植物乳杆菌代谢低聚果糖的多效调控蛋白及其调控方法与流程
本发明属于基因工程领域,具体涉及多效调控蛋白ccpa及其在植物乳杆菌st-ⅲ代谢低聚果糖过程中发挥的负调控作用。
背景技术:
乳酸菌的糖代谢过程常常受到宿主代谢调控的控制,某些糖代谢相关的基因簇在特定糖存在条件下是共转录的,但是葡萄糖会抑制它们的转录和表达。这些现象表明,在上述过程中乳酸菌的代谢调控发挥着重要作用。当外界环境有所变化时,细菌能感受到环境变化并将信号实时传导至胞内,通过转录调控系统来调节相关基因的转录表达,进而引发细胞代谢活动。转录因子、靶基因及启动子区内的结合位点等调控元件构成了转录调控系统,转录因子通过与结合位点的特异性结合来激活或抑制下游靶基因的转录表达。近年来,科学家们对乳酸菌糖代谢过程中的转录调控机制做了大量的研究工作,认为乳酸菌的糖代谢过程常常会受到全局调控和局部调控的双重效应。全局调控通常由一种易被宿主所利用的糖(如葡萄糖)所诱导的多效调控蛋白(catabolitecontrolproteina,ccpa)与cre(cataboliterepressionelement,cre)位点的结合来完成,ccpa与位于启动子区域或其下游的cre位点相结合而阻止该结构基因的转录。
ccpa作为革兰氏阳性菌的全局性调控蛋白,是laci-galr家族中的一个多效调控蛋白,广泛地参与到菌株的多项生理活动,例如参与中心碳、氮代谢的调控、生物被膜的形成和毒性基因的表达等过程。研究表明,ccpa调控蛋白可以通过发挥碳代谢阻遏效应((carboncataboliterepression,ccr)和碳代谢物激活效应(carboncataboliteactivation,cca)来进行碳代谢物代谢调控。然而,在革兰氏阳性细菌中ccpa调控蛋白主要起到一个负调控的作用,在葡萄糖等速效碳源存在的情况下,ccpa在组氨酸磷酸载体蛋白的协助下与抑制非速效碳源代谢基因的启动子区域cre位点结合,抑制其转录基因的转录。
低聚果糖(fos)作为一种常见的益生元,具有选择性地促进肠道中植物乳杆菌的生长从而显著改善肠道菌群平衡的功能,但是关于其代谢通路及调控机理的研究还处于起步阶段,因此目前在植物乳杆菌中ccpa作为全局调控蛋白对fos的代谢调控机制仍不清楚。
技术实现要素:
本发明的目的在于提供一种对植物乳杆菌st-ⅲ代谢fos起负调控作用的多效调控蛋白(即ccpa)的大肠杆菌表达载体(pet-28-ccpa),同时提供该载体的构建方法。此外,本发明还通过emsa实验验证了调控蛋白ccpa与fos代谢相关基因的启动子区域cre位点的特异性结合。
本发明提供的技术方案之一是:
一种分离的核酸,其编码一种分离的蛋白质,所述的分离的蛋白质的氨基酸序列如seqidno:1所示;或为由氨基酸序列如seqidno:1所示的蛋白质与蛋白纯化标签组成的融合蛋白。
其中所述核酸的制备方法为本领域常规的制备方法,所述制备方法较佳地包括:从自然界中提取天然存在的编码氨基酸序列如序列表中seqidno:1所示的蛋白质的核酸分子,或者通过基因克隆技术获得编码氨基酸序列如序列表中seqidno:1所示的蛋白质的核酸分子,或者通过人工全序列合成的方法得到编码氨基酸序列如序列表中seqidno:1所示的蛋白质的核酸分子。
本发明中,所述的核酸优选地具有如序列表中seqidno:2所示的核苷酸序列,更优选地,所述核酸的核苷酸序列如序列表中seqidno:2所示。
如本领域技术人员所知:编码氨基酸序列如序列表中seqidno:1所示的蛋白质的核苷酸序列可以适当引入替换、缺失、改变、插入或增加一个或多个碱基的方式来提供一个多聚核苷酸的同系物,只要该同系物编码的蛋白质依然能够保持氨基酸序列组成如序列表中seqidno:1所示的蛋白质的功能即可。
本发明提供的技术方案之二是:一种包含上述核酸的重组表达载体。
其中所述重组表达载体可通过本领域常规方法获得,即:将上述核酸分子连接于各种大肠杆菌表达载体上构建而成。所述的大肠杆菌表达载体为本领域常规的各种载体。所述载体较佳地包括:各种质粒、粘粒、噬菌体或病毒载体等,优选质粒,本发明所述载体更优选为质粒pet-28a(+),即本发明所述的重组表达载体更优选为将上述核酸连接于质粒pet-28a(+)上构建而成。
本发明提供的技术方案之三是:一种包含上述重组表达载体的重组表达转化体。
其中所述重组表达转化体的制备方法为本领域常规,如可以通过将上述重组表达载体转化至宿主微生物中制得。所述的宿主微生物为本领域常规的各种宿主微生物,只要能满足使上述重组表达载体稳定地自行复制,且使所携带的编码氨基酸序列组成如序列表中seqidno:1所示蛋白质的基因可被有效表达即可。其中所述宿主微生物较佳地为:大肠杆菌(e.coli),更佳地为大肠杆菌bl21(de3)或大肠杆菌dh5α。将前述重组表达载体转化至大肠杆菌bl21(de3)中,即可得本发明优选的基因工程菌株。其中所述转化方法为本领域常规的转化方法,较佳地为化学转化法、热激法或电转法。
本发明提供的技术方案之四是:
一种重组菌,其特征在于,其基因组中含有能表达氨基酸序列组成如序列表中seqidno:1所示蛋白质的外源基因。
本发明提供的技术方案之五是:
一种分离的蛋白质,其特征在于,其氨基酸序列如seqidno:1所示;或为由氨基酸序列如seqidno:1所示的蛋白质与蛋白纯化标签组成的融合蛋白。
本发明所述的蛋白质是一种参与乳酸菌代谢fos的多效调控蛋白,名称为ccpa,来源于植物乳杆菌st-iii,其可以通过本领域常规的方法获得,如将编码所述蛋白质的基因导入合适的宿主细胞得到能够正常表达所述蛋白质的转化体,然后培养所述的转化体,从培养物中分离所述蛋白质即可。所述的蛋白质还可以通过人工全序列合成而得。
如本领域技术人员所知:所述蛋白质的氨基酸序列可以适当引入替换、缺失、改变、插入或增加一个或多个氨基酸而得所述蛋白质的同系物,只要该蛋白质的同系物依然能够保持所述蛋白质的功能即可。
