马齿苋中生物碱化合物及其提取分离方法与流程
本发明涉及中药提取、分离领域,尤其涉及从马齿苋药材中提取、分离和鉴别出的一种生物碱化合物及其提取分离方法,特别是马齿苋中生物碱化合物及其提取分离方法。
背景技术:
马齿苋(portulacaoleraceal.),又名长命菜、马苋菜,为马齿苋科植物。马齿苋性喜肥沃土壤,耐旱亦耐涝,生命力强,分布广泛,资源丰富,而以我国东北部的更为常见。马齿苋既可入药,又可食用,是我国卫生部划定的药食同源的野生植物之一。2015版《中华人民共和国药典》中收载马齿苋的干燥地上部分入药,具有清热解毒、凉血止血、止痢等功效,用于热毒血痢、痈肿疔疮、湿疹、丹毒、蛇虫咬伤、便血、痔血、崩漏下血等。
现代药理学研究表明,马齿苋具有降血脂、降血糖、抗炎、抗氧化、抗肿瘤、抗动脉粥样硬化、松弛或兴奋平滑肌及增强免疫力等功效。研究表明马齿苋中所含多种化学成分与其多样的药理作用息息相关,其主要化学成分包括:黄酮类、生物碱类、萜类、香豆素类、有机酸类、挥发油、多糖、氨基酸、各种色素类和矿物质类等。其中生物碱是马齿苋中的一大类活性成分,而酰胺类生物碱又占绝大多数。目前已报道的生物碱类成分有去甲肾上腺素、多巴胺、少量多巴、腺苷、尿嘧啶、腺嘌呤、n,n-二环己基脲、尿囊素、n-反式-阿魏酰基酪胺;还有环二肽生物碱和酰胺类生物碱:马齿苋酰胺a-i、k、l、n-s。
目前从马齿苋中分离出的化学成分大多数是已知的,且结构一种性较低,因此,对马齿苋中新化合物的开发和分离是亟待需要的。
技术实现要素:
针对上述问题,本发明提供从马齿苋中提取的一种生物碱化合物,经研究发现本发明的一种生物碱具有抗炎、神经保护的作用,同时提供一种针对本发明一种生物碱化合物的简便、快速、环保、纯度高的提取分离方法。
为实现本发明的上述目的,本发明提供一种生物碱化合物,分子式为c18h17no4,命名为oleraciamided,化学结构式为。
为实现本发明的上述目的,本发明还提供一种马齿苋中一种生物碱化合物的提取分离方法,具体步骤为。
步骤1、取马齿苋干燥药材,采用水煎煮提取,水提液滤过,合并滤液直接加热浓缩,放凉至室温,得药液备用。
步骤2、将步骤1中药液用乙酸乙酯反复萃取,减压回收乙酸乙酯至浸膏,得到乙酸乙酯萃取物。
步骤3、将步骤2中乙酸乙酯萃取物经硅胶柱层析分离,依次用乙酸乙酯-甲醇梯度洗脱得到若干洗脱部位,经薄层色谱进行检测,显色,合并显色的洗脱部位,将合并后的洗脱部位经减压浓缩至干,得到浓缩物备用。
步骤4、将步骤3中所得物再经预处理的ods柱(octadecylsilyl,十八烷基硅烷键合硅胶填料)层析分离,用甲醇-水梯度洗脱,得到若干洗脱部位,经薄层色谱进行检测,显色,将显色部位减压浓缩至干,得到浓缩物备用。
步骤5、将步骤4中所得浓缩物经预处理的sephadexlh-20(羟丙基葡聚糖凝胶)层析分离,以甲醇-水等度洗脱,经薄层色谱进行检测,显色,将显色的洗脱部位分别减压浓缩至干,得浓缩物备用。
步骤6、将步骤5中所得浓缩物通过hplc(高效液相)分离制备,以乙腈-水为流动相进行等度洗脱,最终得到本发明所述的生物碱。
所述ods和sephadexlh-20凝胶的预处理过程为甲醇浸泡过24小时,上柱,用甲醇洗至滴入水中无混浊,再以初始流动相平衡。
与现有技术相比,本发明的有益效果。
