一种阿尼芬净侧链中间体对戊氧基三联苯甲酸的制备方法与流程
本发明涉及医药合成领域,更具体地,涉及一种阿尼芬净侧链对戊氧基三联苯甲酸的制备方法。
技术背景
阿尼芬净(anidulafungin)是两性霉菌素b的衍生物,该药是由美国vicuron制药公司研制的第三代棘自菌素类半合成抗真菌药,商品名为eraxis,2006年在美国获批上市。与其他棘自菌素类抗真菌药相比,阿尼芬净具有更大的分布容积和更广谱的抗菌活性。
阿尼芬净作为一种半合成抗真菌剂,是由培养各种微生物制得的环肽抗真菌剂衍生而来。所有这些抗真菌剂的结构特征是:包含一个环六肽核,环氨基酸的一个氨基带有一个脂肪酰基,该脂肪酰基形成一个链。通过酶的脱酰基作用将侧链除去得到自由核,再将核的氨基酰化得到半合成抗真菌化合物。
wo09631228a、wo0051564a1、jp2005325142a公开了一种制备对戊氧基三联苯甲酸的方法,具体为:a.芳基卤代物与硼酸三异丙酯在丁基锂作用下制备芳基硼酸;b.芳基硼酸与对碘苯甲酸甲酯经过suzuki偶联制备4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯;c.再经过碱性条件水解得到目标产物。该路线制备硼酸采用的丁基锂与空气易燃烧,且反应体系对水分的要求较高,不适合大规模生产。
cn103570530a、cn106431901a提供了一种通过格氏试剂制备三联苯甲酸甲酯,然后水解得到对戊氧基三联苯甲酸的方法。具体过程为:a.1,4-二溴苯经碘引发与镁进行格氏试剂反应,降温后与硼酸三甲酯反应,酸性条件下水解制备1,4-苯二硼酸;b.1,4-苯二硼酸、4-戊氧基溴苯和4-碘苯甲酸乙酯通过suzuki偶联制备4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸乙酯。该路线通过格氏试剂制备硼酸,同样对溶剂水分有较高的要求,且制备的格氏试剂需要在手套箱中抽滤除去镁屑,操作繁琐,不适合放大生产。
因此,开发一种工艺简单,条件温和,适合大规模生产的阿尼芬净侧链中间体硼酸的制备方法具有重要的研究意义和经济价值。
技术实现要素:
本发明的目的在于克服现有技术中丁基锂或格式试剂反应制备硼酸的方法中存在操作繁琐,不易控制,危险性较高等问题,提供一种阿尼芬净侧链中间体对戊氧基三联苯甲酸的制备方法。本发明采用suzuki偶联制备芳基硼酸再经过suzuki偶联和碱水解的方法制备目标产物,具有成本低,工艺操作简便安全的优点。
为了实现上述发明目的,本发明采用如下技术方案:
一种阿尼芬净侧链中间体对戊氧基三联苯甲酸的制备方法,包括如下步骤:
s1:4羟基-4’-溴联苯和1-溴戊烷发生亲核取代反应得到4’-溴-4-正戊氧基联苯;
s2:在惰性气氛保护下,4’-溴-4-正戊氧基联苯和四羟基二硼在钯催化剂及膦配体的作用下发生suzuki偶联反应得到4-戊氧基-4’-联苯硼酸;
s3:在惰性气氛保护下,4-戊氧基-4’-联苯硼酸和4-碘苯甲酸甲酯在钯催化剂及膦配体的作用下发生suzuki偶联反应得到4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯;
s4:4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯水解即得到所述阿尼芬净侧链中间体对戊氧基三联苯甲酸。
