类美登素衍生物及其制备方法和用途与流程
本发明涉及一种类美登素衍生物及其制备方法和用途。
背景技术:
类美登素是抑制微管蛋白聚合高度细胞毒化合物(Remillard等,Science189,1002-1005(1975))。美登素最早由Kupchan等从东非灌木Maytenusserrata分离得到(J.Am.Chem.Soc.94:1354-1356(1972))。类美登素,包括美登醇及美登醇C-3酯也可由某些微生物产生(U.S.Pat.No.4,151,042)。具有不同细胞毒性的美登醇的各种类似物也可由合成化学制备(forreviewseeChem.Pharm.Bull.52(1)1-26(2004))。类美登素的实例包括美登素﹑DM1﹑DM3及DM4。美登素为一种强有力的有丝分裂抑制剂并且在实体鼠源肿瘤模型中对多种肿瘤,包括路易斯(Lewis)肺癌和B-16黑素瘤展现显著的抑制活性。据报道,美登素在浓度低至10-7μg/mL可以抑制人类急性淋巴细胞白血病C.E.M.系(Wolpert-DeFillippes等,Biochem.Pharmacol.1735-1738(1975))。业已证明,其细胞毒性比传统化疗试剂如甲氨蝶呤(methotrexate)﹑柔红霉素(daunorubicin)和长春新碱(vincristine)高100到1000倍(U.S.Pat.No.3,896,111)。由于其高毒性性质及体外抗多种癌细胞系活性,类美登素有望能治疗多种不同的癌症。然而,其毒性也使得这类化合物在人类临床试验中不是令人满意的,因为其副作用对很多患者是无法忍受的(Issel等,CancerTreat.Rev.199-207(1978))。
技术实现要素:
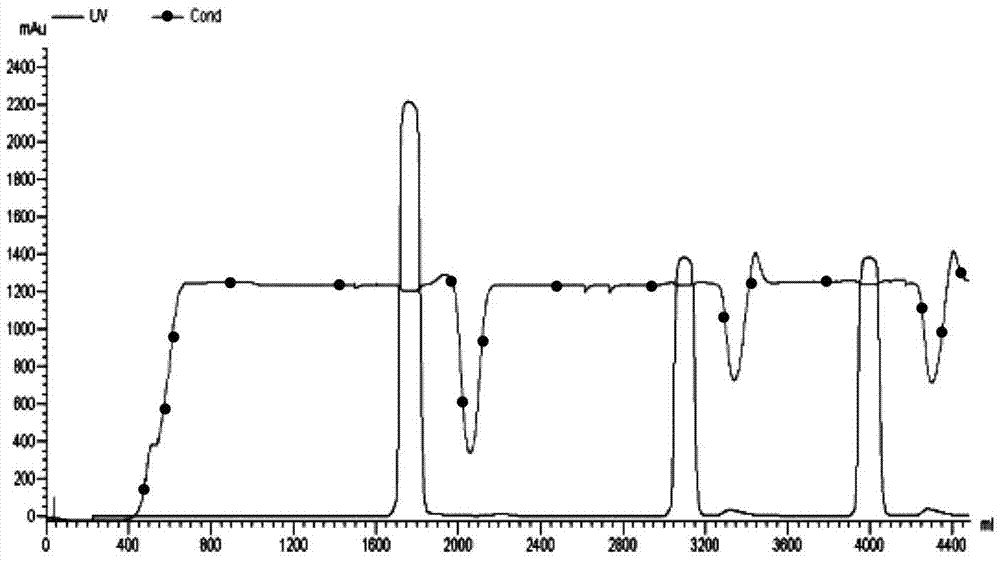
本发明提供了可偶联到抗原结合单元(Abu来自AntigenBindingUnit)的类美登素药物衍生物,以及连有抗原结合单元的类美登素药物(Drug-Linker-AntigenbindingUnit:D-L-Abu)。抗体药物偶联物(ADC)由三个关键元素组成:抗体﹑连接子和药物。特定抗体和药物的选择取决于特定疾病,其对药物的有效性和安全性有重大影响。连接子的稳定性及药物偶联到抗体的方法对ADC药物开发成功或失败起决定性作用。抗体药物偶联物的疗效部分取决于各种参数的组合,不仅包括抗体的特异性和药物的药效强度,而且包括连接子的稳定性或其对断裂的敏感性﹑细胞表面激发内化﹑转运和随后有效细胞毒性“弹头”的释放。因此,即便针对同一靶点,不同的药物,或不同的连接子,或针对同一靶标的不同抗体,ADC的应用性和有效性显著不一样。本发明也提供D-L-Abu、D-L-Abu衍生物以及此类药物偶联物在治疗癌症和免疫失常中抗原阳性细胞的用途。抗原结合单元(Abu)如抗体或D-L-Abu中其它靶向部分能结合至致病细胞或组织的抗原上,偶联至Abu的药物对抗原表达细胞产生细胞毒﹑抑制细胞生长或免疫抑制影响而治疗或阻止抗原阳性癌症或免疫异常的复发。本发明提供了类美登素衍生物化合物,其可通过对酸不敏感﹑对肽酶组织蛋白酶不敏感以及不含二硫键的连接子偶联到抗原结合单元。此类衍生物也被称为“药物-连接子”(“drug-linkers”)。在一些实施方案中,连接子修饰的类美登素化合物为N2’-去乙酰基-N2’-(6-马来酰亚胺基-1-氧代-己基)美登素(N2’-deacetyl-N2’-(6-maleimido-1-oxo-hexyl)maytansine)或其衍生物,其中连接子对酸不敏感﹑对肽酶组织蛋白酶不敏感以及不含二硫键。此类连接子预期可为D-L-Abu在内吞之前提供稳定性,例如在体循环中,阻止D-L-Abu过早降解和释放毒性药物,因而将药物的毒性作用最小化。本发明提供了式I或I-1,II或II-1的类美登素衍生物或其药学上可接受的盐:其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基﹑苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;以及L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为(-CH2-)m,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基。本发明提供了包含上述类美登素化合物以及能被类美登素化合物偶联的抗原结合单元的组合物。本发明提供了连有类美登素化合物的抗原结合单元,其中类美登素化合物通过对酸不敏感﹑对肽酶组织蛋白酶不敏感以及不含二硫键的连接子修饰并连接到抗原结合单元。本发明提供了式Ia或Ia-1,IIa或IIa-1的类美登素连接子抗原结合单元偶联物:或其药学上可接受的盐或溶剂合物,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基﹑苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1﹑2﹑3﹑4﹑5﹑6﹑7﹑8﹑9以及10;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为(-CH2-)m,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及Abu为抗原结合单元。在一些实施方案中,本发明提供了如下式的化合物:或其药学上可接受的盐或溶剂合物。本发明提供了一种包含如上所述类美登素连接子抗原结合单元偶联物的组合物。本发明提供了一种制备如上所述类美登素连接子抗原结合单元偶联物的方法,该方法包括连接抗原结合单元与一个或一个以上本文所述的可偶联到抗原结合单元的类美登素化合物。本发明提供了式Ib或Ib-1,IIb或IIb-1的化合物:或其药学上可接受的盐,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基、苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为(-CH2-)m,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及AA为氨基酸,如半胱氨酸或巯基化的氨基酸如巯基化赖氨酸。在一些实施方案中,本发明提供了如下式的化合物:或其药学上可接受的盐或溶剂合物。本发明涉及一种包含上述式Ib﹑Ib-1﹑IIb或IIb-1的类美登素化合物的组合物。本发明涉及一种将类美登素送达抗原阳性细胞或组织的靶向给药方法,所述美登素与抗原结合单元偶联。本发明涉及一种抗原结合单元为BAT0206,其包含一个氨基酸序列为SEQIDNO:1的抗EGFR轻链和氨基酸序列为SEQIDNO:2的抗EGFR重链。本发明涉及一种美登素与抗原结合单元偶联的连接子,所属抗原结合单元为BAT0206。本发明涉及所述类美登素化合物用于制备治疗一些疾病的药物的用途,这些疾病包括:增生性疾病,如肿瘤,感染或自身免疫疾病,如移植排斥。以及其他可以用靶疗法治疗的疾病,这些疾病的特征为:细胞生成一种抗原或靶点,该抗原能与抗原结合单元特异结合;所述化合物由抗原结合单元与一个或多个美登素的偶联而成。附图说明图1显示了SephadexG25(M)柱分离抗体药物偶联物Batansine-0206的图谱。图2显示了SephadexG25(M)柱分离抗体药物偶联物Batansine-1206的图谱。图3显示了PhenylSepharoseFF柱(GE公司XK26/15)分离抗体药物偶联物Batansine-0206的图谱。图4显示了SephadexG25(M)柱分离抗体药物偶联物Batansine-0606的图谱。图5显示了PhenylSepharoseFF柱(GE公司XK26/15)分离抗体药物偶联物Batansine-0606的图谱。图6显示了前体药物抗体美登素偶联物其代谢产物对微管蛋白聚合的抑制作用。图7显示了Bat0206和Batansine-0206对EGFR阳性乳腺癌细胞系(MDA-MB-468)的生长抑制作用。图8显示了未偶联的抗体Bat0206、cetuximab(西妥昔单抗)、药物偶联的Batansine-0206对EGFR阴性细胞系BT474均无抑制作用。图9显示了药物偶联物D-Lmcc-Bat0206对EGRR阳性乳腺癌细胞系(MDA-MB-468)增殖的抑制作用。图10显示了未偶联的抗体Bat0206、cetuximab(西妥昔单抗)、药物偶联物D-Lmcc-Bat0206对EGFR阴性细胞系BT474均无抑制作用。图11和12显示了D-Lmcc-Bat0206根除A431异种移植动物模型肿瘤的结果。图13显示了D-Lmcc-Bat0206和D-Lspp-Bat0206对A431肿瘤细胞抑制作用。图14显示了D-Lmcc-Bat0206和D-Lspp-Bat0206对A549肿瘤细胞抑制作用。图15A和15B显示了抗EGFR抗体的氨基酸序列。图16和17分别显示了D-Lmcc-bat0606和Batanine-0606在制备过程中,通过SephadexG-25树脂洗脱交换的纯化过程。图18和19分别显示了D-Lmcc-bat0606和Batanine-0606通过尺寸排阻色谱测定聚集率。图20显示了抗Bat0606和Batanine-0606对HER2阳性乳腺癌细胞系(SK-BR-3)的生长抑制作用。与未偶联的抗Bat0606相比,药物偶联的Batanine-0606显示出对HER2阳性细胞增殖显著抑制作用增强。图21显示了未偶联的抗体Bat0606与药物偶联的Batanine-0606,对HER2阴性细胞系A549均无抑制作用。图22显示了药物偶联物D-Lmcc-Bat0606抗体对HER2阳性乳腺癌细胞系(SK-BR-3)增殖的抑制作用。与未偶联的Bat0606相比,D-Lmcc-Bat0606在较低的浓度即显示出对HER2阳性细胞增殖显著的抑制作用。图23显示了未偶联的抗体Bat0606与药物偶联物D-Lmcc-Bat0606,对HER2阴性细胞系A549均无抑制作用。图24显示了抗CD20Bat1206和抗Batansine-1206对CD20阳性淋巴癌细胞Raji的生长抑制作用。图25显示了Bat1206和Batansine-1206对CD20阴性细胞系A431均无抑制作用。图26显示了药物偶联物D-Lmcc-Bat1206对CD20阳性淋巴癌细胞Raji的生长抑制作用。图27显示了用SEC-HPLC检测纯化后的Batansine-1206纯度。图28显示了Bat1206、D-Lmcc-Bat1206非还原的SDS-PAGE图。图29显示了Batansine0606根除NCI-N87异种移植动物模型肿瘤的结果。图30显示了Batansine0606根除BT474异种移植动物模型肿瘤的结果。图31显示了前体药物Batansine-0606的代谢产物Batansine-Cysteine的质谱图。图32显示了前体药物D-Lmcc-Bat0606的代谢产物MDC-MCC-Lysine的两个非对映异构体的质谱图。具体实施方式定义除非另作说明,正如于此使用的那样,单数形式“一(a,an)”和“所述(the)”,包括复数例证。因而,如提到“一个化合物”也包括其复数“一些化合物”。正如于此使用的那样,“大约”为本领域普通技术人员所理解并且依据使用其上下文可在某种程度发生变化。如果有条款不为本领域普通技术人员所清楚,考虑其使用的上下文,“大约”意味着个别条款的加或减10%﹑或加或减5%﹑或加或减1%。正如于此使用的那样,术语“包括”意指组合物和方法包括列举的元素,但未将其他的排除在外。当用来定义组成和方法时,“基本由组成”意指排除对这一组合具有本质意义的其他元素。例如,正如于此定义的那样,基本由某些元素组成的成分并不排除其它对所要求发明的基本和新颖特点没有本质影响的其它元素。“由组成”意味着排除超过所列举的衡量的其它组分和本质意义的方法步骤。每个过渡条款所界定的实施方案中在本发明范围内。。正如于此使用的那样,“类美登素”指的是具美登素类似物,包括其立体异构体。美登素可从美登木属植物分离得到(参见美国专利3896111),其结构式为:类美登素为具有美登素的环结构且其环上具有一个或一个以上取代基修饰的化合物。“烷基”指的是具有1到10个碳原子(优选为1到6个碳原子)的单价饱和脂肪烃基。Cv烷基,其中v为整数表示具有v个碳原子的烷基。这一术语包括,例如,直链和支链的烃基如甲基(CH3-)﹑乙基(CH3CH2-)﹑正丙基(CH3CH2CH2-)﹑异丙基(CH3)2CH-)﹑正丁基(CH3CH2CH2CH2-)﹑异丁基((CH3)2CHCH2-)﹑仲丁基((CH3)(CH3CH2)CH-)﹑叔丁基((CH3)3C-)﹑正戊基(CH3CH2CH2CH2CH2-)和新戊基((CH3)3CCH2-)。“亚烃基”是具有1到10个碳原子(优选为1到6个碳原子)的二价饱和脂肪烃基。