铂配合物及发光器件的制作方法
本发明涉及有机发光材料
技术领域:
,尤其涉及一种铂配合物及发光器件。
背景技术:
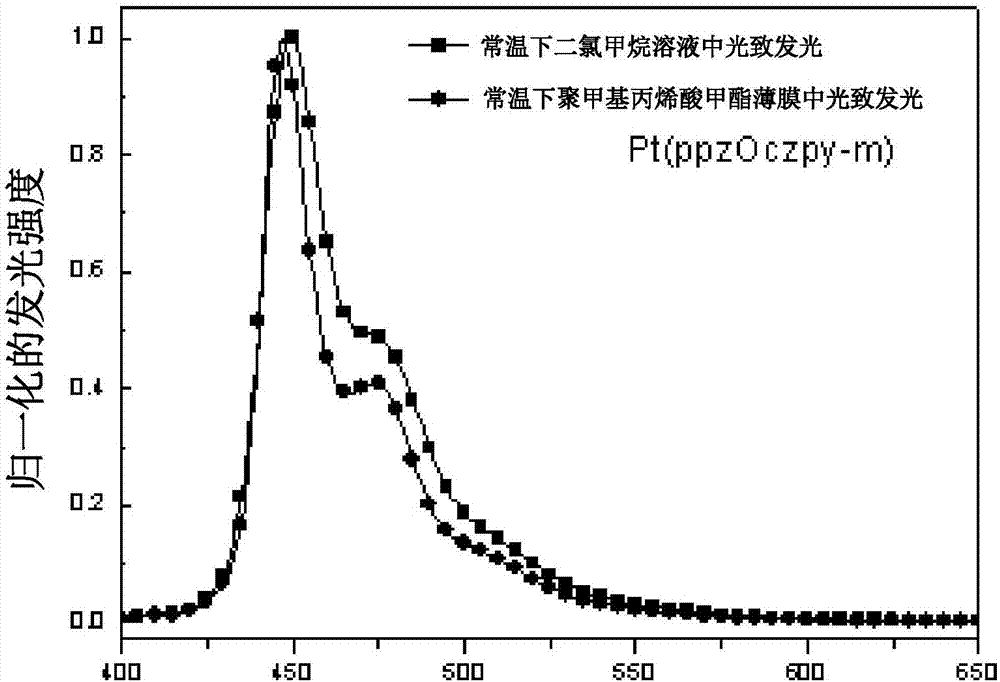
:有机电致发光是指有机材料在电场激发作用下发光的现象,可将电能直接转化为光能。有机发光二极管(OrganicLight-EmittingDiodes,OLEDs)则指利用有机发光材料制成的二极管,又被称为有机电致发光器件(OrganicElectroluminescentDevices)。OLEDs是全固态的自发光,具有宽视角、可弯曲、轻薄以及发光效率和分辨率高,驱动电压低,响应速度快,成本低、生产工艺简单、可进行大面积生产等优点,在显示和照明领域有着广阔而巨大的应用前景。有机电致发光现象和技术可以追溯到20世纪50年代。1953年,Bernanose第一次报道了有机电致发光现象(J.Chem.Phys.1953,50:64)。1963年Pope报道了蒽单晶片在400V电压的作用下的发光现象(J.Chem.Phys.1963,38(8),2042),因驱动电压太高,发光亮度低,并没有引起科学界的广泛关注。直到1987年,美国柯达公司的C.W.Tang等人以Alq3作为电子传输层和发光层成功制成了低电压、高亮度、高发光效率的双层OLEDs器件(Appl.Phys.Lett.1987,51(12),913)。自此,有机电致发光领域得到了迅速的发展。1998年,S.R.Forrest和M.E.Thompson等人报道了磷光电致发光现象,他们使用主客体掺杂的方式,通过重原子旋轨耦合作用使材料三线态激子辐射跃迁允许,同时利用单线态和三线态激子,突破了荧光有机发光材料内量子效率最高25%的限制,使内量子效率在理论上达到100%,该工作开创了磷光电致发光领域(Nature,1998,395,151-154)。磷光有机发光二极管的概念提出之后,OLEDs的效率有了极大的提高。红光和绿光的效率和寿命都已经达到了可产业化的阶段,但是蓝光的效率和寿命一直是OLEDs产业发展的短板,优良的蓝色发射体尤其稀少。因此,蓝光磷光材料一直是OLEDs研究的重点和难点之一。2010年ThomasStrassner报道了发蓝绿光的双齿配体Pt配合物Pt(2,3-OC6H4pmi)(acac),其具有非常好的发光效率(Angew.Chem.Int.Ed.2010,49,10214–10216)。2013年,JianLi组报道了一组高效发光的深蓝光四齿铂配合物(Angew.Chem.Int.Ed.2013,52,6753-6756)。此外,其课题组还发现在吡啶4位引入叔丁基后整个分子的发色光谱明显变窄,其中蓝光分子PtON7-t-Bu最高外量子效率17.6%,光色指数CIE(0.14,0.09)(Adv.Mater.2014,26,7116–7121)。研究发现,PtON1基础上进行取代基修饰可以改变发射光谱的性质(US9673409B2&Inorg.Chem.2017,56,8244.8256),光谱曾现各种型貌。我们发现在络合物分子中的特定位置引入烷基后,可以得到高效的深蓝光磷光材料。因此,有必要提供一种新型的铂配合物及发光器件以解决上述问题。技术实现要素:本发明的目的在于提供一种铂配合物,其分子刚性强,可用作制造蓝光磷光材料。本发明的技术方案如下:一种铂配合物,通式(Ⅰ)为:其中,Ra、Rb、Rc至少一个表示碳数为C1-C6的烷基取代基。优选的,所述铂配合物具有通式(II)所示的结构:其中,Ra、Rb、Rc各自独立的表示氢原子、氘原子、碳数为C1-C6的烷基取代基或芳基取代基;Rd、Re各自独立的表示可以存在或者不存在的取代基,存在时表示一个或者多个氢原子或碳数为C1-C6的烷基取代基和芳基取代基;当Re表示一个或者两个烷基存在时,则Ra和Rd分别存在,其中Ra表示碳数为C1-C6的烷基取代基或芳基取代基;Rd可以存在或者不存在,存在时表示一个或者多个氢原子或碳数为C1-C6的烷基取代基和芳基取代基;当Re不存在时,Ra、Rb、Rc至少有一个表示碳数为C1-C6的烷基取代基或芳基取代基,Rd可以存在或者不存在。优选的,所述铂配合物选自以下结构式所示的化合物:优选的,所述铂配合物应用于纯蓝光发光材料。优选的,所述铂配合物用于制作电致发光材料。优选的,所述铂配合物作为电致发光材料应用于光电电器中。优选的,所述铂配合物用于改善蓝光色纯度器件。本发明还提供了一种发光器件,所述发光器件包括阳极、阴极以及设置于所述阳极和所述阴极之间的至少一个有机层,所述有机层包括所述的铂配合物。与相关技术相比,本发明提供的铂配合物及发光器件具有如下有益效果:分子刚性强,可以有效减少由于分子振动所消耗的能量,磷光发光强度高;产物可作为一种有机磷光发光材料;对比叔丁基,甲基取代的金属铂配合物合成更容易,成本更低。通过在配体的咔唑吡啶上位引入甲基,能明显窄化发光光谱,大部分光谱位于深蓝光区域,为窄蓝光磷光材料的开发提供了一种途径,具有重要意义。【附图说明】为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图,其中:图1为分子改进前后三线态分布和发光光谱对比图;图2为Pt(ppzOczpy-m)在室温下二氯甲烷溶液和5%PMMA(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜中的发光光谱图;图3为Pt(ppzOczpy-2m)在室温下二氯甲烷溶液和5%PMMA(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜中的发光光谱图;图4为Pt(ppzOczpy-2m′)在室温下二氯甲烷溶液和5%PMMA(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜中的发光光谱图;图5为Pt(ppzOczpy-3m)在室温下二氯甲烷溶液和5%PMMA(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜中的发光光谱图;图6为Pt(ppzOczpy-4m)在室温下二氯甲烷溶液和5%PMMA(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜中的发光光谱图;图7为OLED器件结构示意图;图8为Pt(ppzOczpy-m)的器件的电致发光光谱图。【具体实施方式】下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。请参阅图1,本发明实施例提供了一种铂配合物,通式(Ⅰ)为:其中,Ra、Rb、Rc至少一个表示碳数为C1-C6的烷基取代基。优选的,所述铂配合物具有通式(II)所示的结构:其中,Ra、Rb和Rc中的每一种都是要么独立存在的或不存在的,如果是存在的,那么Ra-Rc中的每一种分别代表单取代,两种表示双基取代,也可以是三基取代,并且其中Ra-Rc中的每一种是独立的氢、氘和烷基取代或未取代的。Rd和Re为独立存在的或不存在的,当Re存在时其为各种稳定的取代基,可以是烷基和苯基,或者烷基取代的苯基。Ra为烷基取代基时,Re和Rd同时存在或者Re不存在。