本发明中所述马齿苋一种生物碱化合物的分离和药理活性研究未被现有论文期刊所报道;本发明提供来源于马齿苋的一种生物碱化合物及一种针对本发明新化合物的提取分离方法,依次采用水煎煮提取、乙酸乙酯萃取、硅胶柱层析、ods中压柱、sephadexlh-20及hplc进行分离纯化与制备,成功提取分离出一种新的生物碱化合物,该方法操作步骤仅为六步,操作方法简便及快速,提取分离过程主要采用水提取及乙酸乙酯萃取,工艺方法环保,且经该方法分离得到的化合物纯度较高均大于90%,此外经研究表明以上化合物具有抗炎和神经保护作用,因此本发明一种生物碱化合物及其盐和衍生物可以作为其他化合物合成先导物,以及新药开发和药理活性研究的原料,亦可用于制备抗炎和神经保护的药物。
附图说明
图1为本发明新生物碱化合物oleraciamided的紫外光谱图。
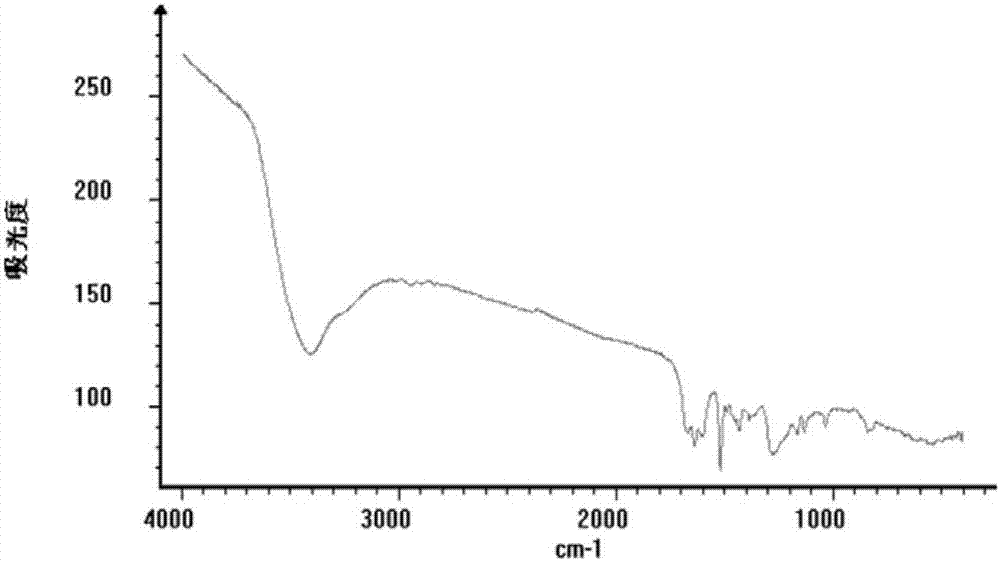
图2为本发明新生物碱化合物oleraciamided的红外光谱图。
图3为本发明新生物碱化合物oleraciamided的1h-nmr光谱图。
图4为本发明新生物碱化合物oleraciamided的13c-nmr光谱图。
图5为本发明新生物碱化合物oleraciamided的核磁共振碳谱(dept)光谱图。
图6为本发明新生物碱化合物oleraciamided的核磁共振1h-1hcosy光谱图。
图7为本发明新生物碱化合物oleraciamided的核磁共振hmbc光谱图。
图8为本发明新生物碱化合物oleraciamided的核磁共振hsqc光谱图。
图9为本发明新生物碱化合物oleraciamided的核磁共振noesy光谱图。
具体实施方式
下面结合具体实施例对本发明做详细的说明。
实施例1。
本发明提供一种生物碱化合物,分子式为c18h17no4,命名为oleraciamided,化学结构式为。
所述一种生物碱化合物根据结构命名为oleraciamided,表1为该一种生物碱化合物的核磁数据:1h-nmr与13c-nmr在meod中。
表1:本发明新生物碱化合物oleraciamided的核磁数据。
本发明一种生物碱化合物oleraciamided的结构鉴定与推导。
oleraciamided:黄色粉末状物,易溶于甲醇,不溶、微溶于水。点样于硅胶薄层板后,喷稀碘化铋钾试液斑点显橘黄色,提示该化合物为生物碱成分。uv(meoh)λmax:320nm,irνn-h3415,νc=o1670,νc=c1634,1594,νc-n1515,νc-o1428,
1h-nmr谱显示一个abx系统,对应信号δ6.84(d,1h,j=1.85),δ6.70(d,1h,j=8.20)以及δ6.89(dd,1h,j=1.9,8.20).同时,1h-nmr信号δ6.73(d,2h,j=8.6),δ7.09(d,2h,j=8.5)以及3c-nmr谱信号δc129.10(c-2”,c-6”,重叠),δc=116.85(c-3”,c-5”,重叠)显示一个aa’bb’系统的存在。依据hmbc谱的相关性,h-3/4与c-2,c-5相关,h-5与c-2,c-6相关,h-6与c-2,c-5相关,并结合一个由δc175.07酰胺特征峰和红外特征吸收峰共同确定的酰胺基团,进一步确定一个5,6-二氢吡啶-2(1h)-酮的存在。由hmbc谱的相关峰,h-5与c-1”,c-2”,c-6”相关,h-6与c-1”相关,h-2”与c-5相关,h-6”与c-5相关,说明c-1”与c-5相连。同时,由h-3/4与c-2’,c-6’相关,h-2’与c-3相关,h-6’与c-3相关可知c-1’与c-3或c-4相连。考虑到δh7.41(d,1h,j=2.05)的耦合常数,可知此氢不会产生邻位耦合作用,因此确定c-1’与c-4相连。此外,考虑到h-och3(s,3.54,3h)与c-3’在hmbc相关性,并结合noesy谱上h-2’与h-och3的相关峰,说明甲氧基与c-3’相连。最后,结合化合物分子式以及处于低场化学位移的c-4’(δc=149.30)和c-4”(δc=157.60)质子,可说明两个羟基的存在,并且它们分别处于c-4’和c-4”位上。