“烯基”指的是具有2到6个碳原子(优选为2到4个碳原子)并且具有至少1个(优选为1到2个)位置的不饱和烯基(>C=C<)直链或支链烃基。这些基团实例包括乙烯基﹑烯丙基和3-丁烯-1-基。顺式和反式异构体或这些异构体的混合物包括在该范围内。“炔基”指的是具有2到6个碳原子(优选为2到3个碳原子)并且具有至少1个(优选为1到2个)位置的不饱和炔基(-C≡C-)的直链或支链烃基。这些炔基的实例包括乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。“氨基”指的是-NR’R”基团,其中R’和R”独立选自氢﹑烷基﹑取代烷基﹑烯基﹑取代烯基﹑炔基﹑取代炔基﹑芳基﹑取代芳基﹑环烷基﹑取代环烷基﹑环烯基﹑取代环烯基﹑杂芳基﹑取代杂芳基﹑杂环﹑取代杂环,以及如果R’和R”都不是氢,其中R’和R”可选地与结合的氮原子共同连接形成杂环或取代杂环(其中烷基﹑取代烷基﹑烯基﹑取代烯基﹑炔基﹑取代炔基﹑环烷基﹑取代环烷基﹑芳基﹑取代芳基﹑杂芳基﹑取代杂芳基﹑杂环﹑取代杂环定义如上)。当R’为氢和R”为烷基,取代氨基有时称作烷氨基。当R’和R”为烷基,取代氨基有时称作二烷氨基。-NH2有时称作未取代氨基。当提及单取代氨基,意为R’和R”两者中任一个为氢,但并非两者都是。当提及二取代氨基,意为R’和R”两者都不是氢。“氨基酸”指包含氨基和羧基的任何化合物,不论其为天然,非天然还是合成的。氨基酸的实例包括,但不限于甘氨酸(NH2CH2COOH)﹑半胱氨酸﹑丙氨酸﹑N-甲基丙氨酸,其包括D和L光学异构体。“氨基酸侧链”指取代甘氨酸亚甲基上一个氢原子的取代基。氨基酸侧链实例包括,但不限于天然氨基酸侧链﹑烷基﹑取代烷基﹑烯基﹑取代烯基﹑炔基﹑取代炔基﹑芳基﹑取代芳基﹑环烷基﹑取代环烷基﹑环烯基﹑取代环烯基﹑杂芳基﹑取代杂芳基﹑杂环基﹑取代杂环基。“芳基”指具有6到14碳原子的单价芳香族碳环基团,其可为单一环系(如苯基)或多稠环(如萘或蒽),只要连接点在芳香碳原子上,这些稠环可以是或者不是芳香族的(如2-苯并噁唑酮﹑2H-1,4-苯并噁嗪-3(4H)-酮-7-基等)。优选的芳基包括苯基和萘基。“羰基”指二价基团-C(O)-,其等价于-C(=O)-。“羧基”指的是-COOH或CO2H,或其盐。“羧酸”指含有至少一个羧基的化合物。“氰基”指–CN基团。“环烷基”指具有3到10个碳原子的环状烷基,可以是单个或多个环状环,包括稠环﹑桥环和螺环系统。只要连接点是通过非芳香﹑非杂环碳环形的环,这些环中一个或多个可以是芳基﹑杂芳基或杂环基。合适的环烷基的实例包括,如:环丙基﹑环丁基﹑环戊基和环辛基。其它环烷基的实例包括双环[2,2,2]辛基﹑降莰基和螺双环如螺[4.5]癸-8-基:环烯指的是环状的烯烃。“环烯基”指的是具有单个或多个环且含有至少一个>C=C<(优选1到2个位置为>C=C<)的具有3到10个碳原子的非芳香环状烷基。“卤原子”或“卤素”指的是氟﹑氯﹑溴和碘,优选为氟或氯。“卤代烷基”指的是被1到5﹑1到3或1到1个卤原子取代的烷基,其中烷基和卤原子定义如上。“羟基”指的是-OH基团。“杂芳基”指的是环中具有6到10个碳原子和1到4个杂原子(从氧﹑氮和硫原子选择)的芳基。这些杂芳基可以是单一环(如吡啶基或呋喃基)或多个稠环(如吲哚基或苯并噻吩基),其中只要连接点是通过杂芳基上的原子,稠环可以是或不是芳香族的和/或含有一个杂原子。在一个实施方案中,杂芳基为5或6元杂芳基,其具有5或6个环原子并具有1到4个杂原子。在一个实施方案中,杂芳基上的氮和/或硫环原子另外可氧化成氮-氧(N→O),或磺酰部分。优选的杂芳基包括吡啶基﹑吡咯基﹑吲哚基和呋喃基。“杂环”或“杂环的”或“杂环烷基”或“杂环基”指的是饱和的或部分饱和的,但不是芳香族的基团,其具有3到10个环碳原子和1到4个环杂原子(由氮﹑硫或氧基团选择)。在一个实施方案中,杂环为5﹑6或7元杂环,其具有5﹑6或7个环原子,其中有1到4个杂原子。这些杂环包括单一环或多个稠环(包括稠桥和稠螺环系)。在稠环系中,只要连接点是通过非芳香环,一个或多个环可以是环烷基﹑芳基或杂芳基。在一个实施方案中,杂环基上的氮和/或硫环原子另外可氧化成氮-氧(N→O),或磺酰部分。杂环和杂芳基的实例包括但不限于吖丁啶﹑吡咯﹑吡唑﹑吡啶﹑吡嗪﹑嘧啶﹑哒嗪﹑吲嗪﹑异吲哚﹑吲哚﹑二氢吲哚﹑吲唑﹑嘌呤﹑喹嗪﹑异喹啉﹑喹啉﹑酞嗪﹑萘吡啶﹑喹喔啉﹑喹唑啉﹑噌啉﹑碟啶﹑咔唑﹑咔啉﹑菲啶﹑吖啶﹑菲啰啉﹑异噻唑﹑吩嗪﹑异噁唑﹑吩噁嗪﹑吩噻嗪﹑咪唑啉﹑哌啶﹑哌嗪﹑吲哚啉﹑邻苯二甲酰亚胺﹑1,2,3,4-四氢异喹啉﹑4,5,6,7-四氢苯并[b]噻吩﹑噻唑﹑噻唑烷﹑噻吩﹑苯并[b]噻吩﹑吗啉基﹑硫代吗啉基﹑1,1-二氧代硫代吗啉基﹑哌啶基,吡咯烷和四氢呋喃基。“取代烷基”﹑“取代烯基”﹑“取代炔基”﹑“取代环烷基”﹑“取代环烯基”﹑“取代芳基”﹑“取代杂芳基”或“取代杂环基”指的是各自被1到5个﹑特别是1到3个﹑优选是1到2个取代基取代的烷基﹑烯基﹑炔基﹑环烷基﹑环烯基﹑芳基或杂环基,这些取代基选自烷基﹑卤代烷基﹑-O-R20﹑-S-R20﹑烯基﹑炔基﹑氧代﹑-C(=O)R20﹑-C(=S)R20﹑-C(=O)OR20﹑-NR20C(=O)R21﹑-OC(=O)R21﹑-NR20R20﹑-C(=O)NR20R20﹑-C(=S)NR20R20﹑-NR20C(=O)NR20R20﹑-NR20C(=S)NR20R20﹑-OC(=O)NR20R20﹑-SO2NR20R20﹑-OSO2NR20R20﹑-NR20SO2NR20R20﹑-C(=NR20)NR20R20﹑芳基﹑-NR20C(=O)OR21﹑-OC(=O)OR21﹑氰基﹑环烷基﹑环烯基﹑-NR20C(=NR20)NR20R20﹑卤素﹑羟基﹑杂芳基﹑杂环基﹑硝基﹑-SO3H﹑-SO2R21和-OSO2R21,其中R20独立地为氢﹑烷基﹑烯基﹑炔基﹑芳基﹑环烷基﹑环烯基﹑杂芳基和杂环基,或两个R20与其结合的原子形成一个杂环,R21为烷基﹑烯基﹑炔基﹑芳基﹑环烷基﹑环烯基﹑杂芳基和杂环基。“硝基”指-NO2基团。“氧代”指的为原子(=O)或(-O)。“螺环系”指的是双环系,其中有一个单一环原子为两个环所共有。“巯基”指的是-SH基团。“硫羰基”指的是二价-C(S)-基团,等同于-C(=S)-。“硫酮”指的是原子(=S)。如上使用的“化合物”应包括指明的分子式的立体异构体和互变异构体。“立体异构体”指的是一个或多个立体中心的手性不同的化合物。立体异构体包括对映异构体和非对映异构体。“互变异构体”指的是一个化合物其质子的位置不同的变化形式,如烯醇-酮和亚胺-烯胺互变异构体,或者杂芳基的互变异构形式包括环原子连接到环-NH-部分和环=N-部分如吡唑﹑咪唑﹑苯并咪唑﹑三氮唑和四氮唑。“溶剂合物”指的是溶剂以晶型方式和化合物缔合。溶剂缔合通常由于在化合物的合成﹑结晶和/或重结晶中使用溶剂。“溶剂合物”包括水以晶型方式和化合物缔合的水合物。“患者”或“接受实验的对象”指的是哺乳动物,其中包括人和非人类哺乳动物。“药学上可接受的盐”指的是化合物药学上可接受的盐,所述盐产生该技术领域众所周知的各种各样的有机和无机抗衡离子,仅仅示例性盐包括,当分子含有酸性官能团时,有机或无机盐如钠盐﹑钾盐﹑钙盐﹑镁盐﹑铵盐﹑异丙胺﹑三甲胺﹑二乙胺基﹑三乙胺﹑三丙胺﹑乙醇胺﹑2-二甲胺基乙醇﹑2-二乙胺基乙醇﹑二环己基胺﹑赖氨酸﹑精氨酸﹑组氨酸﹑咖啡因﹑普鲁卡因﹑hydrabamine﹑胆碱﹑甜菜碱﹑乙二胺﹑葡糖胺﹑甲葡糖胺﹑可可碱﹑嘌呤﹑哌嗪﹑哌啶﹑N-乙基哌啶﹑多胺树脂及四烷基铵盐等;以及当分子含有碱性官能团时,有机或无机酸盐如盐酸盐﹑氢溴酸盐﹑酒石酸盐﹑甲磺酸盐﹑醋酸盐﹑马来酸盐和草酸盐。酸的其它非限制例子包括硫酸﹑硝酸﹑磷酸﹑丙酸﹑乙醇酸﹑丙酮酸﹑丙二酸﹑琥珀酸﹑富马酸﹑酒石酸﹑柠檬酸﹑苯甲酸﹑肉桂酸﹑扁桃酸﹑甲磺酸﹑乙磺酸﹑对甲苯磺酸﹑水杨酸等。患者疾病“治疗”指的是(1)阻止疾病在有倾向性或还没表现疾病症状的患者中出现;(2)抑制疾病或阻止其发展;或(3)减轻疾病或致其退化。“有效量”意指活性化合物或药剂的量,其导致研究人员﹑兽医﹑医生或其他临床医生正寻求的组织﹑系统﹑动物﹑个体以及人的生物或药用响应,这包含治疗一种疾病。与抗原结合单元偶联的药物衍生物本发明公开了具有连接基团的类美登素衍生物,其通过形成对酸不敏感﹑对肽酶组织蛋白酶不敏感以及不含二硫键的连接子可偶联到抗原结合单元(Abu)。适于偶联连接基团的类美登素包括美登醇和美登醇类似物以及可以按照已知方法从天然源分离﹑使用生物技术制造(见如:Yu等,99PNAS7968-7973(2002))﹑或按照已知方法合成制备(见如:Cassady等,Chem.Pharm.Bull.52(1)1-26(2004))。适合的美登醇类似物的实例包括:(1)C-19-去氯(参见美国专利4256746)(由安丝菌素P2经氢化锂铝还原制备);(2)C-20-羟基(或C-20-去甲基)+/-C-19-去氯(参见美国专利4361650和4307016)(使用链霉菌(Streptomyces)或放线菌(Actinomyces)去甲基或使用氢化锂铝(LAH)去氯制备);(3)C-20-去甲氧基、C-20-酰氧基(-OCOR)、+/-去氯(参见美国专利4294757)(使用酰氯通过酰化制备);(4)C-9-巯基(美国专利号4424219)(通过美登醇与H2S或P2S5反应制备);(5)C-14-羟甲基(CH2OH)或酰氧甲基(CH2OC(=O)苯基或CH2OC(=O)(C1-C5烷基))(参见美国专利4331598)(从诺卡氏菌(Nocardia)制备);(6)C-15-羟基/酰氧基(参见美国专利4364866)(美登醇经由链霉菌(Streptomyces)转化而成);(7)C-15-甲氧基(参见美国专利4313946和4315929)(从Trewianudlflora分离得到);(8)C-18-N-去甲基(参见美国专利4362663和4322348)(美登醇经由链霉菌(Streptomyces)去甲基制备);以及(9)4,5-去氧(参见美国专利4371533)(美登醇经由三氯化钛/氢化锂铝还原制备)。取决于连接子类型,美登醇上的很多位置可用作连接位置。例如,对于形成酯键,C-3位置具有羟基﹑C-14位置为羟甲基修饰﹑C-15位置为羟基修饰以及C-20位置具有羟基都是合适的。在一些实施方案中,连接处为C-3位置。在一些实施方案中,本发明提供了式I或I-1的类美登素衍生物:或其药学上可接受的盐,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基﹑苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;以及L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基。在一些实施方案中,式I的化合物为在一些实施方案中,式I-1的化合物为或在一些实施方案中,式I的化合物为异构体:在一些实施方案中,式I-1的化合物为异构体:在一些实施方案中,本发明提供了式II或II-1的类美登素衍生物:或其药学上可接受的盐,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基﹑苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;以及L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基。在一些实施方案中,式II的化合物为在一些实施方案中,式II-1的化合物为或在一些实施方案中,式II的化合物为异构体:在一些实施方案中,式II-1的化合物为异构体:在一些实施方案中,X为氢。在一些实施方案中,X为氯。在一些实施方案中,Y为氢。在一些实施方案中,R1为氢。在一些实施方案中,R2为甲基。在一些实施方案中,R3为甲基。在一些实施方案中,R4为羟基。在一些实施方案中,R7为氨基酸侧链。在一些实施方案中,R7为甲基。在一些实施方案中,R8为甲基。在一些实施方案中,L为未被取代的C1-C20亚烃基。在一些实施方案中,L为被取代的C1-C20亚烃基。在一些实施方案中,L为未被取代的C1-C10亚烃基。在一些实施方案中,L为被取代的C1-C10亚烃基。在一些实施方案中,L为-(CH2)5-。在一些实施方案中,L为未被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,L为被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,提供式III或III-1的化合物:或其药学上可接受的盐,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;以及L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基。