本发明选用特定的原料,采用suzuki偶联制备芳基硼酸再经过suzuki偶联和碱水解的方法制备目标产物,具有成本低,工艺操作简便安全的优点。
本发明s1中的亲核取代反应为常规的反应类型,其条件控制(如催化剂、反应温度、ph等)也可参照现有技术。
优选地,s1中所述亲核取代反应选用四丁基溴化铵为催化剂。
更为优选地,所述四丁基溴化铵和4-羟基-4’-溴联苯的摩尔比0.1~0.5:1。
优选地,s1中所述亲核取代反应的ph为8~11。
优选地,s1中所述亲核取代反应选用无机碱调节ph。
本领域中常规的无机碱均可用于本发明中。
更为优选地,所述无机碱为氢氧化钠。
优选地,s1中所述亲核取代反应的温度为60~95℃。
优选地,s1中1-溴戊烷与4-羟基-4’-溴联苯的摩尔比为1.1~2.0:1。
优选地,s1的具体过程为:以4羟基-4’-溴联苯为原料在氢氧化钠条件下经四丁基溴化铵的催化与1-溴戊烷发生亲核取代反应制备得到4’-溴-4-正戊氧基联苯。
优选地,s2中所述四羟基二硼和4’-溴-4-正戊氧基联苯的摩尔比为1.5~4.0:1。
本领域中常规的钯催化剂和膦配体均可用于本发明中。
优选地,s2中所述钯催化剂为醋酸钯pd(oac)2,二亚苄基丙酮钯pd2(dba)3或氯化钯pdcl2中的一种或几种。
优选地,s2中所述膦配体为三苯基膦pph3、三邻甲基苯基膦p(o-mec6h4)3、三环己基膦pcy3或三叔丁基膦ptbu3中的一种或几种。
优选地,s2中所述膦配体溶解于四氢呋喃溶液中。
更为优选地,所述四氢呋喃和4’-溴-4-正戊氧基联苯的体积质量比为4~10:1ml/g。
更为优选地,所述钯催化剂为醋酸钯;所述膦配体为三(邻甲基苯基)膦。
发明人对钯催化剂和膦配体进行优选发现,选用醋酸钯和三(邻甲基苯基)膦作为催化剂具有提高转化率,减少副产物的效果。
更为优选地,所述醋酸钯和4’-溴-4-正戊氧基联苯摩尔比为1~8:100。
更为优选地,所述三邻甲基苯基膦和4’-溴-4-正戊氧基联苯摩尔比为1~10:100;
优选地,s2中所述suzuki偶联反应的ph为8~11。
优选地,s2中所述suzuki偶联反应选用无机碱调节ph。
本领域中常规的无机碱均可用于本发明中。
更为优选地,所述无机碱为醋酸钾。
优选地,所述醋酸钾和4’-溴-4-正戊氧基联苯的摩尔比为1.5~3.0:1。
优选地,s2中所述suzuki偶联反应的温度为0~40℃。
优选地,所述四羟基二硼溶于甲醇溶液中。
更为优选地,甲醇和四羟基二硼的体积质量比为10~20:1。
优选地,s2中所述惰性气氛为氮气气氛。
更为优选地,s2的具体过程为:在氮气保护下,向醋酸钯和三(邻甲基苯基)膦的四氢呋喃溶液中加入醋酸钾和4’-溴-4-正戊氧基联苯,再加入四羟基二硼的甲醇溶液,通过suzuki偶联反应制备得到4-戊氧基-4’-联苯硼酸。
优选地,s3中所述4-碘苯甲酸甲酯和4-戊氧基-4’-联苯硼酸的摩尔比为1.1~1.5。
优选地,s3中所述钯催化剂为醋酸钯pd(oac)2,二亚苄基丙酮钯pd2(dba)3或氯化钯pdcl2中的一种或几种。
优选地,s3中所述膦配体为三苯基膦pph3、三邻甲基苯基膦p(o-mec6h4)3、三环己基膦pcy3或三叔丁基膦ptbu3中的一种或几种。