具体来说,Ra、Rb、Rc各自独立的表示氢原子、氘原子、碳数为C1-C6的烷基取代基或芳基取代基。Rd、Re各自独立的表示可以存在或者不存在的取代基,存在时表示一个或者多个氢原子或碳数为C1-C6的烷基取代基和芳基取代基。当Re表示一个或者两个烷基存在时,则Ra和Rd分别存在,其中Ra表示碳数为C1-C6的烷基取代基或芳基取代基;Rd可以存在或者不存在,存在时表示一个或者多个氢原子或碳数为C1-C6的烷基取代基和芳基取代基;当Re不存在时,Ra、Rb、Rc至少有一个表示碳数为C1-C6的烷基取代基或芳基取代基,Rd可以存在或者不存在。如图1所示,通过计算方法对三线态轨道分布进行比较,发现通过在吡啶单元上增加一个甲基的烷基取代基可以大幅度增加咔唑上的三线态激发态的电子分布,从而增加以咔唑为中心的辐射发光的比例,达到控制分子激发态振动的目的,从而得到半峰宽较窄的磷光蓝光发光材料。在CDCl3或DMSO-d6溶液中,通过Varian液态核磁共振仪记录1HNMR(氢核磁共振)和13CNMR(碳核磁共振)光谱为300、400或500MHz,而化学位移是以残余的质子化溶剂为基准。如果使用CDCl3作为溶剂,则采用四甲基硅烷(δ=0.00ppm)作为内参比来记录1HNMR(氢核磁共振)光谱;采用CDCl3(δ=77.00ppm)作为内参比来记录13CNMR(碳核磁共振)光谱。如果使用DMSO-d6作为溶剂,则采用残余的H2O(δ=3.33ppm)作为内参比来记录1HNMR(氢核磁共振)光谱;采用DMSO-d6(δ=39.52ppm)作为内参比来记录13CNMR(碳核磁共振)光谱。以下缩写词(或其组合)是用于解说1HNMR(氢核磁共振)的多样性:s=单线态,d=双线态,t=三线态,q=四线态,p=五线态,m=多线态,br=宽。实施例1铂络合物Pt(ppzOczpy-m)是按照以下方案制备:2-溴-9-(4-甲基-2-吡啶基)-咔唑的合成:向带有磁力转子的48mL封管中依次添加2-溴咔唑(246mg,1mmol),2-溴-4甲基吡啶(516mg,3mmol),碘化亚铜(29mg,0.15mmol),1-甲基咪唑(41mg,0.5mmol),碳酸钾(276mg,2mmol)和甲苯(5mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌3天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=25:1,得到白色固体312mg,收率93%。1HNMR(300MHz,CDCl3):δ8.59(d,J=5.1Hz,1H),8.08(d,J=7.8Hz,1H),8.01–7.92(m,2H),7.73(d,J=8.4Hz,1H),7.51–7.38(m,3H),7.37–7.28(m,1H),7.17(d,J=5.1Hz,1H),2.51(s,3H)。铂络合物Pt(ppzOczpy-m)配体的合成:向带有磁力转子的48mL封管中依次添加2-溴-9-(4-甲基-2-吡啶基)-咔唑(222mg,0.66mmol),3-(1-吡唑基)苯酚(96mg,0.6mmol),碘化亚铜(17mg,0.09mmol),N,N-二甲基甘氨酸(18mg,0.18mmol),碳酸铯(586mg,1.8mmol)和1,4-二氧六环(5mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌3天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体238mg,收率95%。1HNMR(300MHz,DMSO-d6):δ8.58–8.50(m,2H),8.31(d,J=8.5Hz,1H),8.25(d,J=7.7Hz,1H),7.81(d,J=8.3Hz,1H),7.72(d,J=1.7Hz,1H),7.66–7.42(m,6H),7.37(t,J=7.4Hz,1H),7.31(d,J=5.1Hz,1H),7.12(dd,J=8.5,2.2Hz,1H),6.99(dd,J=8.2,2.4Hz,1H),6.55(t,J=2.1Hz,1H),2.45(s,3H)。铂络合物Pt(ppzOczpy-m)的合成:向带有磁力转子的75mL封管中依次添加配体(208mg,0.5mmol),氯亚铂酸钾(228mg,0.55mmol)和乙酸(30mL)。该混合物经氮气鼓泡15分钟后在室温下搅拌一天,再加热到110℃搅拌三天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=2:1,得到浅黄色固体244mg,收率80%。1HNMR(300MHz,DMSO-d6):δ9.11(d,J=6.0Hz,1H),8.90(d,J=2.8Hz,1H),8.19–8.05(m,4H),7.87(d,J=8.3Hz,1H),7.53-7.44(m,2H),7.40(t,J=7.4Hz,1H),7.29–7.23(m,1H),7.21(d,J=3.1Hz,1H),7.19(d,J=3.6Hz,1H),6.97(d,J=8.0Hz,1H),6.90(t,J=2.5Hz,1H),2.45(s,3H)。实施例2铂络合物Pt(ppzOczpy-2m)是按照以下方案制备:1-(3-溴-5-甲基苯)吡唑的合成:向带有磁力转子的48mL封管中依次添加3,5二溴甲苯(600mg,2.4mmol),吡唑(136mg,2mmol),碘化亚铜(58mg,0.3mmol),L-脯氨酸(46mg,0.4mmol),碳酸钾(552mg,4mmol)和二甲基亚砜(8mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌3天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=25:1,得到白色固体271mg,收率57%。1HNMR(300MHz,CDCl3):δ7.88(dd,J=2.5,0.6Hz,1H),7.71(d,J=1.7Hz,1H),7.68–7.63(t,J=2.1Hz,1H),7.47(ddd,J=2.2,1.4,0.8Hz,1H),7.24(dt,J=1.7,0.8Hz,1H),6.46(dd,J=2.6,1.8Hz,1H),2.39(s,3H)。Pt(ppzOczpy-2m)配体的合成:向带有磁力转子的15mL封管中依次添加1-(3-溴-5-甲基苯)吡唑(130mg,0.55mmol),9-(4-甲基-2-吡啶基)-咔唑-2-醇(137mg,0.5mmol),碘化亚铜(14mg,0.075mmol),N,N-二甲基甘氨酸(15mg,0.15mmol),碳酸铯(489mg,1.5mmol)和1,4-二氧六环(2mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌3天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体220mg,收率100%。