在一些实施方案中,化合物具有式IV或IV-1:或其药学上可接受的盐,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;以及m选自1到20之间的一个整数,优选为1-10,更优选为5-10。在一些实施方案中,化合物为N2’-去乙酰-N2’-(6-马来酰基-1-氧代-己基)美登素(命名为batansine)或其盐,其中Batansine用式V描述:在一些实施方案中,式I或I-1的化合物选自:以及或其药学上可接受的盐。药物连接子抗原结合单元偶联物本发明公开了通过连接子偶联至抗原结合单元(Abu)的类美登素化合物(药物连接子抗原结合单元偶联物)。在一些示范性实施方案中,药物连接子抗原结合单元偶联物为类美登素通过对酸不敏感﹑对肽酶组织蛋白酶不敏感以及不含二硫键的连接子偶联到抗体。在一些实施方案中,本发明提供了式Ia或Ia-1的类美登素连接子抗原结合单元偶联物:或其药学上可接受的盐或溶剂合物,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基、苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1、2、3、4、5、6、7、8、9以及10;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及Abu为抗原结合单元。在一些实施方案中,式Ia的化合物为:在一些实施方案中,Ia-1的化合物为:在一些实施方案中,Ia的化合物为异构体:在一些实施方案中,Ia-1的化合物为异构体:在一些实施方案中,本发明提供了式IIa或IIa-1的类美登素连接子抗原结合单元偶联物:或其药学上可接受的盐或溶剂合物,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基、苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1、2、3、4、5、6、7、8、9以及10;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及Abu为抗原结合单元。在一些实施方案中,式IIa的化合物为:在一些实施方案中,IIa-1的化合物为:在一些实施方案中,式IIa的化合物为异构体:式IIa-1的化合物为异构体:在一些实施方案中,X为氢。在一些实施方案中,X为氯。在一些实施方案中,Y为氢。在一些实施方案中,R1为氢。在一些实施方案中,R2为甲基。在一些实施方案中,R3为甲基。在一些实施方案中,R4为–羟基。在一些实施方案中,R7为氨基酸侧链。在一些实施方案中,R7为甲基。在一些实施方案中,R8为甲基。在一些实施方案中,L为未被取代的C1-C20亚烃基。在一些实施方案中,L为被取代的C1-C20亚烃基。在一些实施方案中,L为未被取代的C1-C10亚烃基。在一些实施方案中,L为被取代的C1-C10亚烃基。在一些实施方案中,L为-(CH2)5-。在一些实施方案中,L为未被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基﹑-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,L为被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基﹑-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,本发明提供了式IIIa或IIIa-1的化合物:或其药学上可接受的盐或溶剂合物,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1﹑2﹑3﹑4﹑5﹑6﹑7﹑8﹑9以及10;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及Abu为抗原结合单元。在一些实施方案中,化合物具有式IVa或IVa-1:或其药学上可接受的盐或溶剂合物,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1﹑2﹑3﹑4﹑5﹑6﹑7﹑8﹑9及10;m选自1到20之间的一个整数,优选为1-10,更优选为5-10;以及Abu为抗原结合单元。在一些实施方案中,化合物具有式Va:或其药学上可接受的盐或溶剂合物。在一些实施方案中,式Ia或Ia-1的化合物选自:在一些实施方案中,本发明提供类美登素连接子抗体偶联化合物具有式XIVa:或其药学上可接受的盐。具有连接基团可偶联到抗原结合单元(Abu)的类美登素衍生物或类美登素连接子抗原结合单元偶联物的类美登素组分也可由其它合适的细胞毒试剂例如:auristatin﹑DNA小沟结合试剂﹑DNA小沟烷基化试剂﹑烯二炔(enediyne)﹑lexitropsin﹑duocarmycin﹑紫杉烷(taxane)﹑嘌呤霉素(puromycin)﹑dolastatin及长春花碱(vincaalkaloid)所取代。其它合适的细胞毒试剂包括抗微管蛋白试剂如:auristatin﹑长春花碱(vincaalkaloid)﹑竹叶草霉素(podophyllotoxin)﹑紫杉烷(taxane)﹑浆果赤霉素衍生物(baccatinderivative)﹑cryptophysin﹑类美登素(maytansinoid)﹑combretastatin﹑或尾海兔素(dolastatin)。在一些实施方案中,细胞毒试剂为AFP﹑MMAF﹑MMAE﹑AEB﹑AEVB﹑auristatinE﹑长春新碱(vincristine)﹑长春花碱(vinblastine)﹑长春地辛(vindesine)﹑长春瑞滨(vinorelbine)﹑VP-16﹑喜树碱(camptothecin)﹑紫三醇(paclitaxel)﹑多西紫杉醇(docetaxel)﹑埃博霉素A(epothiloneA)﹑埃博霉素B(epothiloneB)﹑诺考达唑(nocodazole)﹑秋水仙碱(colchicines)﹑colcimid﹑雌氮芥(estramustine)﹑西马多丁(cemadotin)﹑圆皮海绵内酯(discodermolide)﹑美登素(maytansine)﹑DM-1﹑DM-3﹑DM-4或eleutherobin。合适的免疫抑制剂包括如丙氧鸟苷(gancyclovir)﹑依那西普(etanercept)﹑环孢霉素(cyclosporine)﹑他克莫司(tacrolimus)﹑雷柏霉素(rapamycin)﹑环磷酰胺(cyclophosphamide)﹑咪唑硫嘌呤(azathioprine)﹑霉酚酸酯(mycophenolatemofetil)﹑甲氨蝶呤(methotrexate)﹑氢化可的松(cortisol)﹑醛甾酮(aldosterone)﹑地塞米松(dexamethasone)﹑环氧酶抑制剂(cyclooxygenaseinhibitor)﹑5-脂氧合酶抑制剂(5-lipoxygenaseinhibitor)或白三烯受体拮抗剂(leukotrienereceptorantagonist)。在一些实施方案中,细胞毒试剂为AFP﹑MMAF﹑MMAE﹑AEB﹑AEVB﹑auristatinE﹑紫杉醇(paclitaxel)﹑多西紫杉醇docetaxel﹑CC-1065﹑SN-38﹑拓扑替康(topotecan)﹑吗啉代阿霉素(morpholino-doxorubicin)﹑根霉素(rhizoxin)﹑氰基吗啉代阿霉素(cyanomorpholino-doxorubicin)﹑尾海兔素-10(dolastatin-10)﹑棘霉素(echinomycin)﹑combretatstatin﹑卡里奇霉素(calicheamicin)﹑美登素(maytansine)﹑DM-1﹑DM-3﹑DM-4或纺锤霉素(netropsin)。具有连接基团能偶联到抗原结合单元(Abu)的类美登素衍生物或类美登素连接子抗原结合单元偶联物的类美登素组分也可由其它合适的免疫抑制剂例如:丙氧鸟苷(gancyclovir)﹑依那西普(etanercept)﹑环孢霉素(cyclosporine)﹑他克莫司(tacrolimus)﹑雷柏霉素(rapamycin)﹑环磷酰胺(cyclophosphamide)﹑咪唑硫嘌呤(azathioprine)﹑麦考酚酸酯(mycophenolatemofetil)﹑氨甲喋呤(methotrexate)﹑氢化可的松(cortisol)﹑醛固酮(aldosterone)﹑地塞米松(dexamethasone)﹑环氧酶抑制剂(cyclooxygenaseinhibitor)﹑5-脂氧合酶抑制剂(5-lipoxygenaseinhibitor)或白三烯受体拮抗剂(leukotrienereceptorantagonist)所取代。抗原结合单元所述抗原结合单元可以是目前已知的任何一种,或者是将要成为众所周知的,包括肽和非肽。通常,这些可能是抗体(例如,单克隆抗体)、淋巴因子、激素、生长因子、维生素、营养运输分子(转铁蛋白),或其他细胞结合分子或物质,这些物质可以特异性结合靶点。这些与靶点结合的物质可以来自抗体、抗体片段、细胞特异性配体,或者来自肽、碳水化合物、天然或合成的化合物,或者药学上可接受的盐或溶剂化物。所述抗原结合单元包括抗体碎片(多克隆抗体和单克隆抗体),如Fab、Fab’、F(ab’)2和Fv(参见Parham,J.Immunol.131:2895-2902(1983);Springetal.,J.Immunol.113:470-478(1974);Nisonoffetal.,Arch.Biochem.Biophys.89:230-244(1960));单域抗体(dAbs)和其抗原结合片段,包括骆驼抗体(参见Desmyteretal.,NatureStruct.Biol,3:752(1996))、被称作新型抗原受体的鲨鱼抗体(IgNAR)(参见Greenbergetal.,Nature,374:168(1995);Stanfieldetal.Science305:1770-1773(2004))。单克隆抗体技术使得抗原结合单元以特异性单克隆抗体的形式成为可能。用有研究意义的抗原,如从靶细胞中分离的肿瘤特异性抗原,免疫小鼠、大鼠或其他哺乳动物来制备单克隆抗体已成为公知技术。另外一种制备抗原结合单元的方法是利用scFv的噬菌体库(单链可变区),尤其是人scFv(参见Griffithsetal.,US5,885,793和5,969,108;McCaffertyetal.,WO92/01047;Limingetal.,WO99/06587),或者用酵母选择系统制备域抗体(参见US7,195,595)。另外,如美国专利5,639,641中所公布的抗体人源化技术也可以用做嵌合抗体或人源化抗体。特异抗原结合单元的选择要依据疾病的类型作为靶点的细胞和组织。在一些实施例中,抗原结合单元是人单克隆抗体。抗原结合单元可以用于特异性结合肿瘤抗原。本文所述“肿瘤抗原”是指肿瘤细胞中表达的靶点物质。肿瘤抗原可以用于肿瘤细胞的鉴别,也可以成为肿瘤治疗的潜在指示剂,或成为肿瘤治疗的靶点。许多本领域熟知的肿瘤抗原和新的肿瘤抗原可以通过筛查而鉴别。肿瘤抗原的非限定性的例子包括EGFR、Her2、EpCAM、CD20、CD30、CD33、CD47、CD52、CD133、CEA、gpA33、粘蛋白、TAG-72、CIX、PSMA、叶酸结合蛋白、GD2、GD3、GM2、VEGF、VEGFR、整合素、αVβ3、α5β1、ERBB2、ERBB3、MET、IGF1R、EPHA3、TRAILR1、TRAILR2、RANKL、FAP和肌腱蛋白。激素和细胞因子可能是肿瘤抗原。非限制性例子包括淋巴因子,如白介素-2、白介素-3、白介素-4、白介素-6,激素,如胰岛素、促甲状腺激素释放激素、黑色素细胞刺激激素,类固醇激素,如雄激素和雌激素;生长因子和细胞集落刺激因子,如表皮生长因子、TGF-alpha、FGF、VEGF、G-CSF和GM-CSF(参见Burgess,ImmunologyToday5:155-158(1984))、转铁蛋白(参见O'Keefeetal.,J.Biol.Chem.260:932-937(1985))和维生素如叶酸。抗原结合单元对肿瘤细胞表达的相应的靶点或抗原具有特异性结合作用,而一般对相应的非肿瘤细胞没有特异性结合。所述的“相应的非肿瘤细胞”是一种非肿瘤细胞,此细胞与肿瘤细胞的原始细胞属于同一细胞类型。需说明的是这种蛋白并非一定不同于肿瘤抗原。