D‑π‑A型二吡咯荧光染料及其合成方法与流程
本发明公开了一种同时包含电子给体-π-电子受体(d-π-a型)的乙烯基桥连的二聚吡咯类化合物及其合成方法,该类化合物中间体可以广泛应用在环境,分析,材料科学等领域。
背景技术:
在广泛的有机π体系中,电子给体-π-电子受体(d-π-a)型有机化合物由于能带隙较低,具有各种优异的光电性质,近年来已引起人们越来越大的兴趣。这类材料另外一个有吸引力的优点就是可以通过选择合适的荧光团、电子给体和电子受体对其π共轭、能带隙和光电性质进行有效的调控。到目前为止,d-π-a型的染料已被开发并应用在各个领域。现有的合成乙烯基桥连的化合物大致有如下的方法:
1)以2,3,3-三甲基吲哚和不同取代位置的吲哚-3-甲醛为原料,构建乙烯基桥连类化合物的基本骨架。
反应过于复杂,步骤繁琐,时间较长。
2)甲酰醛和溴化苯甲酰溴通过wittig反应,制备一种乙烯基桥连的化合物。
由于wittig反应有一定的局限性,一般只能和醛反应,不与酮反应,因此不利于化合物的底物扩展。
3)原料2-溴苯甲缩醛用叔丁基锂处理得到中间体,然后在脱乙酰化得到化合物3,最后缩合得到目标产物乙烯基桥联化合物。
反应过程较复杂,使用的试剂及反应条件比较苛刻,生产成本较高。
可见,上述的方法普遍的存在一些缺点:反应时间长,条件的苛刻,产率不高,底物不易扩展。
技术实现要素:
本发明针对现有技术的不足之处,提出了一种新的合成乙烯基桥连的二吡咯化合物,提高了产率,降低了生产成本,减少了后处理的难度。
本发明的主要目的在于提供一种d-π-a型乙烯基桥连的二吡咯化合物及其合成方法。
本发明的技术方案如下:
一种d-π-a型乙烯基桥连的二吡咯化合物,所述化合物的化学结构式为:
其中,取代基r1为烷氧基、氨基、羟基供电子基团,r2为酰基、醛基、羧基、酰氨基、磺酸基、腈基、硝基、卤仿基、季胺基吸电子基团。
合成所述的d-π-a型乙烯基桥连的二吡咯化合物的合成方法,所述方法包括以下合成路径:
所述方法包括以下步骤:
1)在室温下向反应瓶中加入化合物1,甲苯,化合物2,有机酸,边搅拌边缓慢滴加有机碱,升温回流,纯化后得到固体化合物;
2)所述化合物1为2,3,3-三甲基吲哚衍生物,所述化合物2为2-甲酰基吡咯衍生物;将所述步骤中的固体化合物纯化得到黄色固体化合物3;
所述步骤1)中化合物1与化合物2的投料比为1:1-100。改变投料比,化合物3的收率有很大提高。
所述步骤1)有机酸为冰乙酸,所述步骤1)有机碱为哌啶。若为其他有机酸或者有机碱,反应不发生或者收率很低。
所述步骤1)投料的顺序为化合物1,化合物2,甲苯,有机酸,有机碱。如果其他的加料顺序会使反应体系升温剧烈,不易于反应底物的拓展。
所述步骤1)回流条件为升温至120℃,回流2-18小时。达到回流温度,反应才能顺利进行,否则反应不充分,收率低,反应时间长。
本发明有益效果如下:
1)通过分别带有供电子基团的原料和拉电子基团的原料进行knoevenagel缩合反应,合成了重要的中间体3。这类染料在给体单元和受体单元之间乙烯基作为桥连中心,增加了分子的共轭性,有利于分子内的电荷传输,使其有很好的光物理性能。
2)本发明的合成反应条件易于控制,生产成本低,简化了反应流程,节约了生产时间,产物纯化较为简单,具有普遍的适用性。