根据以上信息,可确定此新生物碱为上述结构。
本发明还提供上述此一种生物碱化合物的提取分离方法,具体步骤为。
步骤1:称取马齿苋干燥药材150kg,采用水煎煮提取,水用量为药材的8~16倍,煎煮提取两次,每次煎煮2h,水提液滤过,合并滤液,100℃加热浓缩至150l,放凉至室温,得药液备用。
步骤2:将步骤1中所得药液,用乙酸乙酯反复萃取3次,乙酸乙酯与浓缩液的体积比例为1:1(v:v),40℃以下减压回收乙酸乙酯至浸膏,得到乙酸乙酯萃取物。
步骤3:将步骤2中所得乙酸乙酯萃取物干法上样,经硅胶柱层析分离,其中硅胶为200~300目,依次用乙酸乙酯-甲醇(1:1、1:2、1:3、1:5,v:v)梯度洗脱,共得到150个部位(即共得到150个瓶,每瓶400ml),经薄层色谱进行检测,显色,合并显色的90~130洗脱部位(即合并显色的90~130瓶,弃去1~89瓶与131~150瓶),将合并后的90~130部位经40℃以下减压浓缩至干,备用。
步骤4:将步骤3中所得物再经预处理的ods中压柱层析分离,其中填料粒度为20~40μm,用甲醇-水(30/70,50/50,70/30,100/0,v/v)梯度洗脱(加压,使流速为1ml/min,温度为室温),得到10个部位(即梯度洗脱得10个瓶,每瓶200ml),经薄层色谱进行检测,显色,将显色的5~7部位合并,50℃以下减压浓缩至干,备用。所述ods的预处理过程为甲醇浸泡过24h,上柱,用甲醇洗至滴入水中无混浊,再以初始流动相平衡。
步骤5:将步骤4中所得显色部位经预处理的sephadexlh-20柱层析,以甲醇-水(50/50,v/v)等度洗脱,得到35个部位(即梯度洗脱得35个瓶,每瓶20ml),经薄层色谱进行检测,显色,将显色的5~10部位合并,50℃以下减压浓缩至干,备用。所述sephadexlh-20凝胶的预处理过程为甲醇浸泡过24h,上柱,用甲醇洗至滴入水中无混浊,再以初始流动相平衡。
步骤6:将步骤5中所得显色部位经hplc分离制备,以乙腈和水体积比为16:84作为流动相,检测波长为230,280nm,分离制备得到本发明新生物碱化合物,归一法测定纯度均为90~99%。
本发明新生物碱化合物的抗炎作用。
1、主要材料。
1.1、药品和试剂:实验所用新生物碱化合物由上述方法制备,纯度为90~99%,精密称取,用dmso稀释至下述各剂量组所需溶液。dmem高糖培养基、胎牛血清(美国hyclone公司);青霉素、链霉素(杭州四季青公司);lps(美国sigma公司);il-6、tnf-α、pge2的elisa试剂盒(美国cayman公司);细胞裂解液、griess试剂(碧云天生物技术有限公司)。
1.2细胞株:raw264.7巨噬细胞(美国atcc细胞库)
1.3分组:对照组、lps组和实验组,各一组。
2实验方法。
2.1细胞培养,dmem高糖培养基,加入10%的胎牛血清,l%抗菌素(100u/ml青霉素和100μg/ml链霉素),置于37.5%,co2培养箱中培养。