非限定性例子包括癌胚抗原(CEA),其过量表达于多数结肠癌、直肠癌、乳腺癌、肺癌、胰腺癌和胃肠道癌症;heregulin受体(HER-2neu或c-erbB-2),其常在乳腺癌、卵巢癌、结肠癌、肺癌、前列腺癌和子宫颈癌中过量表达;EGFR,其大量表达于许多实体瘤,比如乳腺癌、头颈部肿瘤、非小细胞肺癌和前列腺癌;asialoglvcoprotein受体;转铁蛋白受体;表达于肝细胞Serpin酶复合物受体;过量表达于胰腺导管腺癌细胞和很多其他肿瘤组织的成纤维细胞生长因子受体(FGFR);用于抗血管生成靶点治疗的VEGFR;叶酸受体,其选择性过量表达于90%的非粘液性卵巢癌;类似于Glycocalyx细胞表面的蛋白质复合物;糖类受体;以及聚免疫球蛋白受体,其被用于基因治疗方法以传递到呼吸道上皮细胞和被用于治疗肺部疾病如囊性纤维化。抗原结合单元是可以修饰的。例如一种或多个氨基酸序列改变、增加、或减少,以增强抗体依赖的细胞介导的细胞毒性作用(ADCC)。例如,IgG2抗体可以被修饰成包括IgG1抗体的Fc和/或铰链区以增强抗体依赖的细胞介导的细胞毒性作用。增强IgG1-Fc抗体依赖的细胞介导的细胞毒性作用的例子,以及筛选增强IgG1-Fc抗体依赖的细胞介导的细胞毒性作用的抗体或其片断的方法为已知技术(参见Stewartetal.ProteinEngDesSel.24(9):671-8,2011)。在本发明的一个实施例中,抗原结合单元BAT0206,具有增强了的抗体依赖的细胞介导的细胞毒性作用。BAT0206包含一个抗EGFR轻链氨基酸序列SEQIDNO:1和一个抗EGFR重链氨基酸序列SEQIDNO:2。在本发明的另一实施例中,抗原结合单元是西妥昔单抗,其是一种抗EGFR的人鼠嵌合单克隆抗体。西妥昔单抗的轻链和重链序列为SEQIDNO:3和4。另一种抗EGFR抗体是帕尼单抗,其是一种全人源化单克隆抗体。另一抗EGFR抗体是尼妥珠单抗,其轻链和重链序列为SEQIDNO:7和8。另一抗EGFR抗体是马妥珠单抗,其轻链和重链序列为SEQIDNO:9和10,分别见表1。EGFR过量表达于许多肿瘤组织,如转移性结直肠癌和头颈部癌症。利妥昔单抗是一种抗CD20蛋白的嵌合单克隆抗体,首次发现于B细胞表面。利妥昔单抗特异性结合并破坏B细胞,因此,被用于治疗B细胞过量或B细胞过量活化的疾病。这些疾病包括淋巴瘤、白血病、移植排斥和自身免疫性疾病。其轻链和重链序列为SEQIDNO:13和14,详见表1。同样,曲妥珠单抗(Trastuzumab)是一种抗蛋白Her2的嵌合单克隆抗体,从而抑制Her2/neu受体。曲妥珠单抗的轻链和重链序列分别见表1中SEQIDNO:11和12.在一些癌症中,尤其是乳腺癌,Her2过量表达,并且引起乳腺细胞复制失控。曲妥珠单抗被用于治疗特定乳腺癌和胃癌。在另一个实施方案中,抗原结合单元和序列见表1,但抗原结合单元并非限制于此表。非抗体分子也能用作抗原结合单元与类美登素化合物偶联,将药物靶向特定细胞或疾病组织。例如,粒细胞巨噬细胞集落刺激因子(GM-CSF)与骨髓细胞结合,可以用于细胞结合剂靶向病变细胞的急性髓系白血病。另外,白介素2结合活化的T细胞,可以用于预防移植排斥反应、用于治疗和预防移植物抗宿主病和用于急性T细胞白血病的治疗。黑色素细胞刺激素(MSH)与黑素细胞结合,可用于黑素瘤的治疗。叶酸可用于靶向表达叶酸受体的卵巢癌和其他肿瘤。表皮生长因子(EGF)可用于针对肺癌和头颈部等鳞状癌症的治疗。生长抑素可以用于靶向神经母细胞瘤和其他类型的肿瘤。乳房和睾丸的癌症可以分别用细胞结合剂雌激素(或雌激素类似物)或雄激素(或雄激素类似物)针对性治疗。在本发明的另一实施方案中,所述偶联物D-L-Abu,是一个或多个美登素与BAT0206的偶联而成,所述BAT0206包含一个氨基酸序列为SEQIDNO:1的抗EGFR轻链和一个氨基酸序列为SEQIDNO:2的抗EGFR重链形成的抗EGFR抗体。表1.示例性抗原结合单元的氨基酸序列在一些实施方案中,本发明提供了式Id或IId的类美登素BAT0206偶联物:或其药学上可接受的盐或溶剂合物,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基、苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;p选自1﹑2﹑3﹑4﹑5﹑6﹑7﹑8﹑9及10;以及Lb选自任选被C1-C4烷基﹑-SO3H或-P(O)(OH)2取代的C1-C20亚烃基;任选被C1-C4烷基取代的C3-C8环亚烃基;以及任选被C1-C4烷基﹑-SO3H或-P(O)(OH)2取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的一个或多个基团所替代:C3-C8环亚烃基、任选被一个或多个C1-C4烷基或氧代取代的3到8元杂环亚烃基﹑-O-﹑-S-﹑-S-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2;Bat0206为抗原结合单元。在一些实施方案中,Lb为任选被C1-C4烷基﹑-SO3H或-P(O)(OH)2取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:-S-﹑-S-S-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑以及在一些实施方案中,式Id的化合物选自:以及或其药学上可接受的盐或溶剂合物。在一些实施方案中,式Id的化合物选自:以及或其药学上可接受的盐或溶剂合物,其中X为氢或氯;Y为氢或甲基;R7为氢﹑C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;以及p选自1﹑2﹑3﹑4﹑5﹑6﹑7﹑8﹑9以及10。在一些实施方案中,式Id的化合物选自:(Batansine-0206),(D-Lmcc-BAT0206)以及(D-Lspp-BAT0206),或其药学上可接受的盐或溶剂合物,其中Bat0206为抗原结合单元。药物偶联至抗原结合单元如上所述,化合物(例如:类美登素药物衍生物)可以通过连接子偶联至抗原结合单元。在一些实施方案中,抗原结合单元可以用适当的双官能团修饰试剂修饰。在一些实施方案中,含有巯基(SH)的基团可以引入到抗原结合单元上的氨基酸残基侧链,如赖氨酸侧链。例如,抗原结合单元上赖氨酸残基的氨基可以与2-亚氨基硫烷(Traut’sReagent)或与3-(2-吡啶二硫代)丙酸N-羟基琥珀酰亚胺酯(SPDP)﹑4-(2-吡啶二硫代)丁酸N-羟基琥珀酰亚胺酯(DPDB)等反应接着用还原剂如2-巯基乙醇﹑二硫苏糖醇(DTT)或三(2-羧乙基)膦(TCEP)还原转化成含巯基基团。可取代赖氨酸残基上侧链氨基的含巯基基团的非限制性实例包括-NHC(=NH)(CH2)nSH和-NHC(O)(CH3)nSH,其中n为1﹑2﹑3﹑4﹑5或6。当含巯基基团引入氨基酸残基,氨基酸残基被指为巯基化氨基酸。例如,当赖氨酸侧链氨基转化成含巯基基团,赖氨酸残基被指为巯基化赖氨酸。抗原结合单元上引入的游离巯基个数可以变化,比如在1和大约20之间,或者5到15,以及或者5到12。连接子或药物-连接子可以与抗原结合单元上巯基化赖氨酸残基的游离巯基(SH)成键。在一些实施方案中,与抗原结合单元上巯基化赖氨酸残基成键的连接子或连接子-药物个数在1和大约10之间。在一些实施方案中,如此成键个数至少是1,或至少为2﹑或3﹑或4﹑或5。在一些实施方案中,如此连接的个数为不超过10,或者不超过9﹑或8﹑或7﹑或6﹑或5﹑或4。在一些实施方案中,每个抗原结合单元平均与3到5个药物分子偶联。在另一实施方案中,药物-连接子可以通过结合到半胱氨酸残基上的巯基偶联到抗原结合单元。每个抗原结合单元通常包含许多半胱氨酸,但是它们中很多,如果并非所有,彼此形成二硫键,因此无法用于如此偶联。在一些实施方案中,因此,抗原结合单元上的一个或一个以上的二硫键通过与还原剂如2-巯基乙醇﹑二硫苏糖醇(DTT)或三(2-羧乙基)膦(TCEP)反应而断裂形成游离巯基(SH)。该反应可以跟踪和/或控制以便足够数量的二硫键断裂而能偶联,同时维持足够数量的二硫键而保持抗原结合单元的结构稳定性。在一些实施方案中,药物-连接子和抗原结合单元上的半胱氨酸残基之间成键个数为1到10。在一些实施方案中,此类键的个数至少为1,或至少为2﹑或3﹑或4﹑或5。在一些实施方案中,如此形成的键的个数不超过10,或者不超过9﹑或8﹑或7﹑或6﹑或5﹑或4。在一些实施方案中,每个抗原结合单元通过半胱氨酸平均与3到5个药物分子偶联。在一些实施方案中,药物分子通过赖氨酸和半胱氨酸混合残基偶联到抗原结合单元。抗原结合单元可以通过修饰,引入偶联位点。比如,位点特异突变以便引入额外的巯基化赖氨酸或半胱氨酸残基而允许适当的偶联。氨基酸修饰方法为该技术领域众所周知。修饰过的抗原结合单元然后可以用实验方法考察其稳定性和其抗原结合性能。在一些实施方案中,至少一个巯基化赖氨酸或半胱氨酸通过如此修饰而引入。在另一实施方案中,至少两个巯基化赖氨酸或半胱氨酸通过如此修饰而引入。药物负载抗原结合单元上药物负载可以改变,这取决于很多因素如药物的效力、大小、抗原结合单元的稳定性、抗原结合单元上可用的能偶联的基团等。在一些实施方案中,1到10个类美登素药物分子和1个抗原结合单元分子偶联。在一些实施方案中,平均3到5个类美登素药物分子和1个抗原结合单元分子偶联。在一些实施方案中,平均3.5个类美登素药物分子和1个抗原结合单元分子偶联。药物连接子抗原结合单元偶联物的制备本发明提供了由式I﹑I-1﹑II﹑II-1﹑III﹑III-1﹑IV﹑IV-1和V的任一项的化合物与抗原结合单元制备类美登素药物连接子抗原结合单元偶联物化合物的方法。在一些实施方案中,式I﹑I-1﹑II﹑II-1﹑III﹑III-1﹑IV﹑IV-1和V的任一项的化合物与抗原结合单元溶于水溶液。在一些实施方案中,溶液包含缓冲溶液如磷酸盐缓冲溶液。尽管不希望局限于任何理论,可以预期,式Ia到Va任一项化合物一旦内吞,能被细胞内蛋白酶降解至具有细胞毒的类美登素部分组成的降解产物。本发明公开了式Ib或Ib-1的化合物:或其药学上可接受的盐,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基、苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及AA为氨基酸,如半胱氨酸,或巯基化氨基酸,如巯基化赖氨酸。在一些实施方案中,式Ib或Ib-1的化合物分别为:或或者其药学上可接受的盐。在一些实施方案中,本发明提供了式IIb或IIb-1的化合物:或其药学上可接受的盐,其中X为氢或卤素;Y选自氢﹑C1-C6烷基﹑C3-C6环烷基和-C(=O)R5;R1选自H﹑-OH﹑-OC(=O)R5和-OR5基团;R2为H或C1-C6烷基;R3为甲基﹑-CH2OH或-CH2OC(=O)R6;R4为-OH或–SH;R5为C1-C6烷基或苄基;R6为C1-C6烷基﹑苯基或苄基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;Z独立地为氢或C1-C4亚烃基,或两个Z与其所连接的碳原子形成一个羰基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及AA为氨基酸,如半胱氨酸,或巯基化氨基酸,如巯基化赖氨酸。在一些实施方案中,X为氢。在一些实施方案中,X为氯。在一些实施方案中,Y为氢。在一些实施方案中,R1为氢。在一些实施方案中,R2为甲基。在一些实施方案中,R3为甲基。在一些实施方案中,R4为–羟基。在一些实施方案中,R7为氨基酸侧链。在一些实施方案中,R7为甲基。在一些实施方案中,R8为甲基。在一些实施方案中,L为未被取代的C1-C20亚烃基。在一些实施方案中,L为被取代的C1-C20亚烃基。在一些实施方案中,L为未被取代的C1-C10亚烃基。在一些实施方案中,L为被取代的C1-C10亚烃基。在一些实施方案中,L为-(CH2)5-。在一些实施方案中,L为未被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,L为被取代的C1-C20亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-。在一些实施方案中,本发明提供了如下化合物:在一些实施方案中,本发明提供了式IIIb或IIIb-1的化合物:或其药学上可接受的盐,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;L选自任选被取代的C1-C20亚烃基或C3-C8环亚烃基,其中一个或多个-CH2-基团独立地任选地被选自以下的基团所替代:C3-C8环亚烃基、-O-﹑-S-﹑-NR8-﹑-C(O)-﹑-C(=O)NR8-﹑-NR8C(=O)-﹑-SO2NR8-或-NR8SO2-;优选地,L为-(CH2)m-,其中m=1-20,优选为1-10,更优选为5-10;优选地,其中所述C1-C20亚烃基为被1到4个-SO3H﹑-P(O)(OH)2或R23取代的C1-C20亚烃基,其中R23各自独立地为任选地被独立地选自SH﹑S-C1-4烷基﹑-CONR11R11以及-NR11R11的一个或两个基团取代的C1-6烷基;以及AA为氨基酸,如半胱氨酸,或巯基化氨基酸,如巯基化赖氨酸。