一种新羽扇豆烷型三萜细柱五加苷元S及其制备方法与流程
本发明涉及医药领域,特别涉及一种从细柱五加叶中分离出的新化合物及其制备方法。
背景技术:
细柱五加(acanthopanaxgracilistylusw.w.smith)为五加科五加属植物,其根皮为其主要的药用部分,具有强筋壮骨、祛风除湿、利水消肿、补益肝肾、利水消肿之功效。经过近几十年科学工作者其根皮化学成分的研究,作为药用的细柱五加根皮含有紫丁香苷、β-谷甾醇、五加酸、刺五加苷b1、贝壳烯酸、胡萝卜苷、各种有机酸、色素、挥发油等成分。近年来,国内外正全方位开发该属植物,包括茎、叶和果实等,药理研究结果表明,细柱五加叶中丰富的羽扇豆烷型三萜类化合物可抑制脂多糖诱导的巨噬细胞raw264.7,炎症因子tnf-α、il-1β、hmgb1的分泌,其三萜类化合物在抗炎药物、肝病药物等领域具有很好的开发前景,通过近10多年对细柱五加叶的系统研究,首次从细柱五加叶中分离得到天然新化合物细柱五加苷元s。
技术实现要素:
本发明的目的在于提供一种从细柱五加叶中分离出的羽扇豆烷型新化合物细柱五加苷元s及其制备方法。
本发明从细柱五加叶中分离出的羽扇豆烷型三萜类新化合物,其特征在于,所述化合物的化学名为:1β,3α-二羟基-羽扇豆-20(29)-烯-23,28-二酸,命名为细柱五加苷元s,化学结构式为:
一种从细柱五加叶中分离出的羽扇豆烷型三萜类新化合物的制备方法,其特征在于,包括以下步骤:
(1)称取细柱五加叶,粉碎后加入5-16倍体积的溶剂提取,滤液浓缩后用水分散,低极性溶剂萃取去色素后,用脂肪酸酯萃取,得到脂肪酸酯萃取物;
(2)脂肪酸酯萃取物以体积比0.9-2.1∶1.2-2.7∶0.6-2.8∶0.4-2.1的烷烃∶脂肪酸酯∶甲醇∶水作为两相溶剂体系在高速逆流色谱仪上进行分离,将该溶剂系统混合均匀静置后,分开上下相,超声16-40min,溶剂系统的上相为固定相,下相为流动相,以16-40ml/min的流速将上相泵入管路,待固定相充满整个管路后,停泵,启动主机,然后以1.5-3ml/min的流速泵入流动相,当上下相平衡时,记录固定相流出的体积,计算得到固定相的保留值,取步骤(1)制备的脂肪酸酯萃取物,溶解于固定相中,进样,根据检测器检测谱图,收集组分,浓缩,干燥得粗品。
(3)将上述干燥粗品用制备液相进行分离,色谱柱为制备柱cstc18(300*30mm,10μm),流动相为乙腈-水(30∶70,v/v)等度洗脱,检测波长为210nm,流速20ml/min,收集出峰溶液,溶液浓缩,干燥即得有效成分。
一种从细柱五加叶中分离出的羽扇豆烷型三萜类新化合物的制备方法,其特征在于,步骤(1)中的提取溶剂为甲醇、乙醇、水中的一种或多种。
一种从细柱五加叶中分离出的羽扇豆烷型三萜类新化合物的制备方法,其特征在于,步骤(1)中的脂肪酸酯为甲酸乙酯、乙酸乙酯或乙酸甲酯。