更为优选地,所述钯催化剂为醋酸钯,所述膦配体为三苯基膦。
发明人对钯催化剂和膦配体进行优选发现,选用醋酸钯和三苯基膦作为催化剂具有提高转化率,减少副产物的效果。
更为优选地,所述醋酸钯和4-戊氧基-4’-联苯硼酸的摩尔比为0.5~10:1。
更为优选地,所述三苯基膦和4-戊氧基-4’-联苯硼酸摩尔比为1~15:1。
优选地,s3中所述suzuki偶联反应的ph为8~11。
优选地,s3中所述suzuki偶联反应选用无机碱调节ph。
本领域中常规的无机碱均可用于本发明中。
更为优选地,所述无机碱为碳酸钠。
优选地,所述碳酸钠和4-戊氧基-4’-联苯硼酸的摩尔比为1.0~3.0。
优选地,所述4-碘苯甲酸甲酯溶于甲苯/正丙醇混合溶液中。
更为优选地,所述甲苯/正丙醇混合溶液中正丙醇和甲苯的体积比是1~10:1。
优选地,所述正丙醇/甲苯混合溶液和4-正戊氧基-4’-联苯硼酸的质量比为8~15:1。
优选地,s3中所述所述惰性气氛为氮气气氛。
更为优选地,s3的具体过程为:氮气保护下,向4-戊氧基-4’-联苯硼酸和4-碘苯甲酸甲酯的甲苯/正丙醇混合溶液中依次加入碳酸钠水溶液、醋酸钯和三苯基膦,经过suzuki偶联反应制备得到4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯。
本发明s4中的水解反应为常规的反应类型,其条件控制(如催化剂、反应温度、ph等)也可参照现有技术。
优选地,s4中所述水解反应选用十六烷基三甲基溴化铵作为相转移催化剂。
优选地,s4中所述4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯溶于二甲苯溶液中。
优选地,s4中所述水解反应的ph为12~14。
本领域中常规的无机碱溶液均可用于本发明中。
更为优选地,s4中所述水解反应选用氢氧化钾水溶液调节ph。
更为优选地,s4的具体过程为:4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯的二甲苯溶液与氢氧化钾水溶液混合,在相转移催化剂十六烷基三甲基溴化铵的催化下水解,即得到所述阿尼芬净侧链中间体对戊氧基三联苯甲酸。
本发明提供的制备方法可表示为如下反应式:
与现有技术相比,本发明具有如下有益效果:
本发明采用suzuki偶联制备芳基硼酸再经过suzuki偶联和碱水解的方法制备目标产物,具有成本低,工艺操作简便安全的优点。
附图说明
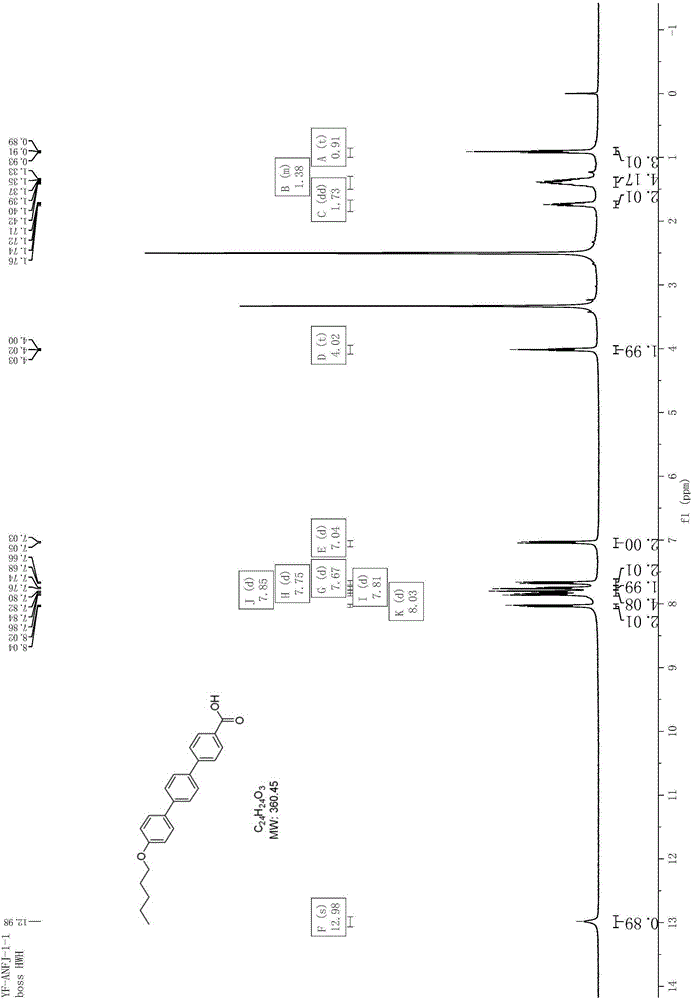
图1为实施例7提供的阿尼芬净侧链中间体对戊氧基三联苯甲酸的核磁谱图氢谱。
具体实施方式
下面结合实施例进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件;所使用的原料、试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
实施例14’-溴-4-正戊氧基联苯的合成