在一些实施方案中,化合物具有式IVb或IVb-1:或其药学上可接受的盐,其中X为氢或氯;Y为氢或甲基;R7为氢、C1-6烷基或氨基酸侧链;R8为氢或者C1-6烷基;m为1到20之间一个整数,优选为1-10,更优选为5-10;以及AA为氨基酸,如半胱氨酸,或巯基化氨基酸,如巯基化赖氨酸。在一些实施方案中,化合物具有式Vb:或其药学上可接受的盐。在一些实施方案中,AA为,但不限于或其中代表与分子其它部分的连接点。在一些实施方案中,式Ib的化合物选自:以及或其药学上可接受的盐。尽管不希望局限于任何理论,可以预期,式Ia到Va,Va到XIIa任一项化合物一旦内吞,能被细胞内蛋白酶降解至具有细胞毒的类美登素部分组成的降解产物。本发明公开了式Vc的化合物:在一些实施方案中,本发明提供了被细胞内蛋白酶降解至具有细胞毒的类美登素部分组成的降解产物的化合物,包括:以及在另一个实施方案中,本发明提供了一种或多种化合物用于制备治疗增殖、炎症或免疫疾病的药物的用途,例如,所述化合物为式Ia-Va、Id和式Va-XIVc中任一种化合物。本发明化合物可以配制成药物组合物和适于所选择的给药途径的各种给药形式,即,口服或静脉内、肌内、局部或皮下等肠胃外给药。化合物的量会有所不同,取决于药物连接体的性质、药物负载、细胞表面引起内化作用的程度、药物的转导和释放、治疗的疾病、病人的情况,如年龄、性别、体重等,并可以通过本领域公知的方法来确定,例如,见美国专利4938949,将最终由主治医生或临床医生来决定。通常,合适的剂量范围是约0.1~200毫克/千克,例如,连续52周中每1-4周每天静脉输注30-90分钟,每千克体重用药量为0.5毫克/千克~50毫克/千克、1.0毫克/千克~25毫克/千克、1.5毫克/千克~15毫克/千克或者1毫克/千克~10毫克/千克在一些实例中,剂量从约1.0毫克/天~100毫克/天,例如,从约2毫克/天至约5克/天,约10毫克/天至约1克/天,约20~500毫克/天,或在约50~100毫克/天。本发明化合物可以每日、每月用药,例如一天一次,持续用药1-3周或一个月。另一方面,本发明化合物可以周期给药,如先每天给药约5~21天,接下来1~7天不用药,如此,循环给药。在另一些实施例中,初始给药量为1-4mg/kg静脉输注30-90分钟,接下来的52周里每1-4周经脉输注30分钟以上,药量为1-2mg/kg。在另一些实施例中,初始给药量为2-10mg/kg静脉输注30-90分钟,接下来的52周里每1-4周经脉输注30-90分钟,药量为1-5mg/kg。在一些实施例中,所述化合物与其他疗法联合应用。例如,所述化合物可与另外一种治疗方法一起应用于肿瘤的治疗,例如,放射治疗或本领域中已知的另一种抗癌剂。在另一个实施例中,本发明提供了式Ib化合物用于制备治疗增生、炎症或免疫疾病的药物的用途。所述式Ib的化合物,其特征在于:作为式Ia的化合物或其药学上可接受的盐给药后的代谢产物。所述代谢化学反应是指身体内部(如,细胞、机体)发生的反应,一种化学物质转化为另一种化学物质。该转化可以通过代谢和/或化学过程,并且可以发生在一个步骤中,或通过两个或多个步骤。代谢的化学反应包括蛋白质或肽的降解反应。组成一个美登木素生物碱连接抗原结合单位结合,如如通过在细胞内的蛋白质的抗体或抗体片段。在一些实施例中,本发明提供了式IIb化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式IIb的化合物是式IIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式IIIb的化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式IIIb的化合物是式IIIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式VIb化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式VIb的化合物是式VIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式Vb化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式Vb的化合物是式Va的化合物或其药学上可接受的盐给药后的代谢产物。在另一些实施例中,本发明提供了式Vc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式Vc的化合物是式Va、VIa或VIIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式VIIIc的化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式VIIIc的化合物是式VIIIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式IXc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式IXc的化合物是式IXa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式Xc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式Xc的化合物是式Xa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式XIc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式XIc的化合物是式XIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式XIIc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式XIIc的化合物是式XIIa的化合物或其药学上可接受的盐给药后的代谢产物。在一些实施例中,本发明提供了式XIVc化合物用于制备治疗增生、炎症或免疫疾病的药物的用途,所述式XIVc的化合物是式XIVa的化合物或其药学上可接受的盐给药后的代谢产物。治疗的疾病,可以通过与抗原结合单元的结合来确定。在一些实施例中,所述疾病为增生性疾病,如,包括黑色素瘤、乳腺癌、膀胱癌、肺癌、甲状腺癌、前列腺癌、卵巢癌、肥大细胞性白血病、生殖细胞瘤,小细胞肺癌、胃肠道间质瘤、急性髓系白血病(AML)、慢性B型淋巴细胞性白血病(B-CLL)和非霍奇金淋巴瘤(NHL)、神经母细胞瘤和胰腺癌等。在一些实施例中所述疾病为炎症性疾病或者免疫疾病,如,移植排斥或自身免疫性疾病,如I型糖尿病、类风湿关节炎、系统性红斑狼疮、炎症性肠。药物组合物本发明提供了一些药物组合物,包括如本文所述的一种或多种组合物,例如,式Ia-Va的任一种化合物的组合物,及其一种或多种药学上可接受的载体。所述组合物应包含至少0.1%的活性化合物。组合物的百分比可以改变,可能是一个给定的单位剂量组成的重量的2~90%。所述治疗效用的组合物中的活性化合物的量需要达到有效剂量水平。所述组合物的口服给药方式包括,但不限于:口服片剂、口含片剂、锭剂、胶囊剂、酏剂、混悬剂、糖浆剂、溶液剂、糯米纸剂等。适合于注射或输液的组合物可以包括药学上可接受的液体载体或赋形剂,如无菌水溶液或分散液,或适于临时配制成无菌注射或难溶溶剂,任选地包封在脂质体中的溶液或分散液含有活性成分的无菌粉末。药物组合物的其他形式包括外用制剂,如凝胶、软膏、霜剂、洗剂或贴剂等所述药物组合物,除本文中提到的以外,还包括本领域公知、药学上可接受的载体;例如,雷明顿,药剂科学和实践,第20版,2000年,利平科特·威廉斯&威尔金斯,(Editors:Gennaro,A.R.,etal.)。在另一个实施例中,本发明提供了一种制备药物组合物的方法,其特征在于:包括所述化合物的混合物,例如,分子式Ia-Va和Id的中任一项的化合物及其药学上可接受的载体。混合的活性成分与药学上可接受的载体的方法在本领域中是公知技术,例如,将活性化合物与液体或细碎的固体载体或两者按规定比例均匀混合,然后,如果有必要,将所得混合物做成所需的形状。在一些实施例中,式Ia-Va和Id任一种化合物制备成注射剂,例如,在药物浓度为2-50mg/ml的水溶液中含有4-10mg/mL的氯化钠和/或5-12mg/ml的醋酸钠,或者在药物浓度为2-50mg/ml的水溶液中含有5-10mg/ml的氯化钠、1-5mg/mL的磷酸氢二钠七水合物、0.1~0.5mg/ml的磷酸二氢钠一水合物。在另一些实施例中,式Ia-Va和Id任一种化合物制备成注射剂,例如,在药物浓度为2-100mg/ml的水溶液中含有0.5-1.0%mg/mL的氯化钠、0.05-0.10%的磷酸二氢钠二水合物、1.0~2.0%的磷酸氢二钠二水合物、0.01~0.05%的柠檬酸钠、0.10-0.20%的柠檬酸一水合物、1.0-2.0%的甘露醇、0.1%~0.2的聚山梨酯80和注射用水(参见USP标准),氢氧化钠调节pH值。方法本发明的化合物可以由容易得到的起始物用如下常规方法和操作加以制备。其中给定的典型或优选方法条件(如反应温度﹑时间﹑反应物摩尔比﹑溶剂﹑压力等),除非另有说明,可以理解为其它方法条件也可以使用。最佳反应条件可能随所用的特定反应物或溶剂而变化,但是这些条件可由熟悉该技术领域的科技人员用常规优化程序确定。此外,显而易见,对熟悉该技术领域的科技人员,常规的保护基对避免某些官能团参与不想要的反应可能是必要的。各种官能团的合适保护基以及特定官能团保护和去保护合适条件为该技术领域的科技人员众所周知。例如由Greene,T.W.和Wuts,G.M.,《有机合成中的保护基团》(ProtectingGroupsinOrganicSynthesis),第三版,1999,Wiley,NewYork中所述的很多保护基及其中所引用的参考文献。此外,本发明的化合物可能包含一个或一个以上手性中心。因此,若需要,此类化合物可以制备或分离得到纯立体异构体,即为单个对映异构体或非对映异构体或为立体异构体富集的混合物。除非另有特别说明,所有这些立体异构体(及其富集的混合物)都包含在本发明的范围之内。纯的立体异构体(或其富集混合物)可以使用如本技术领域熟知的光学活性起始物或立体选择性试剂加以制备。或者,此类外消旋混合物也可使用如手性柱色谱,手性差分试剂等加以分离。下述反应的起始物质通常为已知化合物或者可以通过已知步骤或通过其明显的修改方案进行制备。例如,很多起始物可从供货商如AldrichChemicalCo.(Milwaukee,Wisconsin,USA)﹑Bachem(Torrance,California,USA)﹑Emka-Chemce或Sigma(St.Louis,Missouri,USA)买到。其它起始物可以通过标准参考书如FieserandFieser'sReagentsforOrganicSynthesis,1-15卷(JohnWileyandSons)(1991)﹑Rodd'sChemistryofCarbonCompounds,1-5卷及补卷(ElsevierSciencePublishers)(1989)﹑OrganicReactions,1-40卷(JohnWileyandSons)(1991)﹑March'sAdvancedOrganicChemistry,第四版(JohnWileyandSons)﹑及Larock'sComprehensiveOrganicTransformations(VCHPublishersInc.)(1989)所述的步骤或其明显的修改方案进行制备。于此所述的各种起始物﹑中间体和化合物(包括立体异构体)可以用适当的常规技术如沉淀﹑过滤﹑结晶﹑蒸发﹑蒸馏和色谱分离和纯化。这些化合物可以通过常规方法如熔点﹑质谱﹑核磁共振和各种其它光谱分析方法加以鉴定。偶合试剂包括基于碳二亚胺﹑銨盐和鏻盐试剂。碳二亚胺型试剂包括碳二亚胺如N,N'-二环己基碳二亚胺(DCC)﹑N,N'-二异丙基碳二亚胺(DIC)和1-乙基-3-(3’-二甲氨基丙基)碳二亚胺盐酸盐(EDCI)等。銨盐包括N,N,N',N'-四甲基-O-(7-氮杂苯并三氮唑-1-基)六氟磷酸脲(HATU)﹑N,N,N',N'-四甲基-O-(苯并三氮唑-1-基)六氟磷酸脲(HBTU)﹑N,N,N',N'-四甲基-O-(6-氯苯并三氮唑-1-基)六氟磷酸脲(HCTU)﹑N,N,N',N'-四甲基-O-(苯并三氮唑-1-基)四氟硼酸脲(TBTU)﹑N,N,N',N'-四甲基-O-(6-氯苯并三氮唑-1-基)四氟硼酸脲(TCTU)。鏻盐包括7-氮杂苯并三氮唑-1-基-氧基三(吡咯烷基)磷鎓六氟磷酸盐(PyAOP)和苯并三氮唑-1-基-氧基三(吡咯烷基)磷鎓六氟磷酸盐(PyBOP)。