一种从细柱五加叶中分离出的羽扇豆烷型三萜类新化合物的制备方法,其特征在于,步骤(2)中,两相溶剂系统采用正己烷、乙酸乙酯、甲醇和水四种组分体积比为1.2∶1.8∶2.2∶1.6,临用前分开上下相,超声40min,以溶剂系统的上相为固定相,下相为流动相,以25ml/min的流速将上相泵入管路,带固定相充满整个管路后,停泵,启动主机,把转速调到925r/min,然后以2ml/min的流速泵入流动相,进样量为15ml,紫外检测波长为210nm,根据紫外检测谱图,收集各峰的组分,将各峰对应的收集液分别浓缩,干燥。
附图说明
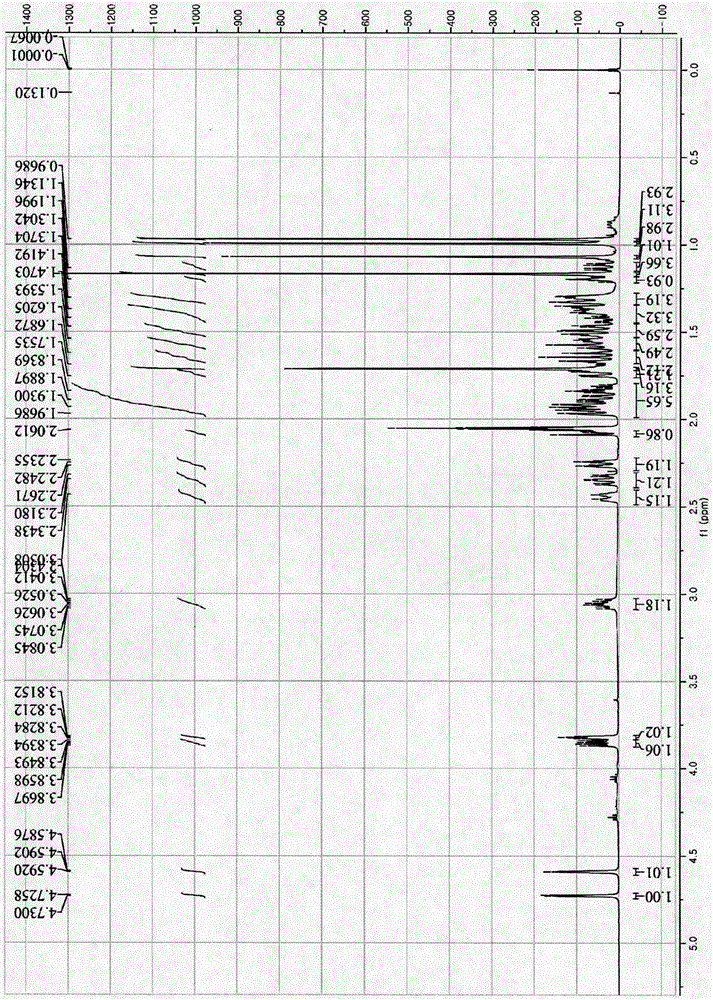
图1细柱五加苷元s的1h-nmr图(400mhz,cd3cocd3)
图2细柱五加苷元s的13c-nmr图(100mhz,cd3cocd3)
图3细柱五加苷元s的主要1h-1hcosy(粗体)和hmbc(箭头)相关
图4细柱五加苷元s的主要noesy相关
具体实施方式
本发明所述的新化合物是以细柱五加叶为原料,粉碎后用有机溶剂提取,滤液浓缩后用水分散,低极性溶剂萃取去色素后,用脂肪酸酯萃取,取脂肪酸酯萃取物在高速逆流色谱仪上进行分离,经紫外在线检测后,用制备液相分离纯化得到。以下结合实际情况,本发明的具体实施方式进行详细说明:
一、化合物的提取和分离
(1)称取细柱五加叶,粉碎后加入5-16倍体积的甲醇提取,滤液浓缩后用水分散,石油醚萃取去色素后,用乙酸乙酯萃取,得到乙酸乙酯萃取物;
(2)乙酸乙酯萃取物以体积比1.2∶1.8∶2.2∶1.6的正己烷∶乙酸乙酯∶甲醇∶水作为两相溶剂体系在高速逆流色谱仪上进行分离,将该溶剂系统混合均匀静置后,分开上下相,超声30min,溶剂系统的上相为固定相,下相为流动相,以24ml/min的流速将上相泵入管路,待固定相充满整个管路后,停泵,启动主机,然后以2.2ml/min的流速泵入流动相,当上下相平衡时,记录固定相流出的体积,计算得到固定相的保留值,取步骤(1)制备的乙酸乙酯萃取物,溶解于固定相中,进样,根据检测器检测谱图,收集组分,浓缩,干燥得粗品。
(3)将上述干燥粗品用制备液相进行分离,色谱柱为制备柱cstc18(300*30mm,10μm),流动相为乙腈-水(30∶70,v/v)等度洗脱,检测波长为210nm,流速20ml/min,收集出峰溶液,溶液浓缩,干燥即得有效成分。
二、化合物结构鉴定
白色粉末(meoh),liebermann-burchard反应呈阳性,ms得出其分子离子峰为m/z501.3[m-h]-1,高分辨质谱esi负离子分子量为501.3221,与分子式c30h45o6[m-h]-1的理论值(501.3222)吻合,推断其分子式为c30h46o6。