2.2mtt比色法测定细胞活力,上述三组分别取对数生长期raw264.7巨噬细胞接种于96孔培养板中,细胞密度为1×104个/ml,每孔100μl,温度37℃,5%co2条件下培养过夜后,实验组加入不同浓度的本发明新生物碱化合物oleraciamided(1-100μm),孵育1h后向lps组和实验组分别加入终浓度为1μg/ml的lps,另设调零组(含dmso溶媒的培养液),每组设3个复孔,考察加入药物后对细胞的影响。上述各组细胞培养24h后,在各孔细胞中加入5mg/mlmtt20μl,温度37℃,5%co2条件下继续孵育4h后,终止培养,吸弃孔内液体,每孔加入100μl二甲基亚砜(dmso),振荡10min,使细胞内结晶充分溶解,酶标仪570nm波长处测定各孔吸光值。
2.3利用格里斯(griess)法测定no的含量,考察本发明生物碱化合物对lps诱导的小鼠巨噬细胞raw264.7的no产生量的抑制作用。小鼠巨噬细胞raw264.7传代后在含10%胎牛血清的高糖细胞培养基dmem中培养,实验组加入不同浓度的本发明新生物碱化合物oleraciamided(1-50μm),在37℃,5%co2条件下孵育1h后用lps(终浓度为1μg/ml)诱导炎症反应,24h后收集上清液,每组处理重复3孔。griess法测定细胞上清液中no的含量,根据不同浓度本发明新生物碱化合物对lps诱导的raw264.7细胞释放no的影响,用以反映no水平。
2.4elisa法测定炎症因子il-6、tnf-α和炎症介质pge2:将对数生长期raw264.7巨噬细胞接种于24孔培养板中,细胞密度为1×105个/ml,每孔1ml,温度37℃,5%co2条件下培养过夜,实验组加入本发明新生物碱化合物oleraciamided(1-50μm),培育1h后,在每孔加入lps(终浓度为1μg/ml),共孵育24h,每组处理重复3孔。elisa法测定马齿苋来源新生物碱处理后的raw264.7巨噬细胞分泌的il-6、tnf-α和pge2的含量。
3实验结果。
实验结果表明本发明新生物碱化合物对lps诱导的巨噬细胞raw264.7的增殖无影响,安全无毒;并可有效抑制lps诱导的巨噬细胞raw264.7所产生过量炎症细胞因子il-6、tnf-α和炎症介质no、pge2,且呈浓度依赖。
细胞相对存活率实验结果如表2所示。
表2:本发明对raw264.7巨噬细胞相对存活率的影响。
注:*p<0.05与对照组比较(高浓度组有显著性差异)。
利用格里斯(griess)法测定no的含量实验结果见表3。
表3:本发明对lps诱导的raw264.7细胞释放no的影响(均数±标准差,n=3)。
注:*p<0.05与对照组比较,#p<0.05与lps组比较。
elisa法测定炎症因子il-6、tnf-α和炎症介质pge2结果如表4所示。
表4:本发明对lps诱导的raw264.7细胞分泌的il-6、tnf-α和pge2含量的影响(均数±标准差,n=3)。
注:*p<0.05与对照组比较,#p<0.05与lps组比较。
本发明新生物碱化合物的神经保护作用。
1主要材料。
1.1药品和试剂:实验所用新生物碱化合物由上述方法制备,纯度为90~99%,精密称取,用dmso稀释至下述各剂量组所需溶液。dmem高糖培养基、胎牛血清(美国hyclone公司);青霉素、链霉素(杭州四季青公司),磷酸盐缓冲液(pbs),(武汉博士德有限公司),ros检测试剂盒(海门碧云天试剂公司)