酰胺形成步骤可以在极性溶剂如二甲基甲酰胺(DMF)中进行并且也可包含有机碱如二异丙基乙胺(DIEA)或二甲氨基吡啶(DMAP)。下面实施例进一步阐述本发明的某些方面和帮助熟悉该技术领域的科技人员实行本发明。这些实施例绝不是被认为限制本发明的范围。实施例在以下实例及贯穿整个专利申请,下述缩略语意义如下。如果没有界定,这些术语具有普遍公认的意义。ACN=乙腈Ala=丙氨酸aq.=水溶液brs=宽的单峰calc.=计算值d=双峰DCM=二氯甲烷dd=双二重峰DIC=N,N'-二异丙基碳二亚胺DMA=N,N-二甲基乙酰胺DMAP=二甲基氨基吡啶DMF=二甲基甲酰胺DMSO=二甲基亚砜DTT=二硫苏糖醇EDTA=乙二胺四乙酸盐Et=乙基EtOAc=乙酸乙酯g=克h=小时HCl=氢氯酸HPLC=高效液相色谱法Hz=赫兹J=偶合常数LC-MS=液相色谱质谱法m=多重峰MDC=美登醇Me=甲基MeOH=甲醇MHz=兆赫min=分钟mL=毫升mm=毫米m.p.=熔点OTf=三氟甲磺酸盐N=标准的r.t.=室温PBS=磷酸盐缓冲液Rt=保留时间s=单峰t=三重峰TLC=薄层色谱法vol=体积μL=微升μm=微米材料和方法:核磁共振谱由BrukerAM400(400MHz)光谱仪测量。CDCl3中的化学位移(ppm)以残留的CHCl3为内标。紫外可见光谱由BeckmanDU-640分光光度计测量。质谱通过ThermoFinniganLCQDECAXP+仪使用电喷雾电离获得。HPLC使用AgilentHPLC1100系统(配置二级阵列检测器和Kromasil反相5μm,250x4.6mm碳-18柱),用乙腈:水进行梯度洗脱(50-95%乙腈0-10min,95%乙腈10-15min,流速=1.0mL/min)。快速柱层析用硅胶来自青岛海洋化工厂分厂。美登醇按以前方法(Widdison等(2006)J.Med.Chem.49:4392-4408)由安丝菌素P-3制备。安丝菌素P-3由微生物珍贵橙色速丝放线菌(Actinosynnemapretiosum)发酵所得。二氯甲烷经由氢化钙蒸馏干燥。二甲基甲酰胺经由氢化钙减压蒸馏干燥。所有其它试剂为试剂级或HPLC级。实施例1美登醇(MDC)与Fmoc-N-甲基-L-丙氨酸的酯化反应(合成Fmoc-N-Me-D/L-Ala-MDC)称取美登醇(0.600g,1.062mmol),Fmoc-N-甲基-L-丙氨酸(6.911g,21.24mmol),三氟甲磺酸钪(0.314g,0.637mmol)和DMAP(0.389g,3.186mmol)置于250毫升Schlenck瓶,氮气保护下加入二氯甲烷(100mL),在-8°C搅拌0.5小时。逐滴加入DIC(2.949g,23.37mmol),继续在-8°C搅拌反应0.5h,慢慢升温至室温,过滤回收催化剂,滤液用稀盐酸淬灭,二氯甲烷萃取,依次用饱和碳酸氢钠和饱和食盐水洗涤,无水硫酸钠干燥,旋干溶剂。柱层析(硅胶,CH2Cl2/MeOH30:1)得到非对映异构体混合物Fmoc-N-Me-D/L-Ala-MDC,白色固体(0.8385g,产率90.5%)。进一步柱层析(硅胶,CH2Cl2/MeOH100:1到20:1)得到两个纯的非对映异构体组分。Rf较大的组分确定是D-氨酰基酯非对映异构体(Fmoc-N-Me-D-Ala-MDC),Rf较小的组分确定是L-氨酰基酯非对映异构体(Fmoc-N-Me-L-Ala-MDC)。Fmoc-N-Me-L-Ala-MDC:白色固体(0.4262g,产率46.0%),1HNMR(400MHz,CDCl3):δ0.77(3H,s),1.22-1.32(6H,m),1.40-1.48(1H,m),1.63(3H,s),2.13(1H,dd,J=14.4,2.8Hz),2.53(1H,dd,J=14.4,10.8Hz),2.64(3H,s),2.88(3H,s),3.00(1H,d,J=9.6Hz),3.07(1H,d,J=12.4Hz),3.35(3H,s),3.48(1H,d,J=8.8Hz),3.59(1H,d,J=11.2Hz),3.97(3H,s),4.13-4.19(1H,m),4.15(1H,s),4.24(1H,t,J=10.8Hz),4.72-4.77(2H,m),5.03(1H,q,J=6.8Hz),5.65(1H,dd,J=15.2,9.2Hz),6.29(1H,br),6.41(1H,dd,J=15.2,11.2Hz),6.52(1H,d,J=1.2Hz),6.70(1H,d,J=10.8Hz),6.79(1H,d,J=1.2Hz),7.33(1H,t,J=7.6Hz),7.36(1H,t,J=7.6Hz),7.39(1H,d,J=7.6Hz),7.49(1H,d,J=7.6Hz),7.70(1H,d,J=7.6Hz),7.72(1H,d,J=7.6Hz)。LC-MS(M+Na+)计算值:894.3,实测值:894.3。Fmoc-N-Me-D-Ala-MDC:白色固体(0.3993g,产率43.1%),1HNMR(400MHz,CDCl3):δ0.84(3H,s),1.22-1.27(3H,m),1.40-1.48(1H,m),1.51(3H,d,J=7.6Hz),1.67(3H,s),2.20(1H,dd,J=14.4,2.8Hz),2.63(1H,dd,J=14.4,12.4Hz),2.85(1H,d,J=9.6Hz),2.96(3H,s),3.17(3H,s),3.20(1H,s),3.24(3H,s),3.40(1H,d,J=9.2Hz),3.51(1H,d,J=12.8Hz),3.99(3H,s),4.20-4.28(2H,m),4.38-4.43(2H,m),4.80-4.98(2H,m),5.80(1H,dd,J=15.2,11.2Hz),6.18(1H,s),6.25(1H,d,J=10.8Hz),6.40(1H,dd,J=15.2,11.2Hz),6.79(1H,d,J=1.6Hz),6.84(1H,d,J=1.6Hz),7.32(2H,t,J=7.6Hz),7.41(2H,t,J=7.6Hz),7.61(2H,d,J=7.6Hz),7.77(2H,d,J=7.6Hz)。LC-MS(M+Na+)计算值:894.3,实测值:894.3。实施例2去保护Fmoc-N-Me-D/L-Ala-MDC(合成N-Me-D/L-Ala-MDC)Fmoc-N-Me-D/L-Ala-MDC(0.463g,0.5307mmol)溶在乙腈(200mL),加入哌啶(0.865g,10.15mmol),室温搅拌4小时,加水淬灭,二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂,干燥得粗品。不需进一步纯化用于下步反应。LC-MS(M+H+)计算值:650.3,实测值:650.3。Rt:3.96min。实施例3去保护Fmoc-N-Me-L-Ala-MDC(合成N-Me-L-Ala-MDC)Fmoc-N-Me-L-Ala-MDC(0.463g,0.5307mmol)溶在乙腈(200mL),加入哌啶(0.865g,10.15mmol),室温搅拌4小时,加水淬灭,二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂,干燥得粗品。不需进一步纯化用于下步反应。LC-MS(M+H+)计算值:650.3,实测值:650.3。Rt:3.96min。实施例4N-Me-D/L-Ala-MDC与6-马来酰亚胺基己酸(MA-ACP)缩合(合成D-3AA-MDC和L-3AA-MDC)将上步制备的粗品N-Me-D/L-Ala-MDC(0.5307mmol)以及MA-ACP(0.448g,2.123mmol)溶于DMF(25mL),冰水浴冷却,加入EDC(0.407g,2.123mmol)。反应混合物室温搅拌过夜,加水淬灭,乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。柱层析(硅胶,CH2Cl2/MeOH30:1)得到粗品。制备HPLC(YMCC-18柱,250x20mm,S10μm)进一步纯化得纯的两个非对映异构体(Rt=6.59min和6.98min)。Rt较大的组分确定是D-氨酰基酯非对映异构体(D-3AA-MDC,45.2%),Rt较小的组分确定是L-氨酰基酯非对映异构体(L-3AA-MDC,54.8%)。L-3AA-MDC:白色固体(0.1364g,两步总产率30.5%),1HNMR(400MHz,CDCl3):δ0.79(3H,s),1.17-1.32(3H,m),1.27(3H,s),1.29(3H,s),1.40-1.76(7H,m),2.12-2.23(2H,m),2.31-2.45(1H,m),2.59(1H,t,J=12.8Hz),2.82(3H,s),3.01(1H,d,J=9.6Hz),3.10(1H,d,J=8.8Hz),3.17(3H,s),3.34(3H,s),3.42(2H,t,J=6.8Hz),3.48(2H,d,J=6.8Hz),3.62(1H,d,J=12.8Hz),3.97(3H,s),4.27(1H,t,J=11.2Hz),4.76(1H,d,J=11.6Hz),5.36(1H,q,J=6.8Hz),5.65(1H,dd,J=15.2,9.2Hz),6.25(1H,s),6.41(1H,dd,J=15.2,11.2Hz),6.64(1H,s),6.65(2H,s),6.72(1H,d,J=11.2Hz),6.82(1H,s)。LC-MS(M+Na+)计算值:865.3,实测值:865.3。Rt:6.59min。D-3AA-MDC:白色固体(0.1128g,两步总产率25.2%),1HNMR(400MHz,CDCl3):δ0.86(3H,s),1.22-1.38(4H,m),1.25(3H,d,J=9.2Hz),1.38-1.45(1H,m),1.48(3H,d,J=7.6Hz),1.56-1.70(4H,m),1.68(3H,s),1.75(1H,d,J=13.6Hz),2.19(1H,dd,J=14.4,2.8Hz),2.28-2.36(2H,m),2.65(1H,dd,J=14.2,12.0Hz),2.80(1H,d,J=9.6Hz),3.01(3H,s),3.19(1H,d,J=13.2Hz),3.32(3H,s),3.42(1H,d,J=9.6Hz),3.47-3.54(3H,m),3.98(3H,s),4.29(1H,t,J=10.4Hz),4.88(1H,dd,J=11.8,3.2Hz),5.07(1H,q,J=7.6Hz),5.84(1H,dd,J=15.2,9.2Hz),6.23(1H,d,J=11.2Hz),6.27(1H,s),6.41(1H,dd,J=15.2,11.2Hz),6.69(2H,s),6.79(1H,d,J=1.2Hz),6.84(1H,d,J=1.2Hz)。LC-MS(M+Na+)计算值:865.3,实测值:865.3。Rt:6.98min。实施例5N-Me-L-Ala-MDC与MA-ACP缩合(合成L-3AA-MDC)将上步制备的粗品N-Me-L-Ala-MDC(0.5307mmol)以及MA-ACP(0.448g,2.123mmol)溶于DMF(25mL),冰水浴冷却,加入EDC(0.407g,2.123mmol)。反应混合物室温搅拌过夜,加水淬灭,乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。柱层析(硅胶,CH2Cl2/MeOH30:1)得到预期的产品L-3AA-MDC:白色固体(0.280g,两步总产率62.6%)1HNMR(400MHz,CDCl3):δ0.79(3H,s),1.17-1.32(3H,m),1.27(3H,s),1.29(3H,s),1.40-1.76(7H,m),2.12-2.23(2H,m),2.31-2.45(1H,m),2.59(1H,t,J=12.8Hz),2.82(3H,s),3.01(1H,d,J=9.6Hz),3.10(1H,d,J=8.8Hz),3.17(3H,s),3.34(3H,s),3.42(2H,t,J=6.8Hz),3.48(2H,d,J=6.8Hz),3.62(1H,d,J=12.8Hz),3.97(3H,s),4.27(1H,t,J=11.2Hz),4.76(1H,d,J=11.6Hz),5.36(1H,q,J=6.8Hz),5.65(1H,dd,J=15.2,9.2Hz),6.25(1H,s),6.41(1H,dd,J=15.2,11.2Hz),6.64(1H,s),6.65(2H,s),6.72(1H,d,J=11.2Hz),6.82(1H,s)。LC-MS(M+Na+)计算值:865.3,实测值:865.3。Rt:6.59min。