本发明中,所述的蛋白纯化标签为本领域常规所述,较佳地为his6纯化标签,即本发明所述的融合蛋白较佳地为由氨基酸序列如序列表中seqidno:1所示的蛋白质与his6纯化标签组成的融合蛋白。
本发明所提供的技术方案之六是:
上述的蛋白质在调控乳酸菌代谢fos中的作用,所述的蛋白质与fos相关基因启动子区域cre位点结合,以实现对fos代谢的负调控。
本发明所提供的技术方案之七是:蛋白免疫印迹(westernblot),一种鉴定重组蛋白表达产物的方法。
其中所述的鉴定重组蛋白表达产物的方法为本领域常规方法,即采用聚丙烯酰胺凝胶电泳分离蛋白质样品,以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。若在凝胶上形成明显的条带则证明蛋白已表达。
本发明所提供的技术方案之八是:一种验证转录因子与相关基因启动子区域特异性结合的方法,即凝胶迁移实验(electrophoresismobilityshiftassay,emsa)。
利用特异性标记的dna探针与纯化的蛋白相结合,在非变性的聚丙烯酰胺凝胶上形成滞后带,从而验证转录因子与相关基因启动子区域的特异性结合。
其中所述验证转录因子与相关基因启动子区域特异性结合的方法为本领域常规方法,即纯化的dna特异结合蛋白(ccpa)和经标记的dna探针一同孵育,在非变性的聚丙烯酰胺凝胶电泳上分离蛋白质-dna复合物和游离的探针,若在凝胶上形成滞后带则证明转录因子与启动子区域已结合。
为了再次验证所述ccpa与相关基因启动子区域潜在的cre位点的特异性结合,对每个cre位点进行点突变,然后再次按照上述方法进行emsa实验,发现转录因子与突变后的启动子区域没有特异性结合。
本发明所提供的技术方案之九是:一种基于cre-lox系统进行植物乳杆菌基因敲除的方法,其特征在于,包括以下步骤:
步骤1:以植物乳杆菌的基因组为模板,将ccpa基因的上游片段(约1000bp)、下游片段(约1000bp)进行pcr扩增;
步骤2:将上游、下游扩增子分别插入到pnz5319的xhoi、pmei和eco53ki酶切位点中,得到重组质粒pnz5319-up-down,为了获得单基因(ccpa)敲除的突变株,上述重组质粒电转入st-iii,挑选具有氯霉素抗性同时不具有红霉素抗性的重组子,命名为δccpa::cat;
步骤3:通过电转化的方法将pnz5348转入δccpa::cat中,涂布于含红霉素的lb平板上,37℃过夜培养。挑选具有红霉素抗性而不具有氯霉素抗性的菌株,利用pcr进行鉴定,在37℃无抗生素存在的条件下连续传n代使pnz5348丢失,通过抗生素抗性分析和pcr鉴定,最终得到双基因敲除突变株δccpa。然后比较野生型和突变型菌株在混合碳源下的生长情况。
通过生长实验发现植物乳杆菌在fos和葡萄糖同时存在时,会产生二次生长的现象;当ccpa基因敲除之后,二次生长现象消失,在整个生长过程中,尽管葡萄糖以较快的速率被消耗,但组成fos的主要成分也同时被利用,从而表明该基因在fos代谢负调控中的重要作用。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料除特别说明之外,均市售可得。
与现有技术相比,本发明的有益效果是:
本发明通过emsa实验证明了多效调控蛋白ccpa与fos相关基因启动子区域的结合;同时,基因敲除实验验证了多效调控蛋白ccpa在代谢fos时所发挥的调控作用,为构建植物乳杆菌代谢fos的整体调控网络提供了理论依据与实验支持。
附图说明
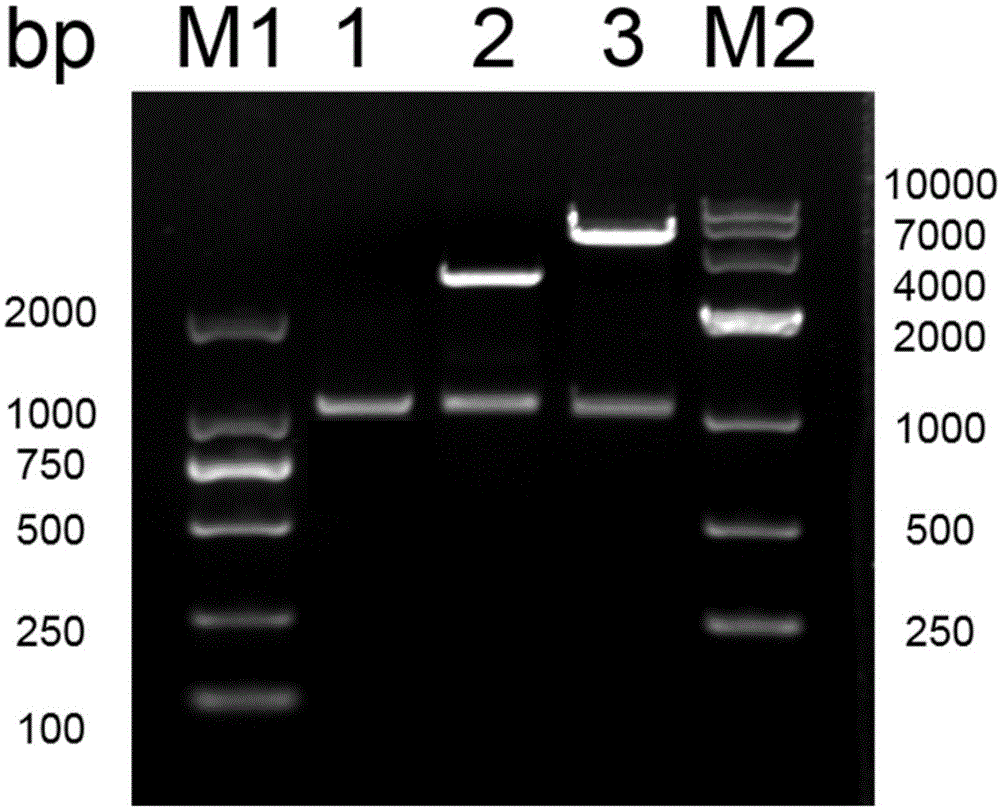
图1中编号“1”的泳道为pcr产物;编号“2”的泳道为pucm-t-ccpa经限制性内切nhei和hindⅲ酶切后的电泳图;编号“3”的泳道为pet-28-ccpa经限制性内切nhei和hindⅲ酶切后的电泳图;m1为dl2000dnamarker;m2为dl10000dnamarker。
图2为重组蛋白表达鉴定及纯化后的sds-page凝胶电泳图,其中,m为蛋白marker;1为大肠杆菌bl21-ccpa诱导前细胞的破碎上清;2为大肠杆菌bl21-ccpa诱导后的细胞破碎上清;3为目的蛋白经ni2+柱纯化后的电泳结果
图3为表达载体pet-28-ccpa的构建示意图。
图4为westernblot的实验结果图,其中,m为蛋白marker;编号“1”的泳道为目的蛋白ccpa纯化后的电泳结果。
图5为验证四个启动子区域ccpa-dna结合的emsa实验结果图,其中,m为蛋白marker;(a)his6标记的ccpa的emsa结果,是含有完整cre区的四个启动子区域的dna片段;(b)his6标记的ccpa的emsa结果,是含有突变的cre位点的四个启动子区域的dna片段;c表示ccpa-dna复合物;f表示游离的dna探针。
图6为混合碳源下发酵过程中野生型菌株的生长曲线和糖消耗量。
图7为混合碳源下发酵过程中δccpa菌株的生长曲线和糖消耗量。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1重组表达质粒的构建