向2l的三口瓶中加入1450ml水,21.2g氢氧化钠,搅拌溶清后加入120g4-羟基-4’-溴联苯和6g四丁基溴化铵,室温下搅拌10min后滴加87.2g1-溴戊烷,滴加完成后回流5h,待反应降至室温后抽滤,滤饼用400ml水洗涤,所的固体用600ml正庚烷/水(1:1)混合溶液热打浆,抽滤并用60ml正庚烷洗涤滤饼,所得固体在60℃下真空干燥5小时,收率87%。1hnmr(d6dmso,400mhz)δ8.04-8.02(m,2h),7.85-7.83(m,1h),7.63-7.57(m,3h),7.02-7.00(m,2h),4.01-3.98(m,2h),1.75(t,j=8.0hz,2h),1.41-1.36(m,4h),0.92-0.89(m,3h).
实施例24’-溴-4-正戊氧基联苯的合成向2l的三口瓶中加入1200ml水,17.7g氢氧化钠,搅拌溶清后加入100g4-羟基-4’-溴联苯和5g四丁基溴化铵,室温下搅拌10min后滴加72.7g1-溴戊烷,滴加完成后回流5h,待反应降至室温后抽滤,滤饼用300ml水洗涤,所的固体用320ml正庚烷/水(1:1)混合溶液热打浆,抽滤并用60ml正庚烷洗涤滤饼,所得固体在60℃下真空干燥5小时,收率87%。1hnmr(d6dmso,400mhz)δ8.04-8.02(m,2h),7.85-7.83(m,1h),7.63-7.57(m,3h),7.02-7.00(m,2h),4.01-3.98(m,2h),1.75(t,j=8.0hz,2h),1.41-1.36(m,4h),0.92-0.89(m,3h).
实施例34-戊氧基-4’-联苯硼酸的合成氮气保护下,向100ml三口瓶中加入5g4’-溴-4-正戊氧基联苯,88mg醋酸钯,4.6g醋酸钾,144mg三(邻甲基苯基)膦和15ml四氢呋喃,冰水浴降温至0-10℃,1.84g四羟基二硼溶于10ml甲醇后滴加到反应中,室温下搅拌反应,待原料消失后加入30ml乙醇稀释,抽滤,滤饼用10ml乙醇洗涤,滤液旋干后加入80ml二氯甲烷/水混合溶剂(1:1)在40℃下热打浆1h,冷却至室温后抽滤,滤饼依次用5ml水和5ml二氯甲烷洗涤,50℃下真空干燥得白色固体2.84g,收率64%。1hnmr(d6dmso,400mhz)δ7.98-7.96(m,2h),7.85-7.83(m,2h),7.62-7.57(m,4h),7.02-7.00(m,2h),4.02(t,j=6.0hz,2h),1.73-1.72(m,2h),1.39-1.37(m,4h),0.92(t,j=6.0hz,3h).
实施例44-戊氧基-4’-联苯硼酸的合成氮气保护下,向2l三口瓶中加入1.41g醋酸钯,2.30g三(邻甲基苯基)膦和480ml四氢呋喃,冰水浴降温至0-10℃,加入80g4’-溴-4-正戊氧基联苯和74g醋酸钾,氮气置换三次,29.4g四羟基二硼溶于400ml甲醇后滴加到反应体系中,升温至35-40℃搅拌反应30min,待原料反应完全后加入500ml乙醇稀释,抽滤,滤饼用100ml乙醇洗涤,滤液旋干后加入1200ml二氯甲烷/水混合溶剂(1:2)热浆洗1h,冷却至10℃后抽滤,滤饼用少量水二氯甲烷洗涤,50℃下真空干燥5小时得白色固体45.2g,收率63%。1hnmr(d6dmso,400mhz)δ7.98-7.96(m,2h),7.85-7.83(m,2h),7.62-7.57(m,4h),7.02-7.00(m,2h),4.02(t,j=6.0hz,2h),1.73-1.72(m,2h),1.39-1.37(m,4h),0.92(t,j=6.0hz,3h).
