具体是将本发明所述的喜树碱类-阿霉素前药和聚乙二醇-b-聚乳酸以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。
本发明的另一种优选的实施方式中,使用聚乙二醇-b-聚磷酯作为药用载体对所述的喜树碱类-阿霉素前药进行物理包裹;具体是将本发明所述的喜树碱类-阿霉素前药和聚乙二醇-b-聚磷酯以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒;或者,将本发明所述的喜树碱类-阿霉素前药和聚乙二醇-b-聚磷酯以一定比例一起溶于有机溶剂(如四氢呋喃),然后挥发掉有机溶剂,再加入水溶液,超声或者搅拌制得纳米颗粒。
本发明的再一种优选的实施方式中,使用聚乙二醇-b-聚己内酯作为药用载体对所述的喜树碱类-阿霉素前药进行物理包裹;具体是将本发明所述的喜树碱类-阿霉素前药和聚乙二醇-b-聚己内酯以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。
在本发明中,喜树碱和阿霉素通过连接键链接在一起形成前药,特别是两者同二硫键链接在一起,能够在肿瘤细胞选择性的释放喜树碱和阿霉素。目前,国内外尚无这种前药被报道。本发明所述的前药及其纳米颗粒制剂具有如下优点:
(1)喜树碱和阿霉素均为拓扑异构酶抑制剂,具有协同作用;
(2)喜树碱和阿霉素均被化学修饰,在二硫键断裂前,对细胞的毒性小;
(3)由于喜树碱和阿霉素被二硫键连接,因此该前药对细胞内的还原性物质,如谷胱甘肽敏感,接触谷胱甘肽后能迅速释放喜树碱和阿霉素;
(4)现有技术中,喜树碱和阿霉素的均不易被两亲性高分子包裹,且包裹的药物容易从颗粒内释放出来,而本发明中的前药,由于分子量更大,更疏水,导致他们从颗粒中的释放速度大大降低,有利于降低药物的毒副作用,提高疗效;
(5)本发明合成的前药能够非常高效的被包裹,纳米颗粒的载药量也非常高;
(6)前药的合成简单,纳米颗粒的制备容易,有利于临床转化。
附图说明
图1为CPT-ss-OH的1H NMR(300MHz,CD2Cl2)。
图2为CPT-ss-LG的1H NMR(300MHz,CDCl3)。
图3为CPT-ss-DOX的1H NMR(300MHz,CDCl3)。
图4为CPT-cc-OH的1H NMR(300MHz,CDCl3)。
图5为CPT-cc-LG的1H NMR(300MHz,CDCl3)。
图6为CPT-cc-LG的1H NMR(300MHz,CDCl3)。
图7为药物从纳米颗粒的释放实验。
图8为通过HPLC测定前药释放喜树碱和阿霉素。
图9为喜树碱和阿霉素的细胞毒性实验。
图10为前药的细胞毒性实验。
图11为体外细胞摄取实验结果。
具体实施方式
下面通过实施例对本发明进行具体描述,本实施例只用于对本发明作进一步的说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据上述发明的内容做出的一些非本质的改进和调整,均属本发明保护范围。
实施例1:CPT-ss-OH的制备