实施例6前体药物抗体美登素偶联物其代谢产物在体外对微管蛋白聚合的影响3AA-MDC、206-3AA-MDC、以及前体药物抗体美登素偶联物其代谢产物Cys-3AA-MDC和Lys-mcc-MDC体外对微观蛋白聚合的影响是使用HTS-TubulinPolymerizationAssay检测试剂盒(BK004P,美国)评价的。按照试剂盒说明书,检测用96孔板放入37℃烘箱进行预热,同时设置酶标仪(SpectraMax,美国分子仪器)的各项参数(波长:405nm;温度:37℃;每1分钟读一次,持续30分钟)。配制药物稀释缓冲液G-PEM(990μlGeneralTubulinBuffer+10μlGTPStock),并且在冰上预冷。然后用G-PEM配制4mg/mltubulin、1μM3AA-MDC(N2’-去乙酰基-N2’-(6-马来酰亚胺基-1-氧代-己基)美登素)、1μM206-3AA-MDC、1μMCys-3AA-MDC、1μMLys-MCC-MDC、100μMPaclitaxel以及100μMNocodazole。将10μlG-PEM、3AA-MDC、206-3AA-MDC、Cys-3AA-MDC、Lys-MCC-MDC、Paclitaxel以及Nocodazole加入96孔板,然后快速在每个孔内加入100μl4mg/mltubulin,并马上放入酶标仪中读数。与PBS缓冲液相比,3AA-MDC、206-3AA-MDC、Cys-3AA-MDC、Lys-MCC-MDC非常显著地抑制了微管蛋白的聚合。抑制剂Nocodazole在此作为负对照。代谢产物Cys-3AA-MDC通过3AA-MDC与半胱氨酸在碱二异丙基乙胺于二氯甲烷中反应制备。LC-MS(M+H+)计算值:964.5,实测值:964.2。Rt:12.97min。代谢产物Lys-MCC-MDC通过SMCC-MDC与赖氨酸在碱二异丙基乙胺于二甲基甲酰胺中反应制备。LC-MS(M+H+)计算值:1103.7,实测值:1103.2。Rt:13.00以及13.18min。实施例7重组抗体表达和纯化参照Woodetal.,JImmunol.145:3011(1990)等的方法,特异性的单克隆抗体在CHO细胞产生。含抗体基因的表达载体分别用常规的分子生物学方法构建。CHO-k1细胞的一种衍生细胞系作为宿主细胞。高产稳定细胞系的构建过程简单描述如下:宿主细胞悬浮生长于CD-CHO培养基,取处于对数生长期的宿主细胞离心,重悬于新鲜的CD-CHO培养基,计数并调节细胞密度到1.43×107个/毫升,取600μl上述细胞悬液加入电击杯,然后加入已线性化的质粒40μg,用移液枪吸打使细胞与质粒混合均匀。用Bio-rad电转仪电击转化,仪器参数设定为:电容:960μFD,电压:300V。通常电击时间为15-20毫秒为正常。把电击后的细胞立即重悬于37℃预热的CD-CHO培养基,每孔100μl分装于96孔板,2-3天后加入等量的筛选培养基。测定96孔板细胞培养上清来测定抗体的表达水平。表达水平较高的克隆从96孔板转移到24孔板,待细胞生长到一定数量,把细胞转入6孔板,使每孔5ml培养基含2×105个细胞,测定细胞的抗体产量和产率。通常20-30个克隆被转到摇瓶做进一步评价。最后5-8个表达量最高的克隆进行亚克隆和进一步的表达检测。收获料液,通过低速离心使细胞和培养基分离,把离心上清高速离心进一步澄清。用蛋白A亲和纯化和离子交换纯化,使用的介质分别是GE公司生产的MabSelectSuRe和CaptoS。实施例8SMCC-MDC与抗EGFR抗体的偶联抗体抗EGFR抗体(抗表皮生长因子受体的抗体)用溶液A(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为6.5)稀释至2.5mg/mL。加入SMCC-MDC,使SMCC-MDC与抗体的比率为7:1(摩尔当量)。然后,加入DMA至总体积的15%,室温搅拌4小时使反应物混匀。多余的未反应的或水解的试剂和过量的SMCC-MDC使用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱,纯化得到D-Lmcc-抗EGFR抗体。然后将偶联物用pH为7.4的磷酸盐缓冲液(水溶液)中透析过夜,然后0.22微米的过滤器过滤,保存。每个抗表皮生长因子受体的抗体最终偶联SMCC-MDC的分子的数目通过测定偶联物在252和280nm处的吸光度,这两个波长处SMCC-MDC和抗体使用公知的消光系数。美登素药物与抗体的比率为3.5:1。实施例9抗EGFR抗体与3AA-MDC的偶联抗体抗EGFR抗体(抗表皮生长因子受体的抗体)用溶液B(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为8.0)稀释至8.0mg/mL,然后用DTT(6摩尔当量)不完全还原。在37℃孵育60分钟后,用溶液A经SephadexG-25树脂洗脱交换。巯基抗体值通过测定吸光度确定,通过硫基与DTNB(5,5'-二硫代双(2-硝基苯甲酸),Aldrich公司)的反应物,然后测定412nm处的吸收值来确定硫基的浓度。偶联反应时DMA的浓度为10%。3AA-MDC(3AA-MDC)按实例4、5方法制备。3AA-MDC与巯基数的比率为1.5:1.0(摩尔当量)。将3AA-MDC加入已还原的抗体中,室温搅拌3小时后,加入5mM半胱氨酸继续搅拌1小时。反应混合物经超滤后,用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱纯化。然后用0.22微米的滤器过滤,-80°C保存。batansine-0206抗体用DTNB测定未反应的巯基数可得到药物/抗体比率,通常,该方法可获得的药物/抗体比率为3.5:1.0。batansine-0206抗体可以通过紫外吸收测得浓度,通过尺寸排阻色谱测定聚集率,通过反向高效液相色谱法测定残留的游离药物。本发明所用的所有单克隆抗体和ADCs均为超过98%的单体蛋白。实施例10batansine-0206抗体偶联物的特性我们使用EGFR阳性乳腺肿瘤细胞系MDA-MB-468和EGFR阴性细胞系BT474(购自中国科学院上海生命科学研究院细胞资源中心)评价了batansine-0206抗体对肿瘤细胞的生长抑制。简言之,MDA-MB-468和BT474用0.25%(体积/体积)的胰蛋白酶消化,使细胞剥离,然后悬浮于100μL完全培养基,10,000个细胞接种于96孔板进行培养。37℃过夜贴壁生长,然后加入100μL含有不同浓度的抗EGFR抗体Bat0206和batansine-0206抗体的培养基。72小时后,用PBS(pH7.5)洗板两次,用细胞计数试剂盒-8(CCK-8,日本同仁化学)试剂进行相对细胞增殖分析。batansine-0206抗EGFR抗体和已经上市的抗体药Cetuximab能更为有效的抑制细胞的增殖(图7)。而无论是裸抗抗EGFR抗体、Cetuximab、还是抗体药物偶联物batansine-0206抗体对EGFR表达阴性的BT474细胞没有生长抑制作用(图8)。实施例11抗体药物偶联物:D-Lmcc-Bat0206抗体的制备抗体抗EGFR抗体bat0206用溶液A(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为6.5)稀释至2.5mg/mL。加入SMCC-MDC,使SMCC-MDC与抗体的比率为7:1(摩尔当量)。然后,加入DMA至15%,室温搅拌4小时使反应物混匀。多余的未反应的或水解的试剂和过量的SMCC-MDC使用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱,纯化得到D-Lmcc-抗EGFR抗体。下一步是将偶联物在pH为7.4的磷酸盐缓冲液中透析过夜,然后0.22微米的过滤器过滤,保存。利用SMCC-MDC和抗体在两个不同波长的消光系数,通过测定偶联物在252和280nm处的吸光度来测定每个抗表皮生长因子受体的抗体最终偶联SMCC-MDC的分子的数目。通常得到的抗体与美登素药物的比率为1比3-5。D-Lmcc-Bat0206对肿瘤细胞生长的抑制作用是通过使用EGFR阳性乳腺肿瘤细胞系MDA-MB-468和EGFR阴性细胞系BT474来评价的。简言之,MDA-MB-468和BT474用0.25%(体积/体积)的胰蛋白酶消化,使细胞剥离,然后悬浮于100μL完全培养基,10,000个细胞接种于96孔板。37℃过夜贴壁生长,然后加入100μL含有不同浓度的Bat0206和D-Lmcc-Bat0206的培养基。72小时后,用PBS(pH7.5)洗板两次,用细胞计数试剂盒-8(CCK-8)试剂进行相对细胞增殖分析。药物偶联物D-Lmcc-Bat0206与非偶联的Bat0206相比,能更为有效的抑制表皮生长因子受体(EGFR)阳性细胞的增殖(图9)。无论是未偶联的抗体抗EGFR抗体Bat0206或者西妥昔单抗,还是药物偶联的D-Lmcc-Bat0206对表皮生长因子受体阴性细胞系BT474的生长均无抑制作用(图10)。实施例12D-Lmcc-Bat0206抗体抗肿瘤特性D-Lmcc-Bat0206的抗肿瘤特性是通过对人A431肿瘤细胞在裸鼠体内的生长抑制效果来评价的。简单描述如下:A431细胞(ATCC,CRL-7907)生长于含10%的胎牛血清和添有2mM谷氨酰胺的DMEM培养基。收集A431细胞重悬于PBS,使100μl体积含有4x106细胞,取4-6周龄的雌性裸鼠,在其右腋注射100μl上述细胞悬液。当肿瘤体积达到80-200mm3时开始分组给药,每组12只。抗EGFR抗体Bat0206和D-Lmcc-Bat0206抗体按每公斤体重5mg抗体量的剂量每三天一次总共给药8次,给药方式是腹腔注射,注射体积为每次100μl。肿瘤体积每三天测定一次。最后一次给药并测定肿瘤体积大小后(第一次给药24天后),动物被立即以无痛至死,肿瘤被剥离并称重。如图11和12所示,在5mg/kg剂量下,Bat0206和D-Lmcc-Bat0206都显著抑制了肿瘤细胞的生长。但与未偶联的Bat0206相比,D-Lmcc-Bat0206抑制肿瘤细胞生长的效果更明显。实施例13抗体药物偶联物:D-Lspp-Bat0206的制备抗体抗EGFR抗体(抗表皮生长因子受体的抗体)用溶液A(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为6.5)稀释至8mg/mL。加入N-琥珀酰亚胺基-4-(2-吡啶基二硫代)戊酸酯(SPP),SPP与抗体的比率为8:1(摩尔当量)。反应在5%DMA(v/v)中室温搅拌2小时使反应物混匀,反应液呈白色悬浊。使用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的脱盐柱纯化抗体。抗体的巯基化程度通过测定加入DTT前后,抗体在280nm和343nm吸光值的变化来测定。加入1.7倍摩尔量的N2'-脱乙酰基-N2'-(3-巯基-1-氧代丙基)-美登素,调整巯基化的Bat0206浓度到2.5mg/mL。反应在5%DMA(v/v)中室温搅拌17小时。偶联物通过离心澄清,使用SephadexG25在pH6.5的磷酸盐缓冲液(水溶液)平衡的脱盐柱纯化抗体。纯化的抗体经0.22μM滤膜除菌(Millex-GV)。通过测定280nm和252nm的吸光值,每个抗EGFR抗体分子偶联药物的数大约为4.5。分子筛测定抗体偶联物单体含量超过96%。D-Lspp-Bat0206和Bat0206对肿瘤细胞的生长抑制特性是使用EGFR阳性的A431和带有KRAS突变的A549细胞来评价的。简言之,A431和A549细胞用0.25%(体积/体积)的胰蛋白酶消化,使细胞剥离,然后重悬于完全培养基,2000/孔的A431细胞和1500/孔的A549细胞分别接种于96孔板,每孔100μL培养基,37℃过夜贴壁生长,然后加入100μL含有不同浓度的抗Bat0206和D-Lspp-Bat0206的培养基。72小时后,用PBS(pH7.5)洗板两次,用细胞计数试剂盒-8(CCK-8)试剂进行相对细胞增殖分析。药物偶联物D-Lspp-Bat0206比Bat0206更加显着地抑制EGFR阳性细胞A431的增殖(图13)。此外,D-Lspp-Bat0206抗体也抑制k-ras突变的EGFR阳性的A549细胞,而未偶联的抗EGFR抗体对A549细胞没有抑制作用(图14)。应用了几种不同的细胞来验证抗EGFR抗体药物偶联体的细胞杀伤功能,惊奇的发现,这些美登素化合物以不同的连接体通过不同的偶联方法偶联在同一抗体的不同部位,这些抗EGFR抗体药物偶联体对不同的肿瘤细胞有不同的选择性和敏感性。例如,3AA-MDC-抗EGFR抗体对A431细胞比D-Lmcc-抗EGFR抗体提高100倍以上。而在HCT116细胞,D-Lspp-抗EGFR抗体比D-Lmcc-抗EGFR抗体提高50倍以上。实施例14Batansine-0606抗体抗乳腺癌细胞系BT474特性Batansine-0606的抗乳腺癌特性是通过对Her2表达阳性的人BT474肿瘤细胞在裸鼠体内的生长抑制效果来评价的。