实施例54″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯的合成氮气保护下,向100ml三口瓶中加入2g4-戊氧基-4’-联苯硼酸,1.75g4-碘苯甲酸甲酯20ml甲苯/正丙醇混合溶液(1:8)室温下搅拌均匀,依次加入4ml碳酸钠溶液(2mol/l),18mg醋酸钯和50mg三苯基膦,氮气置换3次,85℃下反应过夜,待原料反应完全后冷却至室温并抽滤,滤饼依次用2ml甲苯,4ml水和4ml甲基叔丁基醚淋洗,滤饼在50℃下真空干燥5小时得白色固体1.92g,收率71%。1hnmr(cdcl3,400mhz)δ8.13(d,j=8.0hz,2h),7.71-7.66(m,6h),7.53-7.52(m,2h),7.00(d,j=8.0hz,2h),4.02(t,j=8.0hz,2h),3.95(s,3h),1.84-1.80(m,2h),1.49-1.40(m,4h),0.97(t,j=8.0hz,3h).
实施例64″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯的合成氮气保护下,向500ml三口瓶中加入20g4-戊氧基-4’-联苯硼酸,17.5g4-碘苯甲酸甲酯150ml甲苯/正丙醇混合溶液(1:8)室温下搅拌均匀,依次加入40ml碳酸钠溶液(2mol/l),150mg醋酸钯和526mg三苯基膦,氮气置换3次,85℃下反应过夜,待原料反应完全后冷却至室温并抽滤,滤饼依次用10ml甲苯,20ml水和15ml甲基叔丁基醚淋洗,滤饼在50℃下真空干燥5小时得白色固体20.1g,收率80.4%。1hnmr(cdcl3,400mhz)δ8.13(d,j=8.0hz,2h),7.71-7.66(m,6h),7.53-7.52(m,2h),7.00(d,j=8.0hz,2h),4.02(t,j=8.0hz,2h),3.95(s,3h),1.84-1.80(m,2h),1.49-1.40(m,4h),0.97(t,j=8.0hz,3h).
实施例7阿尼芬净侧链中间体对戊氧基三联苯甲酸的合成
向500ml三口瓶中加入20g4″-(戊氧基)-[1,1',4',1″-三联苯]-4-甲酸甲酯,1.17g十六烷基三甲基溴化铵,12g氢氧化钾和60ml二甲苯,85℃下反应6小时,冷却至室温后抽滤,滤饼用水洗涤(3x100ml),所得率并抽干后加入160ml乙二醇二甲醚和100ml稀盐酸(1mol/l),升温至85℃搅拌反应1h,冷却至室温后抽滤,滤饼依次用50ml甲基叔丁基醚和50ml甲醇淋洗,率并在50℃下真空干燥5小时,得白色固体18.4g,收率95.6%。图1为对戊氧基三联苯甲酸的核磁谱图氢谱,1hnmr(d6dmso,400mhz)δ12.98(s,1h),8.04(d,j=8.0hz,2h),7.86-7.74(m,6h),7.68(d,j=8.0hz,2h),7.05(d,j=8.0hz,2h),4.03(t,j=8.0hz,2h),1.76-1.71(m,2h),1.42-1.33(m,4h),0.93(t,j=8.0hz,3h).
以上所述是本发明的特定示例实施方式,对于本领域的技术人员,在不脱离本发明的原理下,还可以做出若干的改进与修辞。事实上,本发明的范围由所附的权利要求及其等效限定。
- 还没有人留言评论。精彩留言会获得点赞!