在搅拌下,将4-二甲氨基吡啶(1.05g,8.60mmol)在10mL二氯甲烷中的溶液滴加到喜树碱(1.0g,2.87mmol)和三光气(0.315g,1.06mmol)的无水二氯甲烷(200mL)的混合物悬浮液中。搅拌30分钟后,加入在无水四氢呋喃(25mL)中的2,2'-二硫代二乙醇(8.60g,55.8mmol),将反应混合物在室温下搅拌过夜。将混合物用50mM盐酸水溶液(2×100mL),水(1×100mL)和饱和盐水(1×100mL)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱分离纯化。产量:1.05g(69%产率)。1H NMR(300MHz,CD2Cl2)δ8.46(s,1H),8.22(d,J=8.3Hz,1H),7.99(dd,J=8.2,1.1Hz,1H),7.86(ddd,J=6.9,6.5Hz,1H),7.70(ddd,J=8.1,6.9,1.2Hz,1H),7.42(s,1H),5.64(d,J=18Hz,1H),5.36(m,1H)4.37(t,J=6.0Hz,2H),3.93-3.81(m,2H),3.03-2.82(m,4H),2.19(tdd,J=14.0,12.3,7.5Hz,3H)1.01(t,J=7.5Hz,3H)。图1为CPT-ss-OH的1H NMR谱图,证明了制备该化合物成功。
实施例2:CPT-ss-LG的制备
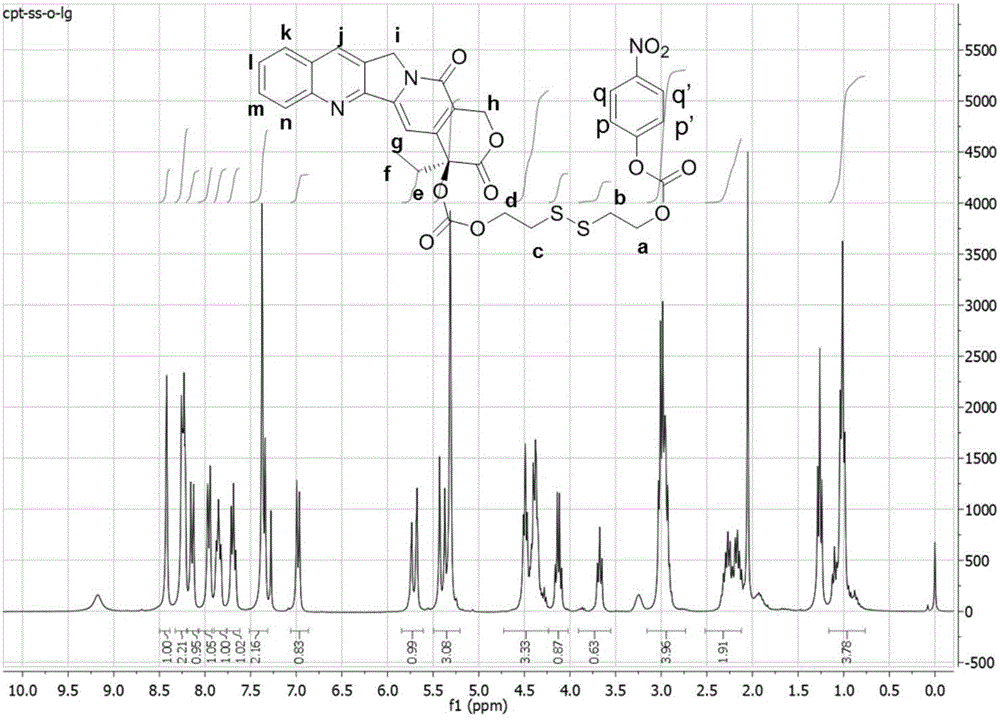
在搅拌下将氯甲酸4-硝基苯酯(372mg,1.85mmol)、实施例1制备的CPT-ss-OH(100mg,0.184mmol)和三乙胺(TEA,224mg,2.22mmol)溶于无水DCM,将反应混合物在室温下搅拌过夜,然后使用预填充二氧化硅柱通过快速色谱(Teledyne ISCO CombiFlash)纯化。使 用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:102mg(80%产率)。1H NMR(300MHz,CDCl3)δ8.40(s,1H),8.27-8.16(m,3H),8.12(d,J=9.0Hz,1H),7.94(d,J=8.0Hz,(t,J=7.3Hz,1H),7.67(t,J=7.5Hz,1H),7.34(d,J=10.2Hz,J=17.2Hz,1H),5.43-5.32(m,1H),5.29(s,2H),4.55-4.41(m,2H),4.42-4.31 2H,1H),2.95(td,J=13.9,6.3Hz,4H),2.35-2.09(m,2H),1.00(t,J=7.3Hz,3H)。图2为CPT-ss-LG的1H NMR谱图,证明了制备该化合物成功。
实施例3:CPT-ss-DOX的制备
将实施例2制备的CPT-ss-LG(43mg,0.062mmol)、DOX.HCl(38mg,0.066mmol)、TEA(63mg,0.62mmol)在3mL DMF中混合,并在氮气下搅拌1天。通过加入过量的乙酸淬灭反应,并通过制备型HPLC使用乙腈和0.1%三氟乙酸的水溶液(梯度:20-95%的乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:35mg(51%产率)。1H NMR(300MHz,CDCl3)δ8.36(s,1H),8.20(d,J=8.3Hz,1H),8.04(d,J=7.0Hz,1H),7.92(d,J=7.7,1H),7.86-7.73(m,3H),7.71-7.58(m,2H),7.43-7.36(m,1H),7.33(s,1H),5.70(d,J=17.2Hz,2H)2H),5.45-5.14(m,7H),4.77(d,J=4.3Hz,2H),4.66(s,1H),4.37(s,2H),4.18-4.01(s,2H),3.65(s,2H),3.32(s,1H),3.26(s,1H),3.07(s,1H),3.01(s,1H),2.89(d,J=2H),2.81(dd,J=10.7,6.2Hz,2H),2.43-2.09(m,7H),2.02-1.75(m,5H),1.36-1.25(m,10H),1.02(t,7.5Hz,3H)。图3为CPT-ss-DOX的1H NMR谱图,证明了制备该化合物成功。