以seqidno:3(上游引物-ccpaf):5’-ctagctagcggctatttttatatggaaaaacaaac-3’和seqidno:4(下游引物-ccpar):5’-cccaagcttcgaacttaatcagcagacttgg-3’为引物,上游5’端加有一段与表达载体末端同源互补的序列,该载体的末端是含nhei酶切口;下游引物5’端加有一段与表达载体末端同源互补的序列,该载体的末端是含hindⅲ酶切口。以植物乳杆菌st-iii基因组为模板,进行pcr反应。pcr反应体系为50μl,各反应物的量为:2μldna模板(9.652ng/μl)、2μlccpaf(0.2μm)、2μlccpar(0.2μm)、25μl2×goldstarbestmastermix,用ddh2o补足至50μl。pcr反应参数为:95℃预变性5min,94℃变性30s,52℃退火30s,72℃延伸1min,30个循环;72℃延伸5min,4℃下保存。扩增产物用1%琼脂糖凝胶电泳检测,并对扩增产物进行纯化。将已纯化的目的基因片段在t4dna连接酶的作用下连接至pucm-t载体上,连接体系:2μl产物(90.131ng/μl)、1μl载体(50ng/μl)、1μl10×ligationbuffer,1μlt4dnaligase(5u/μl),5μlddh2o,反应条件:22℃下连接2.5h。将连接产物转化至大肠杆菌dh5α,通过蓝白斑筛选阳性克隆,并进行pcr鉴定及测序鉴定。抽提含有正确插入片段的重组质粒pucm-t-ccpa与表达质粒pet-28a(+)进行双酶切(nhei、hindⅲ),酶切体系参照限制性内切酶说明书,回收纯化酶切产物。将纯化后的酶切产物进行连接,连接体系:2μl产物(123.159ng/μl)、1μl载体(174.206ng/μl)、1μl10×ligationbuffer,1μlt4dnaligase(5u/μl),5μlddh2o,反应条件:4℃下连接过夜。采用热刺激转化法将连接反应液转化到大肠杆菌bl21感受态细胞中,经37℃恒温振荡培养(180rpm)、60min后,室温4000rpm离心5min,再涂布到含150μg/ml硫酸卡那抗性的lb培养基上,37℃培养24h。筛选阳性克隆并进行测序鉴定,得到正确的目标重组表达载体,将其命名为pet-28-ccpa,该重组表达载体包含一种核酸,其碱基序列如序列表中seqidno:2所示。该核酸编码一种分离的蛋白质,所述的分离的蛋白质为由氨基酸序列如seqidno:1所示的蛋白质与蛋白纯化标签his6组成的融合蛋白,相应的包含该载体的重组菌命名为bl21-ccpa。
结果分析
以植物乳杆菌st-iii基因组作为模板,用seqidno:3和seqidno:4所示序列为引物进行pcr扩增反应,得到1kb左右的基因片段,大小与预期一致,结果如图1编号“1”泳道所示,目标片段中即包含了目的基因ccpa。将扩增后回收纯化的pcr产物片段与对应单酶切载体pet-28a(+)进行连接,构建表达载体pet-28-ccpa,表达载体构建示意图如图3所示。将重组质粒连接物转化至e.colibl21(de3),37℃平板培养24h,挑选阳性转化子接种至lb液体培养基中培养过夜,离心收集菌体,提取重组质粒,用对应的限制性内切酶对重组质粒进行鉴定。pcum-t-ccpa经nhei和hindⅲ双酶切,得到2kb左右的pucm-t线性片段和1kb左右的目的基因片段,如图1编号“2”泳道所示。pet-28-ccpa经nhei和hindⅲ双酶切,得到5kb左右的pet-28a(+)线性片段和1kb左右的目的基因片段,如图1编号“3”泳道所示,证明表达载体pet-28-ccpa成功转入大肠杆菌bl21(de3)宿主菌中。阳性表达宿主命名为bl21-ccpa。
实施例2目的蛋白的诱导表达及sds-page电泳鉴定
挑取大肠杆菌bl21-ccpa单菌落于20ml含150ng/μl的硫酸卡那霉素的液体lb培养基中,37℃,200rpm摇瓶培养过夜。将过夜培养液,接种至200ml含有15μg/ml卡那霉素的液体lb培养基中,接种量为2%,培养温度37℃,摇床振荡速率为200rpm,在od600达到0.4-0.6左右时,加入诱导剂iptg至终浓度为0.1mm,分别在37℃、3h、25℃、6h、和16℃过夜振荡培养,根据sds-page蛋白胶电泳结果选择最佳的诱导条件,结果表明在25℃6h的培养条件下诱导效果最好。诱导完毕后,于4℃,10000rpm/min离心10min,收集湿菌体细胞。每克湿菌体细胞加入5ml细胞裂解缓冲液(0.2mpb,5mnacl,1m咪唑,ph7.4)重悬菌体细胞,加入溶菌酶至1mg/ml,和苯甲基磺酰氟(pmsf)至终浓度1mm,冰浴30min后,进行超声破碎。破碎条件如下:超声200w,超声2s,间歇2s,作用10min,整个过程于冰浴中进行。然后加rnase至10μg/ml和dnase至5μg/ml混匀,细胞破碎液于4℃,12000rpm/min离心10min,收集上清,即蛋白样品。
将10μl蛋白样品与6μl的6×上样缓冲液混合,于漩涡振荡器上振匀,电加热器100℃加热3min后,采用5%的浓缩胶和12%分离胶进行跑胶,电极缓冲液为ph8.3tris-甘氨酸缓冲液。当样品处于浓缩胶时的电压设定为80v,待溴酚蓝指示剂进入分离胶后改电压为120v;电泳完后,用考马斯亮蓝染色液于室温染色2h后,用脱色液脱色至蛋白条带清晰。染色与脱色过程中均在脱色摇床上完成。通过凝胶成像仪灰度扫描电泳胶板,分析蛋白分布与浓度。
结果分析
大肠杆菌bl21-ccpa经iptg诱导后,菌体量明显增多,胞内上清在约39kda处有一条明显的重组蛋白表达条带,诱导蛋白的电泳结果如图2所示。
实施例3蛋白纯化
将每克实施例2所得的湿菌体加入5ml细胞裂解缓冲液(0.2mpb,5mnacl,1m咪唑,ph7.4)重悬菌体细胞,加入溶菌酶至1mg/ml,和苯甲基磺酰氟(pmsf)至终浓度1mm,冰浴30min后,进行超声破碎。破碎条件如下:超声200w,超声2s,间歇2s,作用10min,整个过程于冰浴中进行。然后加rnase至10μg/ml和dnase至5μg/ml混匀,细胞破碎液于4℃,12000rpm/min离心10min,收集上清。将上清装入亲和层析柱后用10倍体积的结合缓冲液(0.2mpb,5mnacl,1m咪唑,ph7.4)洗去杂蛋白后,不同浓度的洗脱缓冲液(0.2mpb,5mnacl和不同浓度的咪唑(浓度分别为:100mmol/l、200mmol/l、300mmol/l、400mmol/l、500mmol/l、600mmol/l))梯度洗脱目标蛋白。取10μl洗脱蛋白液与6μl的6×上样缓冲液混合,用以进行蛋白电泳分析。
