一种基于咔唑的碳硼烷衍生物材料及其制备方法与应用与流程
本发明涉及一种基于咔唑的碳硼烷衍生物材料及其制备方法与应用,属于光电材料技术领域。
背景技术:
有机分子具有结构多样性和易裁剪性,可以结合分子设计和有机合成,得到具有某些特定性能的新型化合物。由于有机分子可设计性好,易于功能化,近些年来,随着分子工程和有机合成的发展,有机分子的电致化学发光(ecl)材料的研究得到了广泛发展,它们在环境污染物检测、生物检测等方面发挥了重大的作用。
电致化学发光是一种独特类型的发光,其中电生成的离子自由基之间的电子转移在电极附近产生电子激发产物,从而发射光。电致化学发光和电化学是常用且灵敏的工具,可用于获得关于自由基离子形成和稳定性以及分子内微妙相互作用的有价值的信息。这种信息在有机发光器件(oled)的设计中是有用的,其中电荷产生,迁移和重组通过活性有机层中的类似带电状态来确定。电致化学发光的另一种用途涉及开发用于标记新的ecl发射体在不同波长的生物分析应用,即找到不同于目前使用的物种波长的ecl发射体。因此,ecl是一种重要的光电化学技术,在有机和无机材料的基础分析方面以及生物分析方面具有多种应用。
对于作为ecl发射体,以吡啶(bpy)类染料应用最为广泛。以吡啶为核心,通过引入其它芳烃链提高π共轭性能,进而提高分子间电荷转移的能力。其中吡啶提供稳定的激发态所需的电子自由基,从而增强ecl强度。然而,吡啶类染料的合成方法复杂、产率低,限制了其在ecl领域更深入的发展应用。由于碳硼烷类化合物中包含稳定的碳硼烷单元,所以它们同样可以稳定地提供因电离产生的激发态电子自由基。而碳硼烷化合物大多都可以通过相对简单的合成方法获得,而且对碳硼烷进行结构上的修饰也非常容易实现。但是,在有机或者无机溶液中,ecl发射体脱去一个质子后,很难发生氧化还原反应,为了使电化学过程顺利循环进行,通常需要将三丙胺(tpra)与ecl发射体共同涂在电极的阳极上。其中三丙胺在这个过程中作为共反应剂,电极表面的tpra通过释放电子发生氧化反应而成为阳离子激发态的tpra+,并迅速自发脱去一个质子而形成激发态三丙胺,这样,在反应体系中就存在具有强氧化性的ecl发射体阳离子和具有强还原性的激发态三丙胺。当具有强氧化性的ecl发射体阳离子和具有强还原性的激发态三丙胺发生氧化还原反应,激发态的ecl发射体通过荧光衰减机制从而产生荧光。由于上述反应体系中仍存在稳定的ecl发射体和三丙胺,使得电极表面的电化学反应和化学发光过程可以继续进行,这样,整个反应过程可以循环进行。通过上述的循环过程,测定信号不断的放大,从而使检测灵敏度大大提高。尽管含硼的有机化合物作为ecl发射体已经有报道;但是,含有碳硼烷基团的材料在ecl上的应用,还未见报道。
技术实现要素:
技术问题:本发明的目的是提供一种基于咔唑的碳硼烷衍生物材料及其制备与应用方法。该发明首次通过合成方法的改良提高了芳烃碳硼烷衍生物的产率,并且将含碳硼烷单元的材料用于ecl发射体,通过引入碳硼烷基团增加电化学过程中的稳定性。提供了一种基于高强度,高稳定性的电致化学发光发射体,且易于制备的咔唑的碳硼烷衍生物的制备方法,解决了目前ecl发射体材料制备复杂的问题。
技术方案:本发明提供一种基于咔唑的碳硼烷衍生物材料及其制备方法与应用。本发明的一种基于咔唑的碳硼烷衍生物材料是含有咔唑结构体系,通过碳硼烷骨架与不同位点苯基单元链接;该类材料具有以下的化合物i或ii式结构:
其中:化合物i是在咔唑中氮原子9-位上的苯基咔唑的4-位与碳硼烷连接;化合物ii是在苯基咔唑的苯环的3-位与碳硼烷连接;c为碳,n为氮。
其中,
所述化合物i和化合物ii的材料均含有一个咔唑单元和一个碳硼烷单元。
所述苯基咔唑上苯环的3-位或者4-位与碳硼烷上的碳原子通过碳碳键链接。
本发明的基于咔唑的碳硼烷衍生物材料的制备方法中,所述化合物i制备方法包括以下步骤:
步骤一:9-(4-乙炔基苯)-9-咔唑的制备:氮气保护条件下,将9-(4-溴苯)-9-咔唑,乙炔基苯,四(三苯甲基)膦钯和将碘化亚铜加入干燥的反应瓶中,在氮气和避光氛围下,然后向混合物中注入三乙胺溶液,在100℃~120℃回流10~14小时,终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取,剩余的有机层用无水硫酸镁干燥并过滤;将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化,最终得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末;
步骤二:化合物i的制备:氮气氛围中,向的反应瓶中加入干燥的蒸馏甲苯以溶解十硼烷和氮,氮-二甲基苯胺,在35℃~45℃搅拌,然后将温度升至100℃~120℃保持,然后冷却至35℃~45℃后,注入用蒸馏甲苯溶剂溶解的化合物9-(4-乙炔基苯)-9-咔唑,将反应混合物在100℃~120℃条件下回流10~12小时,终止反应后,将混合物冷却至室温并用甲醇淬灭,使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤,溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化,得到黄色粉末状的化合物i。
其中,
所述乙炔基苯与9-(4-溴苯)-9-咔唑摩尔比为1:1~1.2:1;所述四(三苯甲基)膦钯与9-(4-溴苯)-9-咔唑摩尔比为0.05:1~0.1:1,所述碘化亚铜与9-(4-溴苯)-9-咔唑摩尔比为0.1:1~0.2:1;每毫摩尔9-(4-溴苯)-9-咔唑加入三乙胺溶剂10~15ml。
所述步骤二中,所述十硼烷与9-(4-乙炔基苯)-9-咔唑摩尔比为1:1~1.1:1,氮,氮-二甲基苯胺与9-(4-乙炔基苯)-9-咔唑摩尔比为1.5:1~1.7:1,每毫摩尔9-(4-乙炔基苯)-9-咔唑加入蒸馏甲苯溶剂10~15ml。
本发明的基于咔唑的碳硼烷衍生物材料的制备方法中,所述化合物ii的制备方法包括以下步骤:
步骤一:9-(3-乙炔基苯)-9-咔唑的制备:氮气保护条件下,将9-(3-溴苯)-9-咔唑,乙炔基苯,四(三苯甲基)膦钯和将碘化亚铜加入干燥的反应瓶中,在氮气和避光氛围下,然后向混合物中注入三乙胺溶液,在100℃~120℃回流10~14小时,终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取,剩余的有机层用无水硫酸镁干燥并过滤;将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化,最终得到9-(3-乙炔基苯)-9-咔唑,为微黄色粉末;
步骤二:化合物ii的制备:氮气氛围中,向的反应瓶中加入干燥的蒸馏甲苯以溶解十硼烷和氮,氮-二甲基苯胺,在35℃~45℃搅拌,然后将温度升至100℃~120℃保持,然后冷却至35℃~45℃后,注入用蒸馏甲苯溶剂溶解的化合物9-(3-乙炔基苯)-9-咔唑,将反应混合物在100℃~120℃条件下回流10~12小时,终止反应后,将混合物冷却至室温并用甲醇淬灭,使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤,溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化,得到黄色粉末状的化合物ii。
其中,
所述乙炔基苯与9-(3-溴苯)-9-咔唑摩尔比为1:1~1.2:1;所述四(三苯甲基)膦钯与9-(3-溴苯)-9-咔唑摩尔比为0.05:1~0.1:1,所述碘化亚铜与9-(3-溴苯)-9-咔唑摩尔比为0.1:1~0.2:1;每毫摩尔9-(3-溴苯)-9-咔唑加入三乙胺溶剂10~15ml。
所述步骤二中,所述十硼烷与9-(3-乙炔基苯)-9-咔唑摩尔比为1:1~1.1:1,氮,氮-二甲基苯胺与9-(3-乙炔基苯)-9-咔唑摩尔比为1.5:1~1.7:1,每毫摩尔9-(3-乙炔基苯)-9-咔唑加入蒸馏甲苯溶剂10~15ml。
本发明基于咔唑的碳硼烷衍生物材料应用在传感器、电致发光器件、有机太阳能电池或有机场效应晶体管方面,或作为电致变色材料、光致变色材料、生物传感材料、空穴传输材料、非线性光学材料、防伪材料或伪装材料应用于有机光电领域。