1HNMR(300MHz,CDCl3):δ8.57(d,J=4.5Hz,1H),8.11(d,J=8.2Hz,2H,7.89(d,J=2.4Hz,1H),7.82(d,J=8.1Hz,1H),7.71(s,1H),7.58(d,J=2.1Hz,1H),7.53–7.41(m,2H),7.39–7.30(m,2H),7.23–7.13(m,2H),7.08(dd,J=8.3,1.8Hz,1H),6.80(s,1H),6.45(s,1H),2.49(s,3H),2.40(s,3H)。Pt(ppzOczpy-2m)的合成:向带有磁力转子的48mL封管中依次添加配体(108mg,0.25mmol),氯亚铂酸钾(114mg,0.275mmol)和乙酸(10mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌三天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体133mg,收率85%。1HNMR(300MHz,DMSO-d6):δ9.08(d,J=6.1Hz,1H),8.83(d,J=2.7Hz,1H),8.15-8.01(m,4H),7.82(d,J=8.3Hz,1H),7.51-7.29(m,3H),7.22(d,J=6.3Hz,1H),7.17–7.03(m,1H),6.85(s,1H),6.77(s,1H),2.40(s,3H),2.31(s,3H)。实施例3铂络合物Pt(ppzOczpy-2m′)是按照以下方案制备:1-(3-溴-4-甲基苯)吡唑的合成:向带有磁力转子的48mL封管中依次添加2,4二溴甲苯(1.12g,4.5mmol),吡唑(204mg,3mmol),碘化亚铜(86mg,0.45mmol),L-脯氨酸(41mg,0.6mmol),碳酸钾(829mg,6mmol)和二甲基亚砜(6mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌一天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=30:1,得到白色固体407mg,收率53%。1HNMR(300MHz,CDCl3):δ7.91(d,J=2.3Hz,1H),7.87(d,J=2.5Hz,1H),7.71(d,J=1.8Hz,1H),7.53(dd,J=8.3,2.3Hz,1H),7.29(d,J=8.2Hz,1H),6.49–6.42(m,1H),2.42(s,3H)。Pt(ppzOczpy-2m′)配体的合成:向带有磁力转子的48mL封管中依次添加1-(3-溴-4-甲基苯)吡唑(391mg,1.65mmol),9-(4-甲基-2-吡啶基)-咔唑-2-醇(411mg,1.5mmol),碘化亚铜(43mg,0.225mmol),N,N-二甲基甘氨酸(46mg,0.45mmol),碳酸铯(1.47g,4.5mmol)和1,4-二氧六环(5mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌一天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=10:1,得到白色固体323mg,收率50%。1HNMR(300MHz,DMSO-d6):δ8.46(d,J=5.1Hz,1H),8.39(d,J=2.5Hz,1H),8.19(d,J=8.6Hz,1H),8.15(d,J=7.9Hz,1H),7.72(d,J=8.2Hz,1H),7.58(d,J=1.7Hz,1H),7.55-7.48(m,2H),7.43–7.34(m,4H),7.28(t,J=7.5Hz,1H),7.22(d,J=5.1Hz,1H),6.95(dd,J=8.5,2.2Hz,1H),6.42(t,J=2.1Hz,1H),2.35(s,3H),2.22(s,3H)。Pt(ppzOczpy-2m′)的合成:向带有磁力转子的48mL封管中依次添加配体(215mg,0.5mmol),氯亚铂酸钾(228mg,0.55mmol)和乙酸(25mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体236mg,收率76%。1HNMR(300MHz,DMSO-d6)δ9.13(d,J=6.0Hz,1H),8.86(d,J=2.7Hz,1H),8.19-8.08(m,4H),7.89(d,J=8.3Hz,1H),7.53–7.46(m,1H),7.45-7.38(m,2H),7.30-7.23(m,2H),7.10(d,J=7.8Hz,1H),6.89(t,J=2.5Hz,1H),2.48(s,3H),2.46(s,3H)。实施例4铂络合物Pt(ppzOczpy-3m)是按照以下方案制备:1-(3-溴-4,6-二甲基苯)吡唑的合成:向带有磁力转子的48mL封管中依次添加4,6二溴间甲苯(1.19g,4.5mmol),吡唑(204mg,3mmol),碘化亚铜(86mg,0.45mmol),L-脯氨酸(41mg,0.6mmol),碳酸钾(829mg,6mmol)和二甲基亚砜(6mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌一天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=30:1,得到白色固体330mg,收率44%。1HNMR(300MHz,CDCl3):δ7.70(d,J=2.0Hz,1H),7.57(d,J=2.5Hz,1H),7.52(s,1H),7.17(s,1H),6.43(t,J=2.2Hz,1H),2.41(s,3H),2.17(s,3H)。Pt(ppzOczpy-3m)配体的合成:向带有磁力转子的48mL封管中依次添加1-(3-溴-4,6-二甲基苯)吡唑(304mg,1.21mmol),9-(4-甲基-2-吡啶基)-咔唑-2-醇(302mg,1.1mmol),碘化亚铜(31mg,0.165mmol),N,N-二甲基甘氨酸(34mg,0.33mmol),碳酸铯(1.07g,3.3mmol)和1,4-二氧六环(4mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌36h。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=10:1,得到白色固体447mg,收率80%。1HNMR(300MHz,CDCl3)δ8.53(d,J=5.1Hz,1H),8.02(d,J=6.0Hz,1H),7.99(d,J=8.5Hz,1H),7.74(d,J=8.2Hz,1H),7.62(d,J=1.8Hz,1H),7.49(d,J=2.4Hz,1H),7.46(d,J=2.1Hz,1H),7.41(s,1H),7.39–7.34(m,1H),7.32–7.27(m,1H),7.17(s,1H),7.11(d,J=5.1Hz,1H),6.96–6.85(m,2H),6.34(t,J=2.1Hz,1H),2.46(s,3H),2.32(s,3H),2.19(s,3H)。Pt(ppzOczpy-3m)的合成:向带有磁力转子的150mL封管中依次添加配体(436mg,0.98mmol),氯亚铂酸钾(447mg,1.08mmol)和乙酸(50mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到115℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体480mg,收率77%。