结果分析
重组蛋白因带有his6标签可被特异性地吸附至亲和介质(ni2+)上从而与杂蛋白迅速分离。低浓度的咪唑能起到去除杂蛋白并纯化蛋白的作用,而与介质亲和力强的重组蛋白能在高浓度的咪唑作用下洗脱下来。由大肠杆菌bl21-ccpa表达的重组蛋白ccpa通过ni2+亲和色谱进行纯化,在结合缓冲液中添加终浓度1m咪唑,即可以去除大部分的杂蛋白,使得重组蛋白最大限度地吸附到填料上;之后采用梯度浓度洗脱目标蛋白,结果表明300mmol/l浓度的咪唑能完全洗脱亲和填料上的目标蛋白。
实施例4重组蛋白表达产物的westernblot鉴定
配置12%的分离胶,加异丙醇静置20min,待分离胶凝固,吸除异丙醇,再用水冲洗残余的异丙醇,吸水纸吸去残液。加入配置好的5%的浓缩胶,插入梳子,静置20min,待浓缩胶凝固后拔出梳子;安装好电泳装置,上样,120v,90min;转膜,湿转120v稳压转100min;封闭,将醋酸纤维膜放入5%的脱脂牛奶中,封闭30min;pbs洗膜3次,每次5min;孵育一抗(生工生物工程(上海)股份有限公司,d191001-0100),4℃孵育过夜;pbs洗膜3次,每次10min;孵育二抗(上海翊圣生物科技有限公司,33208es60),室温孵育2h;pbs洗膜3次,每次10min;按1:1的比例加入试剂盒(生工生物工程(上海)股份有限公司,d601017-0010)中的两种发光剂,孵育1min;用保鲜膜包起,放入曝光盒中,在暗室中将胶片放入曝光盒中进行曝光,曝光时间长短根据结果自行调控;显影、定影,扫描结果。
结果分析
westernblot实验结果(如图4)显示ccpa基因在45kda处有一条明显的蛋白质印迹带,由此说明ccpa在大肠杆菌中成功进行异源表达。
实施例5emsa实验
在位于两个基因簇中含6个cre位点(seqidno:27-seqidno:32)的4个启动子区(psack、ppts1-saca、pagl4和psacr2)设计引物(psack引物对应seqidno:5-seqidno:6;ppts1-saca引物对应seqidno:7-seqidno:8;pagl4引物对应seqidno:9-seqidno:10;psacr2引物对应seqidno:11-seqidno:12),进行pcr扩增,得到大小为200bp的pcr片段;按照实施例1的方法分别构建克隆表达载体,提取质粒。
(1)2.0%tbe丙烯酰胺凝胶的配置按照试剂盒(生工生物工程(上海)股份有限公司,c631100-0200)说明书进行。
(2)探针的标记:在位于两个基因簇中含6个cre位点的4个启动子区设计引物,以植物乳杆菌st-iii基因组为模板,进行pcr反应。pcr反应体系为50μl,各反应物的量为:2μldna模板(9.652ng/μl)、2μl引物f(0.2μm)、2μl引物r(0.2μm)、25μl2×goldstarbestmastermix,用ddh2o补足至50μl。pcr反应参数为:95℃预变性5min,94℃变性30s,51℃退火30s,72℃延伸20s,30个循环;72℃延伸5min,4℃下保存,得到大小为200bp的pcr片段;按照实施例1的方法分别构建克隆表达载体,提取质粒;以得到的质粒为模板,采用高保真dpxdnaploymerase,以m13f-47(fam)和m13r-48(seqidno:13、seqidno:14)为引物进行pcr扩增。pcr反应体系为50μl,各反应物的量为:2μldna模板(7.169ng/μl)、2μlm13f-47(fam)(0.2μm)、2μlm13r-48(0.2μm)、25μldpxdnaploymerase,用ddh2o补足至50μl。pcr反应参数为:95℃预变性5min,94℃变性30s,67℃退火30s,72℃延伸1min,30个循环;72℃延伸5min,4℃下保存,得到含fam标记的探针psack、ppts1-saca、pagl4和psacr2;将得到的pcr产物,用pcrclean-upsystem试剂盒按照说明书步骤进行纯化,并用nanodrop2000c进行定量分析;
(3)反应体系:将40ng的被标记的dna探针、不同浓度(0、0.4、0.8、1.2μg)的已纯化his6-ccpa蛋白和结合缓冲液(50mmtris-hcl[ph8.0],100mmkcl,2.5mmmgcl2,0.2mmdtt,2μgpolydidcand10%glycerol),用nucleasewater补足至20μl;30℃下孵育30min;
(4)电泳显影;用预冷的0.5×tbe作为电泳缓冲液,120v恒压预电泳30min;将蛋白-dna结合产物上样,继续低温电泳,150v,30min;电泳后的胶进行曝光显影。
为了验证ccpa与潜在存在的cre位点的特异性结合,对每个cre位点进行点突变,突变的原则是:在兼并序列中被确定的碱基突变成其他三个任何一个碱基;“w”代表碱基a或t,突变成g或c。用点突变试剂盒(上海吐露港生物科技有限公司,24303-2)按照说明书的步骤进行突变,标记为cremut(seqidno:33-seqidno:38),如表1。
表1含潜在突变前后cre位点的寡核苷酸核酸序列
a加粗字体表示被突变的碱基