有益效果:本发明提供一种基于咔唑的碳硼烷衍生物材料,该类化合物制备方法简单,操作方便快捷,反应过程容易控制,产品易于分离、收率高、纯度高,特别是其具有很高的电致化学发光强度。其在传感器、电致发光器件、有机太阳能电池、有机场效应晶体管等方面具有潜在的应用价值,并可作为电致变色材料、光致变色材料、空穴传输材料、非线性光学材料、防伪材料、伪装材料等应用于有机光电领域。
附图说明
图1为材料i连续的ecl光谱图,插图是对应最高强度时,电致化学发光的发射峰。
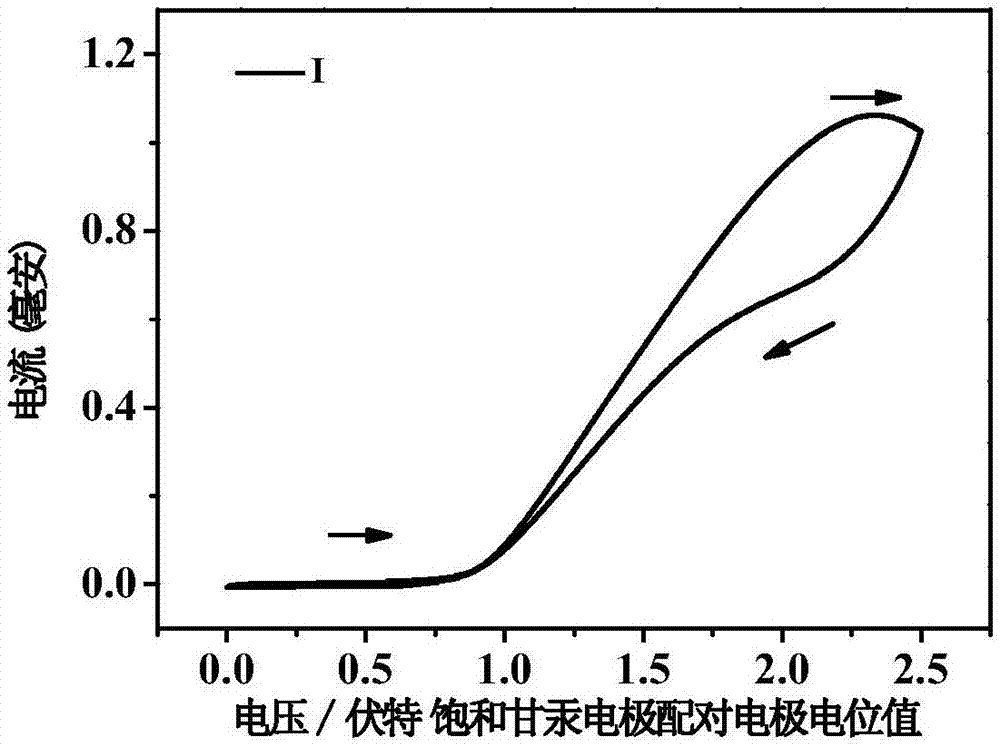
图2为材料i在特定条件下光电倍增管(pmt)获得的循环伏安图,测试范围为0v~2.5v。
图3为材料ii连续的ecl光谱图,插图是对应最高强度时,电致化学发光的发射峰。
图4为材料ii在特性测试条件下光电倍增管(pmt)获得的循环伏安图,测试范围为-1.8v-2.5v。
测试条件:材料i和材料ii均在浓度为0.1毫摩尔的二氯甲烷溶解液中,且在阳电极上涂上20毫摩尔的三丙胺和0.1摩尔的四丁基六氟磷酸铵,以20毫伏每秒的扫描速率进行电致化学发光图谱扫描。
具体实施方式
制备方法包括:9-(4-溴苯)-9-咔唑、9-(3-溴苯基)-9-咔唑通过薗头偶合反应炔基化得到9-(4-乙炔基苯)-9-咔唑、9-(3-乙炔基苯)-9-咔唑,然后与碳硼烷反应合成化合物i、ii。该类碳硼烷衍生物在电致化学发光上有很好的应用,可作为小分子生物荧光探针,该类碳硼烷衍生物具有高的电致化学发光强度与良好的稳定性。
该类咔唑的碳硼烷衍生物的材料的制备方法,包括以下步骤:
步骤一:9-(4-乙炔基苯)-9-咔唑、9-(3-乙炔基苯)-9-咔唑的制备,以9-(4-乙炔基苯)-9-咔唑的制备为例:在氮气氛围和避光条件下,将1毫摩尔的9-(4-溴苯)-9-咔唑、0.05-0.1毫摩尔的四(三苯甲基)膦钯的催化剂、0.1~0.2毫摩尔碘化亚铜以及1.0~1.2毫摩尔的乙炔基苯溶解在10~15ml的三乙胺中,在110℃回流10-14小时。终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取。剩余的有机层用无水硫酸镁干燥并过滤。将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化,最终得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末;
步骤二:化合物i、ii的制备,以化合物i的制备为例:氮气保护条件下,1.0~1.2毫摩尔l的十硼烷和1.5~1.7毫摩尔的氮,氮-二甲基苯胺溶解于5ml的干燥蒸馏甲苯溶剂中,在35℃~45℃搅拌30分钟。然后将温度升至100℃~120℃保持2小时,然后冷却至35℃~45℃后,注入用5~10ml蒸馏甲苯溶剂溶解的化合物9-(4-乙炔基苯)-9-咔唑,将反应混合物在100℃~120℃条件下回流10-12小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物i,用甲醇重结晶得到更为纯净的化合物i。
其中,i是在四(三苯甲基)膦钯、碘化亚铜以及乙炔基苯作用下炔基化反应;ii是在十硼烷和的氮,氮-二甲基苯胺作用下化合物i、化合物ii制备反应。
实施例1:
反应条件一:氮气和避光保护条件下,将9-(4-溴苯)-9-咔唑(322.6毫克,1.0毫摩尔)、的四(三苯甲基)膦钯(57毫克,0.05毫摩尔)、碘化亚铜(19毫克,0.1毫摩尔)以及乙炔基苯(112毫克,1.0毫摩尔)溶解在10ml的三乙胺(et3n)中,在110℃回流12小时。终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取。剩余的有机层用无水硫酸镁干燥并过滤。将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化。最终得到得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末(265毫克),产率为61%