1HNMR(300MHz,DMSO-d6):δ9.07(d,J=5.9Hz,1H),8.59(d,J=2.9Hz,1H),8.12–8.00(m,4H),7.80(d,J=8.3Hz,1H),7.43(t,J=7.6Hz,1H),7.35(t,J=7.4Hz,1H),7.21(d,J=6.3Hz,1H),7.18(d,J=8.3Hz,1H),6.89(s,1H),6.85(t,J=2.6Hz,1H),2.57(s,3H),2.40(m,6H)。实施例5Pt(ppzOczpy-4m)是按照以下方案制备:3,5-二甲基-1-(3-溴-4,6-二甲基苯)吡唑的合成:向带有磁力转子的150mL封管中依次添加3,5二溴甲苯(17.5g,70mmol),3,5-二甲基吡唑(3.36g,35mmol),碘化亚铜(1.0g,5.25mmol),L-脯氨酸(806mg,7mmol),碳酸钾(9.67g,70mmol)和二甲基亚砜(50mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌一天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=30:1,得到白色固体3.06g,收率33%。1HNMR(500MHz,CDCl3):δ7.39(s,1H),7.30(s,1H),7.21(s,1H),5.98(s,1H),2.37(s,3H),2.31(s,3H),2.28(s,3H)。Pt(ppzOczpy-4m)配体的合成:向带有磁力转子的75mL封管中依次添加3,5-二甲基-1-(3-溴-4,6-二甲基苯)吡唑(1.86g,7.04mmol),9-(4-甲基-2-吡啶基)-咔唑-2-醇(1.76g,6.40mmol),碘化亚铜(183mg,0.96mmol),N,N-二甲基甘氨酸(198mg,1.92mmol),碳酸铯(6.26g,19.2mmol)和1,4-二氧六环(25mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌两天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体2.34g,收率80%。1HNMR(300MHz,DMSO-d6):δ8.47(d,J=5.0Hz,1H),8.21(d,J=8.5Hz,1H),8.16(d,J=7.7Hz,1H),7.71(d,J=8.2Hz,1H),7.53(s,1H),7.42–7.40(m,1H),7.40–7.34(m,1H),7.29(d,J=7.5Hz,1H),7.26–7.21(m,1H),7.05–6.98(m,2H),6.86–6.80(m,2H),5.96(s,1H),2.39(s,3H),2.28(s,3H),2.19(s,3H),2.06(s,3H)。Pt(ppzOczpy-4m)的合成:向带有磁力转子的150mL封管中依次添加配体(459mg,1mmol),氯亚铂酸钾(457mg,1.1mmol)和乙酸(50mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体541mg,收率83%。1HNMR(300MHz,DMSO-d6):δ9.09(d,J=6.0Hz,1H),8.16-8.09(m,2H),7.98(s,1H),7.83(d,J=8.3Hz,1H),7.54–7.44(m,1H),7.43–7.36(m,1H),7.19–7.08(m,3H),6.80(s,1H),6.43(s,1H),2.76(s,3H),2.43(s,3H),2.41(s,3H),2.37(s,3H)。实施例6Pt(ppzOczpy-4m-3d)是按照以下方案制备:Pt(ppzOczpy-4m-3d)配体的合成:向带有磁力转子的15mL封管中依次添加2-[5-甲基-3-(3,5-二甲基-1-吡唑基)-苯氧基-9-(4-甲基-2-吡啶基)咔唑(92mg,0.2mmol),乙醇钠(27mg,0.4mmol)和d-乙醇(1mL)。该混合物经氮气鼓泡10分钟后加热到100℃搅拌三天。冷却至室温,加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体92mg,收率100%。1HNMR(300MHz,CDCl3):δ8.54(d,J=5.1Hz,1H),8.07(s,1H),8.04(s,1H),7.76(d,J=8.2Hz,1H),7.53(d,J=2.1Hz,1H),7.46-7.37(m,2H),7.35-7.27(m,1H),7.13(dd,J=5.4,1.5Hz,1H),7.07-6.99(m,2H),6.84(s,2H),5.96(s,1H),2.35(s,3H),2.30(s,3H),2.24(s,3H).Pt(ppzOczpy-4m-3d)的合成:向带有磁力转子的48mL封管中依次添加配体(46mg,0.1mmol),氯亚铂酸钾(42mg,0.11mmol)和乙酸(5mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体42mg,收率65%。1HNMR(300MHz,DMSO-d6):δ9.11(d,J=6.0Hz,1H),8.16(d,J=5.5Hz,1H),8.14(d,J=5.4Hz,1H),8.00(s,1H),7.85(d,J=8.3Hz,1H),7.55–7.47(m,1H),7.42(t,J=7.4Hz,1H),7.21–7.14(m,2H),7.13(s,1H),6.82(s,1H),6.45(s,1H),2.78(s,3H),2.44(s,3H),2.39(s,3H).实施例7Pt(ppzOczpy-mph)是按照以下方案制备:Pt(ppzOczpy-mph)配体的合成:向带有磁力转子的15mL封管中依次添加2-溴-9-(2-吡啶基)-咔唑(178mg,0.55mmol),3-(2-甲基苯基)-5-(1-吡啶基)苯酚(124mg,0.5mmol),碘化亚铜(14mg,0.075mmol),N,N-二甲基甘氨酸(15mg,0.15mmol),碳酸铯(489mg,1.5mmol)和1,4-二氧六环(4mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌两天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体112mg,收率46%。1HNMR(300MHz,DMSO-d6):δ8.71(d,J=4.5Hz,1H),8.64(s,1H),8.33(d,J=8.5Hz,1H),8.27(d,J=7.7Hz,1H),8.12(t,J=2.4Hz,1H),7.83(d,J=3.6Hz,1H),7.80(d,J=3.9Hz,1H),7.74(s,1H),7.65-7.57(m,3H),7.54-7.45(m,2H),7.39(d,J=7.6Hz,1H),7.36-7.27(m,4H),7.20(d,J=8.4Hz,1H),6.91(s,1H),6.56(s,1H),2.25(s,3H)。