根据突变后的6个cremut的dna序列设计引物(cresackmut引物对应seqidno:15-seqidno:16;crepts1mut引物对应seqidno:17-seqidno:18;cresaca1mut引物对应seqidno:19-seqidno:20;cresaca2mut引物对应seqidno:21-seqidno:22;creagl4mut引物对应seqidno:23-seqidno:24;cresacpts26mut引物对应seqidno:25-seqidno:26),以cre位点突变前得到的质粒为模板,pcr反应体系为50μl,各反应物的量为:2μldna模板(7.169ng/μl)、2μl引物f(0.2μm)、2μl引物r(0.2μm)、25μl2×goldstarbestmastermix,用ddh2o补足至50μl。pcr反应参数为:95℃预变性5min,94℃变性30s,51℃退火30s,72℃延伸20s,30个循环;72℃延伸5min,4℃下保存。通过pcr仪扩增并纯化后,用ezmax一步法快速克隆试剂盒进行pcr产物自连,连接体系:2μl产物(20.476ng/μl)、5μl载体(17.281ng/μl)、1μlezmax重组酶、2μl5×buffer,反应条件:37℃反应30min。
筛选阳性克隆,经测序验证后提取质粒,再次按照上述方法进行emsa实验。
结果分析
为验证ccpa是否能够在体外与6个潜在的cre位点结合,用his6标记的ccpa蛋白在大肠杆菌中进行异源表达,用his6-ccpa蛋白进行emsa实验。结果如图5a(泳道1-4)所示,his6-ccpa蛋白与被fam标记的启动子psack、ppts1-saca、pagl4和psacr混合后,凝胶电泳显示出有滞后的条带,形成ccpa-dna复合物,并且随着his6-ccpa蛋白浓度的逐渐增加,滞后的条带颜色越来越深,说明ccpa-dna复合物结合强度越来越强。相反,当标记和未标记的dna探针混合在一起进行竞争性实验时(泳道5),没有发现滞后带的形成,ccpa蛋白与dna探针的结合效应被竞争性抑制,证明了ccpa与这些启动子区域的特异性结合。
emsa实验与含cre位点的启动子区域有特异性结合,但不能精确到具体结合的位置是否是cre位点序列。为验证ccpa蛋白与启动子区域内cre位点的直接结合,对cre位点序列进行点突变后,再次进行emsa实验。结果如图5b所示,蛋白与被突变后的psack、ppts1-saca、pagl4和psacr区域的结合完全消失,结果表明ccpa蛋白与这些启动子区域的6个cre位点之间存在特异性结合。
实施例7敲除ccpa后对植物乳杆菌st-iii利用不同碳源能力的影响
(1)以seqidno:39(正向引物-upf):5’-ccgctcgagataaaaatagccctttcttgcttg-3’和seqidno.40(反向引物upr):5’-gcgtttaaacggcttccagacagacgtcaaag-3’为引物,正向5’端的末端是含xhoi酶切口;反向引物5’端的末端是含pmei酶切口;以seqidno:41(正向引物-downf):5’-gatatctgaaagactttcccacgtttagc-3’和seqidno:42(反向引物downr):5’-gatatcgttcggttgtctgccagctagt-3’为引物,正向及反向5’端的末端均含eco53ki酶切口。以植物乳杆菌st-iii基因组为模板,进行pcr反应。pcr反应体系为50μl,各反应物的量为:2μldna模板(9.652ng/μl)、2μlupf(downf)((0.2μm)、2μlupr(downr)(0.2μm)、25μl2×goldstarbestmastermix,用ddh2o补足至50μl。pcr反应参数为:95℃预变性5min,94℃变性30s,55℃退火30s,72℃延伸1min,30个循环;72℃延伸5min,4℃下保存。扩增产物用1%琼脂糖凝胶电泳检测,并对扩增产物进行纯化。将外源质粒pnz5319用限制性内切酶xhoi和pmei进行双酶切,酶切体系参照限制性内切酶说明书,回收纯化酶切产物。将纯化后的扩增产物(up片段)和酶切产物进行连接,连接体系:2μl产物(12.697ng/μl)、5μl载体(21.356ng/μl)、1μlezmax重组酶、2μl5×buffer,反应条件:37℃反应30min。采用热刺激转化法将连接反应液转化到大肠杆菌dh5α感受态细胞中,经37℃恒温振荡培养(180rpm)、60min后,室温4000rpm离心5min,再涂布到含105μg/ml氯霉素和5005μg/ml红霉素抗性的lb培养基上,37℃培养24h。筛选阳性克隆并进行测序鉴定,得到正确的目标重组质粒,将其命名为pnz5319-up。然后以pnz5319-up为载体,用限制性内切酶eco53ki进行单酶切,酶切体系参照限制性内切酶说明书,回收纯化酶切产物(pnz5319-up)。将纯化后的扩增产物(down片段)和酶切产物进行连接,其他方法均与上述方法相同,筛选阳性克隆并进行测序鉴定,得到正确的目标重组质粒,将其命名为pnz5319-up-down。
(2)抽提含有正确插入片段的重组质粒pnz5319-up-down,电转化入植物乳杆菌st-ⅲ,涂布于含有155μg/ml氯霉素的mrs平板上37℃培养48h,筛选阳性克隆并进行测序鉴定。挑取含有正确重组质粒pnz5319-up-down的植物乳杆菌单菌落于5ml含10μg/ml的氯霉素的mrs培养基液体中,37℃,静置培养过夜。传代直至挑选出具有氯霉素抗性同时不具有红霉素抗性的重组子δccpa::cat。
(3)通过电转化的方法将pnz5348转入δccpa::cat中,涂布于含305μg/ml红霉素的lb平板上,37℃过夜培养。挑选具有红霉素抗性而不具有氯霉素抗性的菌株,利用pcr进行鉴定。在37℃无抗生素存在的条件下连续传n代使pnz5348丢失,通过抗生素抗性分析和pcr鉴定,最终得到双基因敲除突变株δccpa。