简单描述如下:BT474细胞(ATCC,HTB-20)生长于含10%的胎牛血清和添有2mM谷氨酰胺的RPMI-1640培养基。收集BT474细胞重悬于PBS,使100μl体积含有5×107细胞,取8-9周龄的雌性裸鼠,在其右腋注射200μl上述细胞悬液。当肿瘤体积达到150-200mm3时开始分组给药,每组10只。Batansine-0606和BAT0606分别按每公斤体重5、15mg抗体量的剂量每三周一次总共给药3次,对照抗体(利妥昔单抗)按每公斤体重15mg抗体量的剂量每三周一次总共给药3次。给药方式是静脉注射,注射体积为每次100μl。肿瘤体积每周测定两次。最后一次给药继续观察一周并测定肿瘤体积大小后(第一次给药49天后),动物被立即以无痛至死。在同等剂量下,Batansine-0606比BAT0606更能显著抑制肿瘤细胞的生长。至第10天,Batansine-060615mg/kg剂量下已检测不到肿瘤。实施例15SMCC-MDC与抗HER2抗体Bat0606的偶联Bat0606用溶液A(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为6.5)稀释至2.5mg/mL。加入SMCC-MDC,使SMCC-MDC与抗体的比率为7:1(摩尔当量)。然后,加入DMA至总体积的15%,室温搅拌4小时使反应物混匀。多余的未反应的或水解的试剂和过量的SMCC-MDC使用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱,纯化得到D-Lmcc-Bat0606。然后将偶联物用pH为7.4的磷酸盐缓冲液(水溶液)中透析过夜,然后0.22微米的过滤器过滤,保存。每个Bat0606的抗体最终偶联SMCC-MDC的分子的数目通过测定偶联物在252和280nm处的吸光度,这两个波长处SMCC-MDC和抗体使用公知的消光系数。美登素药物与抗体的比率为3.5:1。实施例16Bat0606与Batansine的偶联Bat0606用溶液B(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为8.0)稀释至8.0mg/mL,然后用DTT(6摩尔当量)不完全还原。在37℃孵育60分钟后,用溶液A经SephadexG-25树脂洗脱交换。巯基抗体值通过测定吸光度确定,通过硫基与DTNB(5,5'-二硫代双(2-硝基苯甲酸),Aldrich公司)的反应物,然后测定412nm处的吸收值来确定硫基的浓度。偶联反应时DMA的浓度为10%。Batansine按实例4、5方法制备。Batansine与巯基数的比率为1.5:1.0(摩尔当量)。将Batansine加入已还原的抗体中,室温搅拌3小时后,加入5mM半胱氨酸继续搅拌1小时。反应混合物经超滤后,用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱纯化。然后用0.22微米的滤器过滤,-80°C保存。Batansine-0606用DTNB测定未反应的巯基数可得到药物/抗体比率,通常,该方法可获得的药物/抗体比率为3.5:1.0。Batansine-0606可以通过紫外吸收测得浓度,通过尺寸排阻色谱测定聚集率,通过反向高效液相色谱法测定残留的游离药物。本发明所用的所有单克隆抗体和ADCs均为超过98%的单体蛋白。实施例17Batansine-0606的特征研究我们使用HER2阳性乳腺肿瘤细胞系SK-BR-3和HER2阴性细胞系A549(购自中国科学院上海生命科学研究院细胞资源中心)评价了Batansine-0606对肿瘤细胞的生长抑制。简言之,SK-BR-3和A549用0.25%(体积/体积)的胰蛋白酶消化,使细胞剥离,然后悬浮于100μL完全培养基,10,000个细胞接种于96孔板进行培养。37℃过夜贴壁生长,然后加入100μL含有不同浓度的Bat0606和Batansine-0606的培养基。72小时后,用PBS(pH7.5)洗板两次,用细胞计数试剂盒-8(CCK-8,日本同仁化学)试剂进行相对细胞增殖分析。Batansine-0606比抗Bat0606能更为有效的抑制HER2阳性细胞系SK-BR-3的增殖(图20)。而无论是裸抗Bat0606、还是抗体药物偶联物Batansine-0606对HER2表达阴性的A549细胞没有生长抑制作用(图21).实施例18D-Lmcc-Bat0606的功能D-Lmcc-Bat0606对肿瘤细胞生长的抑制作用是通过使用HER2阳性乳腺肿瘤细胞系SK-BR-3和HER2阴性细胞系A549来评价的。简言之,SK-BR-3和A549用0.25%(体积/体积)的胰蛋白酶消化,使细胞剥离,然后悬浮于100μL完全培养基,10,000个细胞接种于96孔板。37℃过夜贴壁生长,然后加入100μL含有不同浓度的Bat060和D-Lmcc-Bat0606的培养基。72小时后,用PBS(pH7.5)洗板两次,用细胞计数试剂盒-8(CCK-8)试剂进行相对细胞增殖分析。药物偶联物D-Lmcc-Bat0606与抗Bat0606相比,更为有效的抑制HER2阳性细胞系SK-BR-3的增殖(图22)。无论是未偶联的Bat0606,还是药物偶联的D-Lmcc-Bat0606抗体对HER2阴性细胞系A549的生长均无抑制作用(图23)。实施例19大鼠中血清稳定性通过Sprague-Dawley大鼠对Batansine-0606的稳定性进行研究。于第0天,通过一侧尾静脉为动物施用单次10mg/kgBatansine-0606。在给药后0(给药前)、10和30分钟;1、2、4、8、24和36小时;和2、3、4、7、14、21、28天的每个时间点收集大约200μL全血,离心(14,000×g,3分钟)后分离血清。用ELISA方法分别测定血清中抗HER2抗体以及Batansine-0606的浓度。血清中抗HER2抗体测定方法如下:将HER2ECD蛋白用包被液(碳酸钠/碳酸氢钠缓冲液,pH9.6)稀释到一定浓度(2μg/ml)后包被于96孔板上,于4℃放置过夜。弃去孔中液体,甩干后加入封闭液(PBS,1%BSA,0.05%Tween20),室温放置1小时,再用PBST(PBS,0.05%Tween20)清洗,甩干。用稀释液PBS稀释标准品至一定浓度,表达上清根据情况适当稀释,分别加样至96孔板中,37℃孵育2小时后,PBST清洗,甩干。用稀释液PBS稀释酶联抗体(HRP-鼠抗人IgG特异性抗体)至适当浓度,加至96孔板中,37℃下反应2小时,弃液,PBST清洗,甩干。每孔加入四甲基联苯胺显色剂,37℃孵育10min。每孔加入1N硫酸终止液,终止反应。在490nm处读取吸收值OD。血清中Batansine-0606测定方法如下:HER2ECD蛋白包被以及标准品、血清样品的处理如上所述。样品孵育2小时后,PBST清洗,甩干。每孔加入兔抗maytansine抗体,37℃下反应1小时,弃液,PBST清洗,甩干。用稀释液PBS稀释酶联抗体(HRP-羊抗鼠IgG特异性抗体)至适当浓度,加至96孔板中,37℃下反应1小时。显色反应及吸收值OD测定如上所述。用WinNonlin软件的非房室模型模块分析了药物代谢的动力学参数。Batansine-0606的抗原结合部分的半衰期为571.07小时,Batansine-0606抗体偶联的药物部分半衰期为88.36小时。实施例20Batansine-0606抗胃癌特性Batansine-0606的抗胃癌特性是通过对Her2表达阳性的人NCI-N87肿瘤细胞在裸鼠体内的生长抑制效果来评价的。简单描述如下:NCI-N87细胞(ATCC,CRL-5822)生长于含10%的胎牛血清和添有2mM谷氨酰胺的RPMI-1640培养基。收集NCI-N87细胞重悬于PBS,使100μl体积含有5×106细胞,取4-8周龄的雌性裸鼠,在其右腋注射200μl上述细胞悬液。当肿瘤体积达到100-200mm3时开始分组给药,每组10只。Batansine-0606分别按每公斤体重5mg或15mg抗体量的剂量每周一次总共给药5次。把头分别按每公斤体重5mg或15mg抗体量的剂量每周一次总共给药3次,然后每周1次用Batansine-0606分别按每公斤体重5mg或15mg抗体量的剂量给药两次。对照抗体(利妥昔单抗)按每公斤体重15mg抗体量的剂量每周一次总共给药5次。给药方式是静脉注射,注射体积为每次100μl。肿瘤体积每周测定两次。最后一次给药继续观察一周并测定肿瘤体积大小后(第一次给药35天后),动物被立即以无痛至死。Batansine-06065mg/kg或15mg/kg,从第11天开始,该组动物肿瘤体积快速减小。至第24天,Batansine-060615mg/kg已检测不到肿瘤。与未偶联的抗体BAT0606相比,Batansine-0606显著抑制了肿瘤细胞的生长。实施例21SMCC-MDC与抗CD20抗体Bat1206的偶联Bat1206抗体用溶液A(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为6.5)稀释至2.5mg/mL。加入SMCC-MDC,使SMCC-MDC与抗体的比率为7:1(摩尔当量)。然后,加入DMA至总体积的15%,室温搅拌4小时使反应物混匀。多余的未反应的或水解的试剂和过量的SMCC-MDC使用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱,纯化得到D-Lmcc-Bat1206。然后将偶联物用pH为7.4的磷酸盐缓冲液(水溶液)中透析过夜,然后0.22微米的过滤器过滤,保存。每个抗CD20的抗体最终共轭SMCC-MDC的分子的数目通过测定偶联物在252和280nm处的吸光度,这两个波长处SMCC-MDC和抗体使用公知的消光系数。美登素药物与抗体的比率为3.5:1。实施例22Bat1206与Batanine的偶联Bat1206抗体用溶液B(50mM磷酸钾,50mMNaCl和2mMEDTA,pH为8.0)稀释至8.0mg/mL,然后用DTT(6摩尔当量)不完全还原。在37℃孵育60分钟后,用溶液A经SephadexG-25树脂洗脱交换。巯基抗体值通过测定吸光度确定,通过硫基与DTNB(5,5'-二硫代双(2-硝基苯甲酸),Aldrich公司)的反应物,然后测定412nm处的吸收值来确定硫基的浓度。偶联反应时DMA的浓度为10%。Batanine按实例4、5方法制备。Batanine与巯基数的比率为1.5:1.0(摩尔当量)。将Batanine加入已还原的抗体中,室温搅拌3小时后,加入5mM半胱氨酸继续搅拌1小时。反应混合物经超滤后,用G25在pH值7.4的磷酸盐缓冲液(水溶液)平衡的凝胶过滤柱纯化。然后用0.22微米的滤器过滤,-80°C保存。Batanine-1206用DTNB测定未反应的巯基数可得到药物/抗体比率,通常,该方法可获得的药物/抗体比率为3.5:1.0。Batanine-1206可以通过紫外吸收测得浓度,通过尺寸排阻色谱测定聚集率,通过反向高效液相色谱法测定残留的游离药物。本发明所用的所有单克隆抗体和ADC均为超过98%的单体蛋白。实施例23使用CD20阳性Raji细胞系评价Batanine-1206抗体偶联物的生长抑制特性。简言之,10,000/孔的Raji细胞接种于96孔板,每孔100μL培养基,37℃过夜生长,然后加入100μL含有不同浓度的抗Bat1206抗体和Batanine-1206偶联物的培养基。72小时后,用细胞计数试剂盒-8(CCK-8)试剂进行相对细胞增殖分析。如图25所示,Batanine-1206比未偶联的抗Bat1206更有效的抑制了Raji细胞的生长。实施例24药物与抗体的偶联物作为前体药物在细胞内代谢释放有效化合物的检测3AA-MDC与抗体的偶联物在细胞内代谢产物的分析参照Erickson等在CancerRes66:4426-4433(2006)的方法。简单描述如下:离心收集A431、BT474、Raji等细胞,分别重悬于含有10-7mol/L抗体药物偶联物的3ml培养基。置于37℃培养箱3-30小时。通过离心(2,000g,5分钟)使细胞和培养基分离。弃去上清,细胞重悬于3mLHBBS缓冲液,向细胞悬液中加入0.6ml丙酮后,样品放于-80℃至少1小时。离心,小心取上清,弃去管底的蛋白沉淀。向上清中加入少许醋酸(5%v/v),并把酸化了的样品冷冻干燥。把干燥好的样品溶解于0.12ml的含有0.025%的三氟乙酸的乙腈(20%v/v)中。取该溶液0.1ml上样进行LC-MS分析。Batansine-0206的代谢产物:LC-MS(M+H+)计算值:964.5,实测值:964.2。D-Lmcc-Bat0206的代谢产物:LC-MS(M+H+)计算值:1103.7,实测值:1103.3。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1