实施例4:CPT-cc-OH的制备
在搅拌下,将4-二甲氨基吡啶(1.68g,13.8mmol)在10mL二氯甲烷中的溶液滴加到喜树碱(1.5g,4.31mmol)和三光气(0.473g,1.59mmol)的无水二氯甲烷(200mL)的混合物悬浮液中。搅拌30分钟后,加入在无水四氢呋喃(15mL)中的1,6-己二醇(6.64g,43.1mmol),将反应混合物在室温下搅拌过夜。将混合物用50mM盐酸水溶液(2×100mL),水(1×100mL)和饱和盐水(1×100mL)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:1.48g(70%产率)。1H NMR(300MHz,CDCl3)δ8.41(s,1H),8.23(d,J=8.7Hz,1H),7.95(dd,J=8.2,1.1Hz,1H),7.85(ddd,J=6.9,6.5Hz,1H),7.68(ddd,J=8.1,6.9,1.2Hz,1H),7.36(s,1H),5.70(d,J=17.3Hz,1H),5.39(d,J=1H),5.30(s,2H),4.21-4.05(m,2H),3.59(dd,J=10.1,6.1Hz,2H),2.43-2.01(m,2H),1.74-1.65(m,3H),1.59-1.47 (m,2H),1.45-1.32(m,4H),1.00(t,J=7.5Hz,3H)。图4为CPT-cc-OH的1H NMR谱图,证明了制备该化合物成功。
实施例5:CPT-cc-LG的制备
在搅拌下将氯甲酸4-硝基苯酯(372mg,1.85mmol)、实施例4制备的CPT-cc-OH(100mg,0.203mmol)和三乙胺(TEA,224mg,2.22mmol)溶于无水DCM将反应混合物在室温下搅拌过夜,然后使用预填充二氧化硅柱通过快速色谱(Teledyne ISCO CombiFlash)纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:113mg(85%产率)。1H NMR(300MHz,CDCl3)δ8.41(s,1H),8.33-8.19(m,3H),7.95(d,J=8.2Hz,1H),7.84(ddd,J=8.5,6.9,1.5Hz,1H),7.68(ddd,J=8.1,6.9,1.1Hz,1H),7.40-7.31(m,3H),5.70(d,J=17.3Hz,1H),5.39(d,J=17.3Hz,1H),5.30(s,5H),4.15(dddd,J=13.1,11.3,10.7,6.6Hz,4H),2.36-2.07(m,3H),1.81-1.66(m,4H),1.48-1.41(m,3H),1.00(t,J=7.5Hz,3H)。图5为CPT-cc-LG的1H NMR谱图,证明了制备该化合物成功。
实施例6:CPT-cc-DOX的制备