附图说明
图1是化合物3的氢谱。
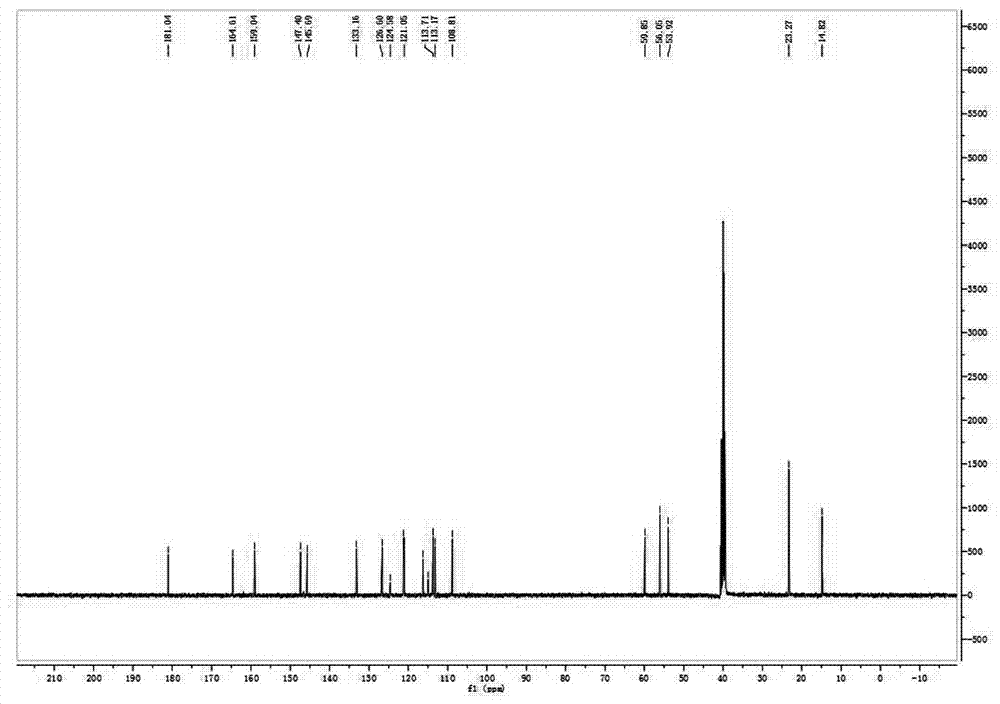
图2是化合物3的碳谱。
图3是化合物3的高分辨质谱。
具体实施方式
下面结合实施例来进一步说明本发明,但本发明要求保护的范围并不局限于实施例表述的范围。
实施例1
取2,3,3-三甲基-6-甲氧基吲哚1(1.89g,9.00mmol),称取2-甲酰基吡咯(1.72g,18.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶0.30ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(949.60mg),产率:31%。
实施例2
取2,3,3-三甲基-6-甲氧基吲哚1(18.00mmol),称取2-甲酰基吡咯(18.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶0.30ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(919.60mg),产率:35%。
实施例3
取2,3,3-三甲基-6-甲氧基吲哚1(9.00mmol),称取2-甲酰基吡咯(36.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶0.30ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(900.60mg),产率:42%:
实施例4
取2,3,3-三甲基-6-甲氧基吲哚1(9.00mmol),称取2-甲酰基吡咯(9.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶0.30ml混合后加热搅拌,于120℃回流8小时,旋干后过滤纯化得到黄色的化合物3(920.60mg),产率:30.5%:
实施例5
取2,3,3-三甲基-6-甲氧基吲哚1(9.00mmol),称取2-甲酰基吡咯(3.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶0.30ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(101.48mg),产率:15.8%:
实施例6
取2,3,3-三甲基-6-甲氧基吲哚1(9.00mmol),称取2-甲酰基吡咯(9.00mmol),量取甲苯溶液0.15l,取冰乙酸2.40ml,哌啶0.30ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(852.16mg),产率:28.5%:
实施例7
取2,3,3-三甲基-6-甲氧基吲哚1(9.00mmol),称取2-甲酰基吡咯(18.00mmol),量取甲苯溶液0.15l,取冰乙酸0.24ml,哌啶3.00ml混合后加热搅拌,于120℃回流2小时,旋干后过滤纯化得到黄色的化合物3(882.59mg),产率:29.3%:
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!