Pt(ppzOczpy-mph)的合成:向带有磁力转子的48mL封管中依次添加配体(108mg,0.22mmol),氯亚铂酸钾(96mg,0.23mmol)和乙酸(11mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=2:1,得到浅黄色固体94mg,收率62%。1HNMR(300MHz,DMSO-d6):δ9.32(d,J=5.4Hz,1H),9.02(d,J=2.8Hz,1H),8.33(d,J=8.4Hz,1H),8.28(d,J=7.4Hz,1H),8.25-8.22(m,1H),8.19(d,J=7.4Hz,1H),8.13(d,J=8.1Hz,1H),7.92(d,J=8.3Hz,1H),7.58(s,1H),7.53(d,J=7.2Hz,1H),7.50–7.43(m,2H),7.42-7.31(m,4H),7.25(d,J=8.3Hz,1H),6.96-6.91(m,2H),2.39(s,3H)。实施例8Pt(ppzOczpy-pr)是按照以下方案制备:Pt(ppzOczpy-pr)配体的合成:向一个25mL史莱克管添加9-(4-溴-2-吡啶基)-2-(3-吡唑-1-基-苯氧基)咔唑(240mg,0.5mmol)。然后在室温下逐滴添加iPrMgBr(3mL,1M)的四氢呋喃(THF)溶液。在搅拌2小时后,添加水(0.6mL)淬灭。30分钟后,用Na2SO4对混合物进行干燥。将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到浅黄色固体44mg,收率20%。1HNMR(300MHz,CDCl3):δ8.57(d,J=5.2Hz,1H),8.08(s,1H),8.06(s,1H),7.88(d,J=2.5Hz,1H),7.76(d,J=8.4Hz,1H),7.68(d,J=1.6Hz,1H),7.51(d,J=2.1Hz,1H),7.47–7.36(m,5H),7.32(s,1H),7.16(d,J=5.3Hz,1H),7.06(dd,J=8.6,2.2Hz,1H),6.96(d,J=7.7Hz,1H),6.43(t,J=2.1Hz,1H),3.05–2.91(m,1H),1.30(s,3H),1.28(s,3H)。Pt(ppzOczpy-pr)的合成:向带有磁力转子的48mL封管中依次添加配体(44mg,0.1mmol),氯亚铂酸钾(46mg,0.11mmol)和乙酸(5mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体13mg,收率20%。1HNMR(300MHz,CDCl3):δ9.14(d,J=6.5Hz,1H),8.13(s,1H),8.09(s,1H),8.04(d,J=7.2Hz,1H),7.90(d,J=8.2Hz,1H),7.85(s,1H),7.79(d,J=7.8Hz,1H),7.43-7.31(m,4H),7.20(d,J=7.7Hz,1H),7.12(d,J=6.1Hz,1H),7.00(d,J=6.1Hz,1H),6.67(d,J=2.5Hz,1H),3.02-2.93(m,1H),1.35(s,3H),1.32(s,3H)。实施例9Pt(ppzOczpy-mesi’)是按照以下方案制备:Pt(ppzOczpy-mesi’)配体的合成:向带有磁力转子的15mL封管中依次添加2-溴-9-(4-甲基-2-吡啶基)-咔唑(185mg,0.55mmol),3-(1,3,5-三甲基苯基)-5-(3,5-二甲基-1-吡啶基)苯酚(153mg,0.5mmol),碘化亚铜(14mg,0.075mmol),N,N-二甲基甘氨酸(15mg,0.15mmol),碳酸铯(489mg,1.5mmol)和1,4-二氧六环(2mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌两天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体228mg,收率81%。1HNMR(300MHz,DMSO-d6):δ8.52(d,J=5.0Hz,1H),8.29(d,J=8.5Hz,1H),8.23(d,J=7.7Hz,1H),7.75(d,J=8.2Hz,1H),7.60(d,J=1.4Hz,1H),7.49(d,J=2.3Hz,1H),7.45(d,J=7.9Hz,1H),7.36(d,J=7.6Hz,1H),7.32(d,J=4.5Hz,1H),7.18-7.13(m,2H),7.00(t,J=1.6Hz,1H),6.93(s,2H),6.77(s,1H),6.07(s,1H),2.47(s,3H),2.31(s,3H),2.26(s,3H),2.15(s,3H),1.99(s,6H)。Pt(ppzOczpy-mesi’)的合成:向带有磁力转子的48mL封管中依次添加配体(225mg,0.4mmol),氯亚铂酸钾(183mg,0.44mmol)和乙酸(20mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=2:1,得到浅黄色固体132mg,收率44%。1HNMR(300MHz,DMSO-d6):δ9.11(d,J=6.0Hz,1H),8.15(d,J=4.1Hz,1H),8.12(d,J=4.4Hz,1H),7.99(s,1H),7.85(d,J=8.2Hz,1H),7.49(t,J=7.7Hz,1H),7.40(t,J=7.2Hz,1H),7.22-7.12(m,2H),6.96(s,3H),6.92(s,1H),6.66(s,1H),6.44(s,1H),2.65(s,3H),2.43(s,4H),2.41(s,3H),2.29(s,3H),2.08(s,6H)。实施例10Pt(ppzOczpy-m’)是按照以下方案制备:3-溴苯基吡唑的合成:向一38mL封管加入1,3-二溴苯(708mg,3.0mmol),吡唑(136mg,2.0mmol),CuI(58mg,0.3mmol),L-脯氨酸(46mg,0.4mmol),碳酸钾(552mg,4.0mmol),DMSO(3mL),氮气鼓泡约5min后升温至120℃,反应过夜,加入水(20mL),用EA(10mLx3)萃取,合并有机相用饱和食盐水(10mLx2)洗涤。用PE:EA=20:1-10:1过柱,得到223mg淡黄色油状物(收率50%)。1HNMR(300MHz,DMSO-d6):δ8.59(d,J=2.6Hz,1H),8.09(d,J=2.0Hz,1H),7.89(dt,J=7.5,1.9Hz,1H),7.78(d,J=1.7Hz,1H),7.56–7.41(m,2H),6.57(d,J=2.2Hz,1H)。2-甲基-7-甲氧基-9-(2-吡啶基)-咔唑的合成:向一38mL封管加入2-甲氧基-7-甲基咔唑(211mg,1.0mmol),2-溴吡啶(174mg,1.1mmol),CuI(29mg,0.15mmol),1-甲基咪唑(42mg,0.5mmol),碳酸钾(345mg,2.5mmol),甲苯(3mL),氮气鼓泡约5min后升温至120℃,反应过夜,加入水(20mL),用EA(10mLx3)萃取,合并有机相用饱和食盐水(10mLx2)洗涤。