将实施例5制备的CPT-cc-LG(41mg,0.062mmol)、DOX.HCl(38mg,0.066mmol)、TEA(63mg,0.62mmol)在3mL DMF中混合,并在氮气下搅拌1天。通过加入过量的乙酸淬灭反应,并通过制备型HPLC使用乙腈和0.1%三氟乙酸的水溶液(梯度:20-95%的乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:32mg(48%产率)。1H NMR(300MHz,CDCl3)δ8.38(s,1H),8.20(d,J=8.3Hz,1H),8.04(d,J=7.1Hz,1H),7.93(d,J=8.2Hz,1H),7.87-7.74(m,3H),7.66(ddd,J=8.1,5.6,1.1Hz,1H),7.39(d,J=8.0Hz,1H),7.33(d,J=2.1Hz,1H,5.69(d,J=17.2Hz,1H),5.51(d,J=2.8Hz,1H),5.42(d,J=2.9Hz,1H)(d,J=8.0Hz,3H),5.02(d,J=8.0Hz,1H),4.77(s,2H),4.60(s,1H),4.19-4.03J=6.2Hz,3H),3.85(dd,J=26.0,19.7Hz,4H),3.66(s,1H),3.29(d,J=17.6Hz,1H),3.06(t,J=14.8Hz,1H),2.40-2.06(m,6H),1.88-1.77(m,2H),1.75-1.42(m,13H),1.26(ddd,J=12.2,8.4Hz,12H),0.99(t,J=7.5Hz,4H)。图6为CPT-cc-DOX的1H NMR谱图,证明了制备该化合物成功。
实施例7:用两亲性高分子聚乙二醇44-b-聚乳酸28(PEG44-b-PLA28)包裹前药
取实施例3制备的前药CPT-ss-DOX或者实施例6制备的前药CPT-cc-DOX和聚乙二醇-b-聚乳酸以一定比例一起溶于四氢呋喃,然后将混合溶液滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。包裹的结果如下表1显示:
表1.纳米颗粒包裹实例
从表1可以看出,PEG44-b-PLA28对阿霉素和喜树碱均不能形成良好的纳米颗粒,而且不稳定。但是对本发明的前药却能有效包裹,且包裹效率非常高。药物载药量几乎达到50%。
实施例8:药物释放实验
通过透析法从纳米颗粒释放药物。通过透析法测量来自mPEG-b-PLA纳米颗粒的DOX、CPT、CPT-ss-DOX和CPT-cc-DOX的释放。简言之,用磷酸盐缓冲盐水(PBS)稀释在水中的含有DOX、CPT、CPT-ss-DOX或CPT-cc-DOX的纳米颗粒,对于CPT-ss-DOX或CPT-cc-DOX的浓度为10μg/mL DOX和5μg/mL的DOX或CPT。然后,将0.8mL的每种制剂转移到预浸渍的透析盒(MWCO:50kDa)中,并分别用25mL,用或不用10mM谷胱甘肽(GSH)透析72小时。对于DOX、DOX-ss-CPT和DOX-cc-CPT,从透析盒中取出0.5mL溶液,并在预定的时间从透析液测量它们在490nm的UV吸收,然后将其放回透析盒。对于装载CPT的纳米颗粒,通过HPLC(条件:30%乙腈,含0.1%TFA,1mL/min,UV检测器,250nm)监测CPT浓度的死亡。每个实验重复3次。