将活化好的ccpa突变前后的两株菌按2%的接种量接种到含0.1%的葡萄糖和0.4%的fos的cdm液体培养基上,37℃下静置培养16h,每隔2h测定od600的值,并绘制其标准曲线。每个样品重复测定3次。为了测定突变型菌株在混合碳源下生长的过程中对碳源的利用情况,在上述发酵过程中,每2h取样一次,将发酵液于25℃,10000g,离心10min后取上清,将上清用0.45μm的滤膜过滤除菌后,在通过hpaec法进行糖残留量的测定。测定条件是:ics5000型色谱柱为carbopacpa20阴离子交换柱(150mm×3mm),检测器脉冲安培检测器;流动相是水和250mm的naoh水溶液,梯度洗脱;流速为0.5ml/min,上样量为20μl。每个样品重复测定3次。
结果分析
以植物乳杆菌st-iii基因组作为模板,用seqidno.5、6和seqidno:7、8所示序列为引物进行pcr扩增反应,得到1kb左右目的基因的上下游片段,大小与预期一致,目标片段中即包含了目的基因ccpa的上游(up片段)和下游(down片段)。将扩增后回收纯化的pcr产物片段与对应酶切载体pnz5319进行连接,构建重组质粒pnz5319-up-down。将重组质粒连接物转化至大肠杆菌dh5α,37℃平板培养24h,挑选阳性转化子接种至lb液体培养基中培养过夜,离心收集菌体,提取重组质粒,用对应的限制性内切酶对重组质粒进行鉴定。抽提含有正确插入片段的重组质粒pnz5319-up-down,电转化入植物乳杆菌st-ⅲ,挑取含有正确重组质粒pnz5319-up-down的植物乳杆菌单菌落于含10μg/ml的氯霉素的mrs培养基液体中,37℃,静置培养过夜。传代直至挑选出具有氯霉素抗性同时不具有红霉素抗性的重组子δccpa::cat。通过电转化的方法将pnz5348转入δccpa::cat中,涂布于含红霉素的lb平板上,37℃过夜培养。挑选具有红霉素抗性而不具有氯霉素抗性的菌株,利用pcr进行鉴定。在37℃无抗生素存在的条件下连续传n代使pnz5348丢失,通过抗生素抗性分析和pcr鉴定,最终得到双基因敲除突变株δccpa。野生型菌株在混合碳源下的生长情况如图6所示,突变型菌株在混合碳源下的生长情况如图7所示。从图中可以看出敲除ccpa基因后植物乳杆菌st-iii的二次生长现象消失,在整个生长过程中,尽管葡萄糖以较快的速率被消耗,但组成fos的主要成分也同时被利用,发酵结束后,所有的糖都被消耗殆尽。
sequencelisting
<110>上海应用技术大学
<120>植物乳杆菌代谢低聚果糖的多效调控蛋白及其调控方法
<130>48
<160>48
<170>patentinversion3.5
<210>1
<211>336
<212>prt
<213>lactobacillusplantarum
<400>1
metglulysglnthrvalthriletyraspvalalaargglualaala
151015
valsermetalathrvalserargvalvalasnglyasnproasnval
202530
lysproalathrarglyslysvalleualavalilegluargleuasp
354045
tyrargproasnalavalalaargglyleualaserlysargserthr
505560
thrvalglyvalileileproaspvalthrasniletyrphealaala
65707580
leualaargglyileaspaspilealametmettyrlystyrasnile
859095
ileleuthrasnseraspaspalaglygluglngluvalasnvalleu
100105110
asnthrleumetalalysglnvalaspglyvalilephemetglyasn
115120125
hisileaspasplysleuargalagluphelysargalalysalapro
130135140
valvalleualaglythrvalaspproasnasngluthrproserval
145150155160
asnileasptyralaalaalavalglugluvalvalthrasnleuile
165170175
alaargglyhislyslysilealaleualaleuglyserleusergln
180185190
serileasnalaglutyrargleuthrglytyrlysargalaleuthr
195200205
lysalalysilepropheaspaspalaleuvaltyrglualaglytyr
210215220
sertyraspalaglyarglysleuglnprovalilealaaspsergly
225230235240
alathralavalphevalglyaspaspglumetalaalaglyleuile
245250255
asnalasermetgluserglyileasnvalproaspaspleugluval
260265270
valthrserasnaspthrileilethrglnilethrargproalaile
275280285
thrserilethrglnproleutyraspileglyalavalalametarg
290295300
metleuthrlysleumetasnasplysgluleuaspglulysthrval
305310315320
thrleuprotyrglyilevalargargglyserthrlysseralaasp
325330335
<210>2
<211>1011
<212>dna
<213>lactobacillusplantarum
<400>2
atggaaaaacaaacagtaacaatttatgatgtggcacgcgaagcggcggtttcaatggcc60
acagtttcacgagtcgtcaatggtaatcccaacgttaaacccgcaacgcgtaaaaaggta120