反应条件二:氮气和避光保护条件下,将9-(4-溴苯)-9-咔唑(322.6毫克,1.0毫摩尔)、的四(三苯甲基)膦钯(104毫克,0.01毫摩尔)、碘化亚铜(19毫克,0.1毫摩尔)以及乙炔基苯(112毫克,1.0毫摩尔)溶解在10ml的三乙胺中,在110℃回流12小时。终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取。剩余的有机层用无水硫酸镁干燥并过滤。将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化。最终得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末(304毫克),产率为70%。
反应条件三:氮气和避光保护条件下,将9-(4-溴苯)-9-咔唑(322.6毫克,1.0毫摩尔)、的四(三苯甲基)膦钯(57毫克,0.01毫摩尔)、碘化亚铜(19毫克,0.1毫摩尔)以及乙炔基苯(123毫克,1.1毫摩尔)溶解在10ml的三乙胺中,在110℃回流12小时。终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取。剩余的有机层用无水硫酸镁干燥并过滤。将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化。最终得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末(325毫克),产率72.9%。
反应条件四:氮气保护条件下,将9-(4-溴苯)-9-咔唑(322.6毫克,1.0毫摩尔)、的四(三苯甲基)膦钯(57毫克,0.01毫摩尔)、碘化亚铜(19毫克,0.1毫摩尔)以及乙炔基苯(134.4毫克,1.2毫摩尔)溶解在15ml的三乙胺中,在110℃回流12小时。终止反应冷却至室温后,反应混合物加入水中并用二氯甲烷萃取。剩余的有机层用无水硫酸镁干燥并过滤。将溶剂旋转蒸发干燥,固体通过硅胶色谱柱纯化。最终得到9-(4-乙炔基苯)-9-咔唑,为微黄色粉末(297毫克),产率65%。
实施例2
反应条件一:氮气保护条件下,将十硼烷(121.3毫克,1.0毫摩尔)和氮,氮-二甲基苯胺(181.4毫克,1.5毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在35℃搅拌30分钟。然后注入10ml蒸馏甲苯溶剂溶解的化合物9-(4-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在100℃条件下回流10小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物i,用甲醇重结晶得到107毫克更为纯净的化合物i,产率为23%。
反应条件二:氮气保护条件下,将十硼烷(133.4毫克,1.1毫摩尔)和氮,氮-二甲基苯胺(181.4毫克,1.5毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在40℃搅拌30分钟。然后将温度升至110℃保持2小时,冷却至40℃后,注入10ml蒸馏甲苯溶剂溶解的化合物9-(4-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在110℃条件下回流12小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物i,用甲醇重结晶得到358.8毫克更为纯净的化合物i,产率为75%。
反应条件三:氮气保护条件下,将十硼烷(145.5毫克,1.2毫摩尔)和氮,氮-二甲基苯胺(205.4毫克,1.7毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在45℃搅拌30分钟。然后将温度升至120℃保持2小时,冷却至40℃后,注入10ml蒸馏甲苯溶剂溶解的化合物9-(4-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在120℃条件下回流14小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物i,用甲醇重结晶得到343毫克更为纯净的化合物i,产率为70%。