用PE:EA=20:1-10:1过柱,得到200mg白色固体(收率69%)。1HNMR(300MHz,DMSO-d6):δ8.74(dd,J=5.0,1.9Hz,1H),8.12(td,J=7.8,2.0Hz,1H),8.04(d,J=8.5Hz,1H),7.99(d,J=7.9Hz,1H),7.77(d,J=8.1Hz,1H),7.53(s,1H),7.48(dd,J=7.4,4.9Hz,1H),7.26(d,J=2.2Hz,1H),7.11(d,J=7.9Hz,1H),6.92(dd,J=8.5,2.3Hz,1H),2.44(s,3H)。7-甲基-9-(2-吡啶基)咔唑基-2-醇的合成:向100mL单口瓶中加入2-甲基-7-甲氧基-9-(2-吡啶基)-咔唑(200mg,0.69mmol),HBr(5mL,40%水溶液),升温至120℃,搅拌过夜,冷却至室温,加入水30mL,EA(20mL×3)萃取,合并有机相,无水硫酸钠干燥,PE:EA=5:1过柱纯化得150mg黄色固体(收率79%)。1HNMR(300MHz,DMSO-d6):δ9.49(d,J=2.2Hz,1H),8.78–8.64(m,1H),8.18–8.06(m,1H),7.92(d,J=8.2Hz,1H),7.72(d,J=7.9Hz,1H),7.49(s,2H),7.13(d,J=2.1Hz,1H),7.07(d,J=8.0Hz,1H),6.75(dd,J=8.4,2.0Hz,1H),2.43(s,3H)。Pt(ppzOczpy-m’)配体的合成:向一38mL封管加入2-羟基-7-甲基-9-(2-吡啶)咔唑(100mg,0.36mmol),3-吡唑溴苯(87mg,0.39mmol),CuI(10mg,0.05mmol),N,N-二甲基甘氨酸(5mg,0.05mmol),碳酸铯(140mg,0.60mmol),DMSO(3mL),氮气鼓泡约5min后升温至120℃,反应过夜,加入水(20mL),用EA(20mLx3)萃取,合并有机相用饱和食盐水(20mLx2)洗涤。用PE,EA=10:1过柱纯化。1HNMR(300MHz,DMSO-d6):δ8.68–8.61(m,1H),8.48(d,J=11.6Hz,2H),8.18(d,J=8.4Hz,1H),8.09–8.01(m,2H),7.70–7.61(m,2H),7.58–7.39(m,2H),7.14(d,J=8.1,1H),7.03(dd,J=8.5,2.1Hz,1H),6.97(dd,J=8.1,1.5Hz,1H),6.90(dd,J=8.1,2.5Hz,1H),6.53–7.45(m,2H),2.44(s,3H)。铂配合物Pt(ppzOczpy-m’)的合成:向一38mL封管中加入配体(100mg,0.24mmol),K2PtCl4(104mg,0.25mmol),CHCl3(2mL),HOAc(6mL),氮气鼓泡5min后,80℃下搅拌过夜,升温至120℃,搅拌48h后将至室温,加入水,过滤,滤饼用二氯甲烷淋洗,淋洗液浓缩,用PE:DCM=3:1过柱纯化,得95mg淡黄色固体(收率67%)。1HNMR(300MHz,DMSO-d6):δ9.25(d,J=5.8Hz,1H),8.89(d,J=2.8Hz,1H),8.30(d,J=8.5Hz,1H),8.27–8.20(m,1H),8.16(d,J=2.3Hz,1H),8.01(d,J=7.9Hz,1H),7.88(s,1H),7.80(d,J=8.3Hz,1H),7.50(d,J=7.7Hz,1H),7.45–7.36(m,1H),7.28–7.15,(m,3H),6.96(d,J=8.0Hz,1H),6.89(t,J=2.5Hz,1H),2.53(s,3H)。实施例11Pt(ppzOczpy-m”)是按照以下方案制备:3-甲基-2-甲氧基-9-(2-吡啶基)咔唑的合成:向一38mL封管加入3-甲基-2-甲氧基咔唑(211mg,1.0mmol),2-溴吡啶(174mg,1.1mmol),CuI(29mg,0.15mmol),1-甲基咪唑(42mg,0.5mmol),碳酸钾(345mg,2.5mmol),甲苯(3mL),氮气鼓泡约5min后升温至120℃,反应过夜,加入水(20mL),用EA(10mLx3)萃取,合并有机相用饱和食盐水(10mLx2)洗涤。用PE:EA=20:1-10:1过柱,得到180mg白色固体(收率63%)。1HNMR(300MHz,CDCl3):δ8.74(dd,J=5.0,1.9Hz,1H),8.10(qd,J=7.6,1.7Hz,2H),7.96(s,1H),7.77(dd,J=14.2,8.1Hz,2H),7.47(dd,J=7.4,4.9Hz,1H),7.33(q,J=3.8,2.6Hz,2H),7.30–7.21(m,1H),3.84(s,3H),2.32(s,3H)。3-甲基-9-(2-吡啶基)咔唑基-2-醇的合成:向100mL单口瓶中加入3-甲基-2-甲氧基-9-(2-吡啶基)咔唑(180mg,0.63mmol),HBr(5mL,40%水溶液),升温至120℃,搅拌过夜,冷却至室温,加入水30mL,EA(20mLx3)萃取,合并有机相,无水硫酸钠干燥,PE:EA=5:1过柱纯化得129mg黄色固体(0.47mmol,收率68%)。1HNMR(300MHz,CDCl3):δ9.54(s,1H),8.72(dd,J=5.2,1.8Hz,1H),8.16–8.06(m,1H),8.02(d,J=7.3Hz,1H),7.87(s,1H),7.71(dd,J=8.1,4.0Hz,2H),7.45(dd,J=7.6,5.0Hz,1H),7.36–7.15(m,3H),2.28(s,3H)。Pt(ppzOczpy-m”)配体的合成:向一38mL封管加入2-羟基-3-甲基-9-(2-吡啶)咔唑(120mg,0.44mmol),3-吡唑溴苯(107mg,0.48mmol),CuI(10mg,0.05mmol),N,N-二甲基甘氨酸(5mg,0.05mmol),碳酸铯(286mg,0.88mmol),DMSO(3mL),氮气鼓泡约5min后升温至120℃,反应过夜,加入水(20mL),用EA(20mLx3)萃取,合并有机相用饱和食盐水(20mLx2)洗涤。用PE,EA=10:1过柱纯化,得160mg黄色固体(0.38mmol,收率87%)。1HNMR(300MHz,DMSO-d6):δ8.66(dd,J=5.2,1.8Hz,1H),8.51(d,J=2.5Hz,1H),8.28-8.21(m,2H),8.06(td,J=7.8,2.0Hz,1H),7.82(d,J=8.3Hz,1H),7.77(d,J=8.2Hz,1H),7.68(d,J=1.7Hz,1H),7.57-7.37(m,7H),6.83(dd,J=7.7,2.4Hz,1H),6.52(t,J=2.1Hz,1H),2.35(s,3H)。