ctagctgtcatcgaacggttagattaccggcctaacgcagttgcacgagggttagcaagt180
aagcgttcaactacggtcggggtaatcatcccggacgtgaccaacatttattttgcggcg240
ttagcacgcgggattgatgatattgcgatgatgtacaagtacaatatcattttaacgaac300
tccgatgatgcgggtgaacaagaagtcaacgtgttgaatacgttgatggctaagcaagtt360
gacggggtcatctttatgggtaaccatattgatgacaagctccgggctgaattcaaacgg420
gccaaggcaccagttgttttagctgggacggttgatccgaacaatgaaacgccaagcgtt480
aatatcgactacgctgcagctgttgaagaagtggtcactaacttaattgcacgtggtcac540
aagaagatcgcgttggctttaggctcattgtcacaatcaatcaacgctgaataccgactg600
acgggttacaaacgggccttaaccaaggctaagattccgtttgacgacgccctcgtttat660
gaagcgggctactcatatgatgctgggcggaagttacaaccagtcattgcggatagtggc720
gctactgcggtattcgttggggacgacgaaatggcggctggtcttatcaacgcaagtatg780
gaatccgggatcaacgttccagatgacttggaagttgtgacgagtaatgatacgattatt840
acccaaatcacgcgtccagccattacctcgattacccaaccattatatgatattggtgcg900
gttgccatgcggatgttaactaagttgatgaacgataaagaacttgacgagaagacggtc960
accttgccatacggcattgttcggcggggctcaaccaagtctgctgattaa1011
<210>3
<211>35
<212>dna
<213>artificialsequence
<400>3
ctagctagcggctatttttatatggaaaaacaaac35
<210>4
<211>31
<212>dna
<213>artificialsequence
<400>4
cccaagcttcgaacttaatcagcagacttgg31
<210>5
<211>23
<212>dna
<213>artificialsequence
<400>5
cagcttcaattgcacctaaaagc23
<210>6
<211>24
<212>dna
<213>artificialsequence
<400>6
aacgactcaaagtcacaaatgtcc24
<210>7
<211>20
<212>dna
<213>artificialsequence
<400>7
taagtacgcggtcggcaact20
<210>8
<211>23
<212>dna
<213>artificialsequence
<400>8
aggggtataacgggttttacgat23
<210>9
<211>21
<212>dna
<213>artificialsequence
<400>9
caaccggggtaacatttggat21
<210>10
<211>22
<212>dna
<213>artificialsequence
<400>10
ggcggtcgatcagctattacat22
<210>11
<211>47
<212>dna
<213>artificialsequence
<400>11
cgccagggttttcccagtcacgacaacactaggccgatggtttaaat47
<210>12
<211>48
<212>dna
<213>artificialsequence
<400>12
agcggataacaatttcacacaggagactaaatgttttaagggcaaacg48
<210>13
<211>24
<212>dna
<213>artificialsequence
<400>13
cgccagggttttcccagtcacgac24
<210>14
<211>24
<212>dna
<213>artificialsequence
<400>14
agcggataacaatttcacacagga24
<210>15
<211>44
<212>dna
<213>artificialsequence
<400>15
aagattgaccgggcgcggaaatttctaattaagagtatactatt44
<210>16
<211>38
<212>dna
<213>artificialsequence
<400>16
gcccggtcaatcttttaactttgacgtaaatgttgcgt38
<210>17
<211>45
<212>dna
<213>artificialsequence
<400>17
aagatttgacgggaggcggctattaaattaaaaataattttagac45
<210>18
<211>41
<212>dna
<213>artificialsequence
<400>18
cctcccgtcaaatcttgacccaataataacaagtttggaaa41
<210>19
<211>38
<212>dna
<213>artificialsequence
<400>19
cgcaaccatcccatgatgatatgtcaatcgtttgacat38