1hnmr(400mhz,cdcl3):δ8.10(d,j=7.6hz,2h),7.64(d,j=8.8hz,2h),7.50(d,j=7.4hz,2h),7.35(ddd,j=25.7,10.1,4.1hz,7h),7.22(t,j=6.9hz,4h);13cnmr(101mhz,cdcl3)δ140.15,139.53,132.20,130.70,130.57,130.34,129.33,128.39,126.27,126.08,123.66,120.46,120.43,109.47,85.39,84.35;11bnmr(128mhz,cdcl3)δ-2.45,-10.52.
实施例3
反应条件一:氮气保护条件下,将十硼烷(121.3毫克,1.0毫摩尔)和氮,氮-二甲基苯胺(181.4毫克,1.5毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在35℃搅拌30分钟。然后注入10ml蒸馏甲苯溶剂溶解的化合物9-(3-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在100℃条件下回流10小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物ii,用甲醇重结晶得到88.6毫克更为纯净的化合物ii,产率为19%。
反应条件二:氮气保护条件下,将十硼烷(133.4毫克,1.1毫摩尔)和氮,氮-二甲基苯胺(181.4毫克,1.5毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在40℃搅拌30分钟。然后将温度升至110℃保持2小时,冷却至40℃后,注入10ml蒸馏甲苯溶剂溶解的化合物9-(3-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在110℃条件下回流12小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物ii,用甲醇重结晶得到330.1毫克更为纯净的化合物ii,产率为69%。
反应条件三:氮气保护条件下,将十硼烷(145.5毫克,1.2毫摩尔)和氮,氮-二甲基苯胺(205.4毫克,1.7毫摩尔)溶解于5ml的干燥蒸馏甲苯溶剂中,在45℃搅拌30分钟。然后将温度升至110℃保持2小时,冷却至40℃后,注入10ml蒸馏甲苯溶剂溶解的化合物9-(3-乙炔基苯)-9-咔唑(345毫克,1.0毫摩尔),将反应混合物在120℃条件下回流14小时。终止反应后,将混合物冷却至室温并用甲醇淬灭。使用旋转蒸发除去溶剂,将残余物加入水中,有机层用二氯甲烷萃取,用无水硫酸镁干燥并过滤。溶剂用旋转蒸发干燥,固体用硅胶柱色谱纯化。得到黄色粉末状的化合物ii,用甲醇重结晶得到299.2毫克更为纯净的化合物ii,产率为61%。
1hnmr(400mhz,cdcl3):δ8.10(d,j=7.6hz,2h),7.65–7.62(m,2h),7.52–7.48(m,2h),7.40–7.28(m,7h),7.24–7.20(m,4h);13cnmr(101mhz,cdcl3)δ140.49,137.77,132.67,130.70,130.41,130.11,129.94,129.22,128.88,128.57,126.08,123.44,120.43,120.32,109.27,85.18,84.10;11bnmr(128mhz,cdcl3)δ-2.39,-10.41.
以上是本发明的实施例,需要说明的是本发明不限于这些实例,这些实例仅为了更好的理解本发明,依据本发明的技术方案所作的任何等效变换,均属于本发明保护范围。
- 还没有人留言评论。精彩留言会获得点赞!