1.2细胞株:人神经母细胞瘤细胞株(sh-sy5y、imr-32)(中科院上海细胞
1.3分组:分为对照组、h2o2损伤模型组和实验组。
2实验方法。
2.1细胞培养,dmem高糖培养基,加入l0%的胎牛血清,l%抗菌素(100u/ml青霉素和100μg/ml链霉素),置于37℃、5%co2培养箱中培养。
2.2mtt比色法测定细胞活力,上述三组分别取对数生长期sh-sy5y细胞和imr-32细胞接种于96孔培养板中,细胞密度为1×104个/ml,每孔100μl,温度37℃,5%co2条件下培养过夜后,实验组加入不同浓度的本发明新生物碱化合物oleraciamided(5-40μm),孵育1h后向h2o2组和实验组分别加入终浓度为800μm/l的h2o2,另设调零组(含dmso溶媒的培养液),每组设3个复孔,考察加入药物后对细胞的影响。上述各组细胞培养24h后,在各孔细胞中加入5mg/mlmtt20μl,温度37℃,5%co2条件下继续孵育4h后,终止培养,吸弃孔内液体,每孔加入100μl二甲基亚砜(dmso),振荡10min,使细胞内结晶充分溶解,酶标仪450nm波长处测定各孔吸光值(a)值,计算细胞存活率,细胞存活率=(ah2o2损伤-a空白)/(a对照-a空白)。
2.3dcfh-da法检测sh-sy5y细胞和imr-32细胞内ros,各组细胞给予相应物质后孵育24h,孵育结束前30min,各孔加入dcfh-da,使终浓度为10μmol/l,于37℃继续孵育30min,收集细胞,pbs洗2次,细胞计数,将各组细胞制成相同浓度的细胞悬液。取100μl细胞悬液检测荧光强度,激发波长485nm,发射波长538nm。以对照组荧光强度为100%,其余各组与对照组荧光强度相比较,计算胞内ros变化。
2.4int显色反应法测定ldh的释放量,除上述对照组、h2o2损伤模型组和实验组外,另设立空白对照组(空白对照组不接种细胞),各组细胞加入相应物质培养24h,取各孔上清120μl至新的96孔板中,加60μl配好的ldh检测工作液,避光室温孵育30min,在490nm处用多功能酶标仪测定a值,计算相对于对照管的ldh释放量百分率。ldh释放率=(a给药-a空白)/(a对照-a空白)。
3实验结果。
细胞相对存活率实验结果如表5所示。
表5:本发明对人神经母细胞瘤细胞株sh-sy5y和imr-32细胞相对存活率的影响。
注:*p<0.05与h2o2损伤模型组比较。
sh-sy5y细胞和imr-32细胞内ros量检测结果如表6所示。
表6:本发明对人神经母细胞瘤细胞株sh-sy5y和imr-32细胞内ros量的影响。
注:*p<0.05与对照组比较,#p<0.05与h2o2损伤模型组比较。
sh-sy5y细胞和imr-32细胞内ldh释放的影响结果如表7所示。
表7:本发明对人神经母细胞瘤细胞株sh-sy5y和imr-32细胞内ldh释放的影响。
注:*p<0.05与对照组比较,#p<0.05与h2o2损伤模型组比较。
综上所述,本发明提供一种生物碱化合物及其提取分离方法,依次采用水煎煮提取、乙酸乙酯萃取、硅胶柱层析、ods中压柱层析、及sephadexlh-20柱层析,hplc分离制备,成功的分离得到新生物碱化合物,该方法简便,快速,环保,且经该方法分离得到的化合物纯度较高,由于所得化合物化学结构独特,从常用中药马齿苋中提取出来,其具有抗炎和神经保护作用,因此本发明新生物碱及其盐和衍生物可以作为天然产物开发中药新药,具有广阔的前景。
- 还没有人留言评论。精彩留言会获得点赞!