Pt(ppzOczpy-m”)的合成:向一38mL封管中加入配体(130mg,0.31mmol),K2PtCl4(139mg,0.33mmol),CHCl3(3mL),HOAc(9mL),氮气鼓泡5min后,80℃下搅拌过夜,升温至120℃,搅拌72h后降至室温,加入水,过滤,滤饼用二氯甲烷淋洗,淋洗液浓缩,用PE:DCM=3:1过柱纯化,得101mg淡黄色固体(收率55%)。1HNMR(300MHz,DMSO-d6):δ9.31(d,J=5.7Hz,1H),8.94(d,J=2.8Hz,1H),8.30(d,J=8.4Hz,1H),8.29-8.21(m,1H),8.20(d,J=1.8Hz,1H),8.15(d,J=7.9Hz,1H),8.11(d,J=8.0Hz,1H),7.80(s,1H),7.58–7.48(m,2H),7.48–7.42(m,2H),7.27(t,J=7.9Hz,1H),7.06(d,J=7.7Hz,1H),6.94(d,J=2.6Hz,1H),2.60(s,4H)。实施例12Pt(ppzOczpy-ph)是按照以下方案制备:Pt(ppzOczpy-ph)配体的合成:向带有磁力转子的15mL封管中依次添加2-溴-9-(2-吡啶基)-咔唑(248mg,0.77mmol),3-苯基-5-(1-吡啶基)苯酚(165mg,0.7mmol),碘化亚铜(21mg,0.11mmol),N,N-二甲基甘氨酸(22mg,0.21mmol),碳酸铯(684mg,2.1mmol)和二甲基亚砜(3mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌两天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体130mg,收率39%。1HNMR(300MHz,DMSO-d6):δ8.71(d,J=2.5Hz,1H),8.35(d,J=8.5Hz,1H),8.29(d,J=7.6Hz,1H),8.12(td,J=7.8,1.9Hz,1H),7.91(t,J=1.8Hz,1H),7.86(d,J=3.9Hz,1H),7.83(d,J=4.0Hz,1H),7.80–7.73(m,3H),7.66(d,J=2.1Hz,1H),7.59–7.46(m,6H),7.44(d,J=7.4Hz,1H),7.39(d,J=7.4Hz,1H),7.29(t,J=1.9Hz,1H),7.20(dd,J=8.4,2.2Hz,1H),6.58(t,J=2.1Hz,1H)。Pt(ppzOczpy-ph)的合成:向带有磁力转子的48mL封管中依次添加配体(130mg,0.27mmol),氯亚铂酸钾(125mg,0.3mmol)和乙酸(14mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌两天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=2:1,得到浅黄色固体150mg,收率83%。1HNMR(300MHz,DMSO-d6):δ9.32(d,J=5.6Hz,1H),9.13(d,J=2.7Hz,1H),8.32(t,J=7.Hz,1H),8.28–8.22(m,2H),8.19(d,J=7.2Hz,1H),8.12(d,J=8.1Hz,1H),7.96–7.84(m,4H),7.58-7.48(m,3H),7.47–7.39(m,3H),7.32(s,1H),7.27(d,J=8.3Hz,1H),6.97(t,J=2.5Hz,1H)。实施例13Pt(ppzOczpy-m-tb)是按照以下方案制备:1-(3-溴-5-叔丁基苯)吡唑的合成:向带有磁力转子的48mL封管中依次添加1,3-二溴-5-叔丁基苯(876mg,3mmol),二甲基吡唑(136mg,2mmol),碘化亚铜(58mg,0.3mmol),L-脯氨酸(46mg,0.4mmol),碳酸钾(552mg,4mmol)和二甲基亚砜(4mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌一天。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=15:1,得到浅黄色液体350mg,收率63%。1HNMR(300MHz,DMSO-d6):δ8.61(d,J=2.6Hz,1H),7.88(t,J=1.9Hz,1H),7.84(t,J=1.8Hz,1H),7.75(d,J=1.7Hz,1H),7.45(t,J=1.7Hz,1H),6.54(t,J=2.1Hz,1H),1.31(s,9H).Pt(ppzOczpy-m-tb)配体的合成:向带有磁力转子的48mL封管中依次添加1-(3-溴-5-叔丁基苯)吡唑(307mg,1.1mmol),9-(4-甲基-2-吡啶基)-咔唑-2-醇(274mg,1mmol),碘化亚铜(29mg,0.15mmol),N,N-二甲基甘氨酸(30mg,0.3mmol),碳酸铯(977mg,3mmol)和1,4-二氧六环(4mL)。该混合物经氮气鼓泡10分钟后加热到120℃搅拌16h。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。加圧蒸留除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/乙酸乙酯=5:1,得到白色固体341mg,收率72%。1HNMR(300MHz,DMSO-d6):δ8.53(d,J=2.5Hz,1H),8.49(d,J=5.1Hz,1H),8.26(d,J=8.5Hz,1H),8.21(d,J=7.9Hz,1H),7.75(d,J=8.2Hz,1H),7.66(d,J=1.7Hz,1H),7.59(t,J=1.8Hz,1H),7.57(s,1H),7.47(m,1H),7.42(d,J=7.7Hz,1H),7.34(d,J=7.6Hz,1H),7.29(m,2H),7.08(dd,J=8.5,2.2Hz,1H),7.04(t,J=1.9Hz,1H),6.49(t,J=2.1Hz,1H),2.41(s,3H),1.30(s,9H).Pt(ppzOczpy-m-tb)的合成:向带有磁力转子的75mL封管中依次添加配体(341mg,0.72mmol),氯亚铂酸钾(330mg,0.79mmol)和乙酸(36mL)。该混合物经氮气鼓泡15分钟后室温下搅拌一天,再加热到110℃搅拌三天。冷却至室温,加水沉淀出深色固体,过滤得粗产品,将所得粗产品通过硅胶柱色谱分离纯化,淋洗剂:石油醚/二氯甲烷=1:2,得到浅黄色固体220mg,收率46%。1HNMR(300MHz,DMSO-d6):δ9.11(d,J=6.0Hz,1H),9.02(s,1H),8.17-8.12(m,3H),8.09(d,J=8.1Hz,1H),7.86(d,J=8.3Hz,1H),7.56(s,1H),7.48(t,J=7.5Hz,1H),7.40(t,J=7.4Hz,1H),7.27(d,J=6.0Hz,1H),7.19(d,J=8.4Hz,1H),6.96(s,1H),6.89(s,1H),2.45(s,3H),1.39(s,9H).实施例铂配合物在CH2Cl2、PMMA中的光谱测试。将实验所制备的Pt配合物置于CH2Cl2、PMMA中进行光谱测试,结果如图2-6所示,具体实验数据如表1和表2所示。