结果发现(图7),单独被包裹的喜树碱和阿霉素从mPEG-b-PLA纳米颗粒的释放极快,半衰期仅为2小时左右。快速释放有可能导致大量药物在血液中释放,从而副作用较强。与之形成鲜明对比,本发明的CPT-ss-DOX和CPT-cc-DOX在不含GSH的PBS中释放非常缓慢,释放半衰期分别为16小时和18小时。但是当GSH存在时,CPT-ss-DOX的半衰期仅为3.7小时,证明本发明制备得到的前药对GSH具有很高的响应性,能够在癌症组织中选择性的一同释放喜树碱和阿霉素。
实施例9喜树碱和阿霉素从载有CPT-ss-DOX的mPEG-b-PLA纳米颗粒释放
用含有10mM GSH的磷酸盐缓冲盐水(PBS)稀释装载CPT-ss-DOX的mPEG-b-PLA纳米颗粒,在20mL小瓶中产生10μg/mL的浓度。将制剂在37℃下在搅拌下温育。然后在0h、1h、3h、5h、8h分别取20μL,并通过HPLC(条件:30%乙腈,含0.1%TFA,1mL/min,UV检测器,250nm)分析。
结果显示(图8)在10mM GSH存在下,喜树碱和阿霉素迅速地从前药中释放出来,证明了通过二硫键这种连接方式是有效的。
实施例10:体外细胞毒性测试
将人结肠癌细胞HCT116接种在96孔板的McCoy's 5a培养基(10%胎牛血清和1%青霉素)中。将细胞在37℃下在含有5%CO2的潮湿气氛中孵育。接种后24小时用新鲜培养基替换培养基。将每种制剂(阿霉素、喜树碱、阿霉素和喜树碱混合物、表1中的DOX-ss-CPT_20%和DOX-cc-CPT_20%)溶解于PBS中并使用细胞培养基稀释。对于每个孔,加入100μL具有不同指定药物浓度的细胞培养基。通过加入100μL培养基产生阴性对照。将细胞孵育48小时,并且在此之后,用含有0.5mg/mL 3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑(MTT)的100μL培养基替换培养基。在细胞与试剂孵育2小时后,小心地除去含有未反应的MTT的培养基。然后,将获得的蓝色晶体溶解在100μL DMSO中,并在BioTek Synergy H4读数器中测量在570nm的波长下吸光度。使用GraphPad Prism 5进行IC50值的计算和统计分析。
如图9显示,喜树碱的IC50为0.22μM,阿霉素为0.66μM。两者对HCT116细胞都具有很好的杀伤力。但两者以1:1混合后,IC50降为0.087μM,细胞毒性大大增强。IC50的协同系数为0.53,证明喜树碱和阿霉素具有很强的协同性。
当前药被PLA包裹以后,CPT-ss-DOX的IC50为0.19μM,CPT-cc-DOX的IC50为4.0μM(图10)。证明前药被PLA包裹以后,仍然具有很强的毒性。二硫键的细胞毒性比非二硫键的高很多,说明二硫键断裂并且释放喜树碱和阿霉素是维持CPT-ss-DOX的药物毒性的关键 所在。
实施例11:体外细胞摄取
将HCT116细胞接种到24孔板中,24小时后,用HCT116细胞孵育DOX、CPT、CPT-ss-DOX和CPT-cc-DOX(10μM)负载的纳米颗粒4小时。然后,使用胰蛋白酶分离细胞,并用Dulbecco'sPBS洗涤三次。使用BD Beckman Coulter流式细胞仪(Brea,CA)分析细胞的阿霉素荧光强度。如图11显示,阿霉素、PLA包裹的阿霉素和PLA包裹的前药Dox-ss-CPT有相似的细胞摄取,且均比Dox-cc-CPT纳米颗粒强很多。很有可能是因为Dox-cc-CPT在纳米颗粒中释放较少,从而导致部分荧光焠灭。
- 还没有人留言评论。精彩留言会获得点赞!