1h-nmr谱中显示含有一对孤立的环外烯氢信号δ4.58和δ4.72,以及与这2个烯氢有偶合作用的甲基δ1.71,可知分子中含有1个烯丙基,δ3.05是连接该烯丙基的氢信号峰,这是羽扇豆烷型三萜的特征峰;δ3.82的三重峰及δ3.84的峰是连接羟基的碳上的质子;δ0.97(3h,s),0.99(3h,s),1.07(3h,s),1.17(3h,s)和1.71(3h,s)为5个与季碳相连的甲基质子信号。
13c-nmr谱显示含有30个碳信号,其中2个烯键碳8151.89和δ110.26,为羽扇豆烷型三萜环外末端双键的特征信号;δ73.99和δ75.46为两个连接有羟基的碳信号峰,δ177.84和178.16为两个羧基的碳信号峰;再结合dept谱,δ13.24,15.40,17.31,17.92,19.72为5个甲基碳信号峰,δ22.09,24.68,26.87,30.75,31.61,33.15,35.19,37.36,37.84和110.26为10个仲碳信号峰,δ39.14,45.25,48.24,50.30,52.98,73.99,75.46为7个叔碳信号峰,其他8个没有在dept图上显示的δ42.76,43.72,44.33,52.27,56.98,151.89,177.84和178.16为季碳信号峰。
将1d-nmr谱与2d-nmr谱结合起来,通过hmqc谱确定了碳和氢之间直接相连的关系。由1h-1hcosy谱显示邻偶相关关系,由hmbc谱确定了碳和氢之间远程关系(如表1,和图3),noesy谱确定了碳和氢之间noe关系(如图4)。noesy谱上1位氢与2位、5位、9位及11位氢发生noe效应,推断1位-oh为β型取代。3位氢与2位、24位氢发生noe效应,而与5位氢没有noe效应,说明3位氢键为e键,推断3位-oh取代应为α型取代(a键);24位h与3位、25位氢有noe效应,故推断c-4位羧基为α型取代。13位氢与19位氢有noe效应,因而c-19位上的异丙烯基为α型取代。
1h-nmr(400mhz,pyrindine-d5)和13c-nmr(100mhz,pyrindine-d5)数据见表1。所有1d-nmr与2d-nmr数据都证实化合物1为羽扇豆烷型三萜皂苷元1β,3a-二羟基-羽扇豆-20(29)-烯-23,28-二酸,命名为细柱五加苷元s,结构如式1。
表1化合物1的1h-nmr(400mhz)和13c-nmr(100mhz)数据(cd3cocd3,δppm)
以上实施例显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,而不是以任何方式限制本发明的范围,在不脱离本发明范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!