在本实验的测量所得到的发射光谱中可以看出,在不同激发光的激发下都可以发射出很强的荧光,薄膜主要发射峰在447-453nm之间,效率在大于94%。Pt配合物在薄膜中的光至发光光谱有微小蓝移主要发射峰在447-447nm之间,半峰宽变窄至20nm以下,颜色纯度提升明显,因此这些Pt配合物可以用来制备高饱和度的深蓝光发光器件。表1室温下二氯甲烷溶液中的各项指数表25%聚甲基丙烯酸甲酯(PMMA)的三氯甲烷溶液制备的薄膜中的各项指数络合物波峰波长/半峰宽CIE指数坐标PtON1449/44.7(0.151,0.148)Pt(ppzOczpy)447/22.9(0.148,0.088)Pt(ppzOczpy-m)447/18.5(0.150,0.089)Pt(ppzOczpy-2m)448/19.0(0.145,0.079)Pt(ppzOczpy-2m′)448/20.0(0.144,0.085)Pt(ppzOczpy-3m)449/20.0(0.144,0.082)Pt(ppzOczpy-4m)448/21.9(0.145,0.082)Pt(ppzOczpy-m-tb)448/19.2(0.146,0.086)Pt(ppzOczpy-m’)451/26.5(0.142,0.109)Pt(ppzOczpy-m”)453/24.4(0.143,0.112)通过表1、表2数据可知,无论是在溶液还是薄膜中,铂配合物发光光谱中445-495nm波长范围的积分面积占比都在65%以上,即大部分光谱处于深蓝光区域,半峰宽都小于30nm,CIE坐标(X≤0.150,Y≤0.120)。因为薄膜中分子振动被限制,所以薄膜中铂配合物深蓝光占比更高,半峰宽更窄,色纯度也更优异。本发明所述多取代甲基四齿配位的铂配合物适用于各种光学和光电器件,例如太阳能和光敏感器这样的光吸收器件、光发射器或既有光吸收又有光发射能力的器件以及用于生物应用的标志物。下面以有机发光二极管(OLED)为例对本发明所述多取代甲基四齿配位的铂配合物在光电器件方面的应用进行描述。如图7所示,显示了OLED1000的断面图,OLED1000包含一阳极1004位于基体1002之上,其材质为透明材料,例如氧化铟锡;空穴传输材料层(HTL)1006与阳极1004相连;发光功能层1008位于空穴传输材料层1006之上,所述发光功能层1008包括发射体和主体的发光材料;电子传输材料层(ETL)1010及一金属阴极层1012依次设置于发光功能层1008之上。所述OLED及类似发光器件可包含单层或叠层。从各方面说,该单或叠层的任何层均可包含氧化铟锡(ITO)、MoO3、Ni2O3、聚(3,4-乙烯二氧噻吩)(PEDOT)、聚苯乙烯磺酸钠(PSS)、4,4',4”-((1E,1'E,1”E)-环丙烷-1,2,3-三亚甲基三(氰基-甲基亚基))三(2,3,5,6-四氟苯腈)(NHT-49)、2,2'-(全氟萘烷-2,6-二基)丙二腈(NHT-51)、2,3,5,6-四氟四氰基-对醌二甲烷(F4-TCNQ)、N,N′-二-1-萘基-N,N′-联苯-1,1′-联苯-4,4′二胺(NPD)、4,4,4-三(N-3-甲基苯基-N-苯基氨基)三苯胺(m-MTDATA)、4,4',4”-三(咔唑-9-基)三苯胺(TCTA)、1,1-顺((二-4-对甲苯氨基)苯基)环己烷(TAPC),2,6-顺(N-咔唑基)-吡啶(mCpy)、1,3-双(3,5-双吡啶基-3-苯基)苯(BmPyPB)、2,8-顺(叠氮磷酸二苯酯)二苯并噻吩(PO15)、LiF、LiQ、Cs2CO3、CaCO3、Al或其组合物。在这一具体实施方式中,发光功能层1008可包含本发明所述多取代喹啉配位的铱杂配化合物中的一或多个化合物,本实验选择使用Pt(ppzOczpy-1m),可选择性地连带一主体材料。ETL层1010和1006也可包含一或多个多取代甲基四齿配位的铂配合物以及与电极接近的另一注入层。注入层的材料可包括(电子注入层)EIL、(空穴注入层)HIL和CPL(盖层),其形式可以是单一层或分散在传输材料中。主体材料可以是本技术中已知的任何合适的主体材料。OLED的发光颜色由发光功能层1008的发光能量(光学能隙)决定,通过对发光化合物进行调谐处理和/或主体材料的电子结构来调谐发光功能层1008的发光能量(光学能隙)。HTL层1006中的空穴传输材料和ETL层1010中的电子传输材料可包含本技术中已知的任何合适的空穴传输体。实施例11本发明所述多取代甲基四齿配位的铂配合物在OLED方面的应用。铂配合物Pt(ppzOczpy-1m)与PtON1(Angew.Chem.Int.Ed.2013,52,6753-6756)作为蓝光磷光发光材料应用于器件后的比较。Pt(ppzOczpy-1m)设计OLED器件结构如下:ITO/PEDOT:PSS/TAPC/8%dopant:2,6mCPy/PO15/BmPyPB/LiF/Al。将装有OLED有机材料的坩埚和装有金属铝粒的坩埚依次放置在有机蒸发源和无机蒸发源位置上。关闭腔体,进行初抽真空和抽高真空步骤,使得OLED蒸镀设备内部蒸镀度达到10E-7Torr。OLED蒸镀成膜方法:打开OLED有机蒸发源,对OLED有机材料进行100℃预热,预热时间为15分钟,保证进一步移除OLED有机材料中的水汽。然后对需要蒸镀的有机材料进行快速升温加热处理,并打开蒸发源上方的挡板,直到该材料的蒸发源有有机材料跑出,同时晶振片检测器检测到蒸发速率时,然后进行缓慢升温,升温幅度为1~5℃,直到蒸发速率稳定在1A/秒时,打开掩膜板板正下方的挡板,进行OLED成膜,当电脑端观测到ITO基板上的有机膜达到预设膜厚时,关闭掩膜板挡板和蒸发源正上方挡板,关闭该有机材料的蒸发源加热器。其它有机材料和阴极金属材料的蒸镀工艺如上所述。封装采用UV环氧树脂进行光固化封装。封装后的样品进行IVL性能测试,IVL设备采用McScienceM6100进行测试。器件数据对比数据测试如下表3所示:表3PtON1和Pt(ppzOczpy-1m)器件数据对比图如表3所示,通过器件数据对比,器件电致发光波长主要由Pt配合物本身光致发光的决定,Pt配合物本身光致发光光谱的纯度和电致发光的光谱纯度直接相关。在同一条件下,器件的效率高低也与Pt配合物本身的发光量子效率趋势一致,器件发光的色纯度与掺杂材料本身光激发下发射光的光谱色纯度直接关联。实施例12如图8所示,Pt(ppzOczpy-m)的器件的电致发光光谱。作为发光材料制备的器件进行电致发光光谱测试,说明配合物属于纯蓝光,此材料可以完全满足显示器中的红光的色度要求。本发明的技术方案至少具有以下有益的效果:分子刚性强,可以有效减少由于分子振动所消耗的能量,磷光发光强度高;产物可作为一种有机磷光发光材料;对比叔丁基,甲基取代的金属铂配合物合成更容易,成本更低。通过在配体的咔唑吡啶上位引入甲基,能明显窄化发光光谱,大部分光谱位于深蓝光区域,为窄蓝光磷光材料的开发提供了一种途径,具有重要意义。以上所述的仅是本发明的实施方式,在此应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出改进,但这些均属于本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1