<210>20
<211>41
<212>dna
<213>artificialsequence
<400>20
ttcgaatcagccatccgctatcagtgtaagcggttttataa41
<210>21
<211>40
<212>dna
<213>artificialsequence
<400>21
tcatgggatggttgcgaatagtgacaattaagtcgaacgt40
<210>22
<211>39
<212>dna
<213>artificialsequence
<400>22
cgcaaccatcccatgatgatacctaccgactaagctttt39
<210>23
<211>44
<212>dna
<213>artificialsequence
<400>23
tacggttaccgtaagggggatataggagtagtaattatgcaaaa44
<210>24
<211>39
<212>dna
<213>artificialsequence
<400>24
ccttacggtaaccgtaggctttaatcaaaagaaaaaggc39
<210>25
<211>35
<212>dna
<213>artificialsequence
<400>25
aatgtgtcagtcggaataggggggtgagcctaaat35
<210>26
<211>41
<212>dna
<213>artificialsequence
<400>26
ccgactgacacattgggtaattaatataacgacctttcaag41
<210>27
<211>16
<212>dna
<213>lactobacillusplantarum
<400>27
tgtaatcgttaacaaa16
<210>28
<211>16
<212>dna
<213>lactobacillusplantarum
<400>28
tgtaaagcgcttgcat16
<210>29
<211>16
<212>dna
<213>lactobacillusplantarum
<400>29
tgtcaaacgattgaca16
<210>30
<211>16
<212>dna
<213>lactobacillusplantarum
<400>30
gtgtaagcggttttat16
<210>31
<211>16
<212>dna
<213>lactobacillusplantarum
<400>31
gtggaatcgattccaa16
<210>32
<211>16
<212>dna
<213>lactobacillusplantarum
<400>32
tggaaacgattccaaa16
<210>33
<211>16
<212>dna
<213>artificialsequence
<400>33
aagattgaccgggcgg16
<210>34
<211>16
<212>dna
<213>artificialsequence
<400>34
aagatttgacgggagg16
<210>35
<211>16
<212>dna
<213>artificialsequence
<400>35
aagcttagtcggtagg16
<210>36
<211>16
<212>dna
<213>artificialsequence
<400>36
tcatgggatggttgcg16
<210>37
<211>16
<212>dna
<213>artificialsequence
<400>37
tacggttaccgtaagg16
<210>38
<211>16
<212>dna
<213>artificialsequence
<400>38
aatgtgtcagtcgggg16
<210>39
<211>33
<212>dna
<213>artificialsequence
<400>39
ccgctcgagataaaaatagccctttcttgcttg33
<210>40
<211>32
<212>dna
<213>artificialsequence
<400>40
gcgtttaaacggcttccagacagacgtcaaag32
<210>41
<211>29
<212>dna
<213>artificialsequence
<400>41
gatatctgaaagactttcccacgtttagc29
<210>42
<211>28
<212>dna
<213>artificialsequence
<400>42
gatatcgttcggttgtctgccagctagt28
<210>43
<211>16
<212>dna
<213>lactobacillusplantarum
<400>43
tgtaatcgttaacaaa16
<210>44
<211>16
<212>dna
<213>lactobacillusplantarum
<400>44
tgtaaagcgcttgcat16
<210>45
<211>16
<212>dna
<213>lactobacillusplantarum
<400>45
gtgtaagcggttttat16
<210>46
<211>16
<212>dna
<213>lactobacillusplantarum
<400>46
gtggaatcgattccaa16
<210>47
<211>16
<212>dna
<213>lactobacillusplantarum
<400>47
tggaaacgattccaaa16
<210>48
<211>16
<212>dna
<213>lactobacillusplantarum
<400>48
tgtcaaacgattgaca16
- 还没有人留言评论。精彩留言会获得点赞!