3-氨基磺酰基丙氨酸的制备方法与流程
本发明属于化合物制备领域,具体地涉及3-磺胺基丙氨酸的一种全新的合成方法。
背景技术:
3-氨基磺酰基丙氨酸是一种具有多种生物活性的化合物,如抗结核活性(j.am.chem.soc.1959,81,19,5125–5128),抗肿瘤活性(j.med.chem.1978,21,1,45-49)。
目前,已报道的3-氨基磺酰基丙氨酸的合成方法如路线一所示(j.am.chem.soc.1959,81,19,5125–5128;j.med.chem.1978,21,1,45-49),该路线中使用到毒性气体氯气,以及具有强烈刺激性气味的氨气;此外,最后一步酸性水解乙酰基有可能生成其他大极性杂质,目标物3-氨基磺酰基丙氨酸的纯化非常复杂,需要经过大孔树脂进行柱层析,再经重结晶获得。整体而言,该合成方法存在操作复杂,条件苛刻,所用试剂毒性较大,且收率较低,副产物较多,分离纯化复杂的特点。研究出合成方法简便,路线较短的合成方法对于3-氨基磺酰基丙氨酸的大量合成以及后期工业化生产具有重要意义。
路线一、文献报道的3-氨基磺酰基丙氨酸的合成路线
技术实现要素:
本发明的主要目的是寻找一条合成路线相对较短,避开使用氯气,氨气等刺激性毒性气体的反应,且后处理操作简单,即可高收率获得3-磺胺丙酸的制备方法。
为了实现上述目的,本申请所采用的技术方案如下:
3-氨基磺酰基丙氨酸的制备方法,所述3-氨基磺酰基丙氨酸具有式i所示结构,其中手性碳原子选自(r)构型、(s)构型或者消旋体:
具体包括如下步骤:
(1)以化合物a为原料,在碱性条件下,在非质子溶剂中,与二碳酸二叔丁酯反应生成化合物b;
(2)化合物b在碳酸铵存在下,经二醋酸碘苯氧化获得化合物c;
(3)化合物c以二氯甲烷为溶剂,在三氟乙酸作用下脱除叔丁氧羰基保护,并同时酸水解叔丁酯获得式i目标物3-磺胺基丙氨酸。
作为优选的一种具体方案,所述步骤(1)的碱性环境由加入三乙胺、n,n二异丙基乙胺、碳酸钾或碳酸钠提供。
作为优选的一种具体方案,所述化合物a与所述二碳酸二叔丁酯的摩尔比为1:2~3.0,优选为1:2.5.
作为优选的一种具体方案,所述步骤(1)的碱性物质选自三乙胺、n,n二异丙基乙胺、碳酸钾或碳酸钠提供,所述碱性物质与所述化合物a的摩尔比为4.5~5.5:1,优选为5:1。
作为优选的一种具体方案,所述步骤(1)的非质子溶剂选自二氯甲烷、乙酸乙酯或乙腈。
作为优选的一种具体方案,所述步骤(1)中,反应完毕后,用二氯甲烷或乙酸乙酯稀释,分别经过酸洗、饱和碳酸氢钠洗、饱和食盐水洗,再经无水硫酸镁或无水硫酸钠干燥,过滤,浓缩既得粗产品化合物a。
作为优选的一种具体方案,所述步骤(2)中化合物b:碳酸铵:二醋酸碘苯的摩尔比为1:1.5~2.5:6~8,优选为1:2:7。
作为优选的一种具体方案,所述步骤(2)为以甲醇为溶剂,在碳酸铵存在下,使用二醋酸碘苯,氧化获得化合物c,该步骤反应完毕后经柱层析获得的纯品,或者经萃取操作获得的粗品可直接用于下一步反应。
作为优选的一种具体方案,所述步骤(3)的酸性条件为以二氯甲烷为溶剂,加入三氟乙酸。
作为优选的一种具体方案,所述步骤(3)中,反应完毕后,用乙醇或者甲醇溶解,加吡啶获得游离的目标化合物。
作为优选的一种具体方案,在步骤(3)后还包括重结晶的步骤,具体为使用95%的乙醇重结晶。
作为优选的一种具体方案,所述步骤(1)~(3)的反应温度为室温,具体为25℃±5℃。
作为优选的一种具体方案,具体路线如路线二所示,以l-胱氨酸双(叔丁酯)二盐酸盐为原料,三乙胺存在下,同二碳酸二叔丁酯在二氯甲烷中反应5小时,二氯甲烷稀释,1n的盐酸洗涤,饱和碳酸氢钠洗涤,饱和食盐水洗,无水硫酸镁干燥,过滤,浓缩制得化合物b;化合物b溶于甲醇中,在二醋酸碘苯,碳酸铵存在下反应12小时,浓缩,硅胶柱层析可得化合物c;化合物c溶于二氯甲烷中,加入三氟乙酸搅拌2小时,浓缩,加入二氯甲烷,再次浓缩,加入工业乙醇(95%)溶清,向反应液中加入吡啶,静置1小时,有白色固体析出,过滤,工业乙醇洗涤,既得目标化合物l-i为白色固体。
采用上述技术方案所产生的有益效果在于:
本发明所提供的制备方法,较已报道的路线具有以下优势:1)本发明避免了使用氯气,氨气等刺激性毒性气体;2)整体路线后处理简单,仅仅在第二步需要简单的柱层析;3)最后一步目标化合物的纯化操作简单,且化合物纯度和收率较高;4)全部过程条件温和,室温下即可反应,无需苛刻条件和环境,可以降低能耗。
本发明所提供的方法三步环环紧扣,反应条件温和,简化了后处理操作,从整个过程中减少了副产物的含量,保证了最终产物的纯度,并且提高了收率,整个过程适合工业化控制,适合于大规模生产。
附图说明
以下,结合附图来说明本发明实施方案,其中:
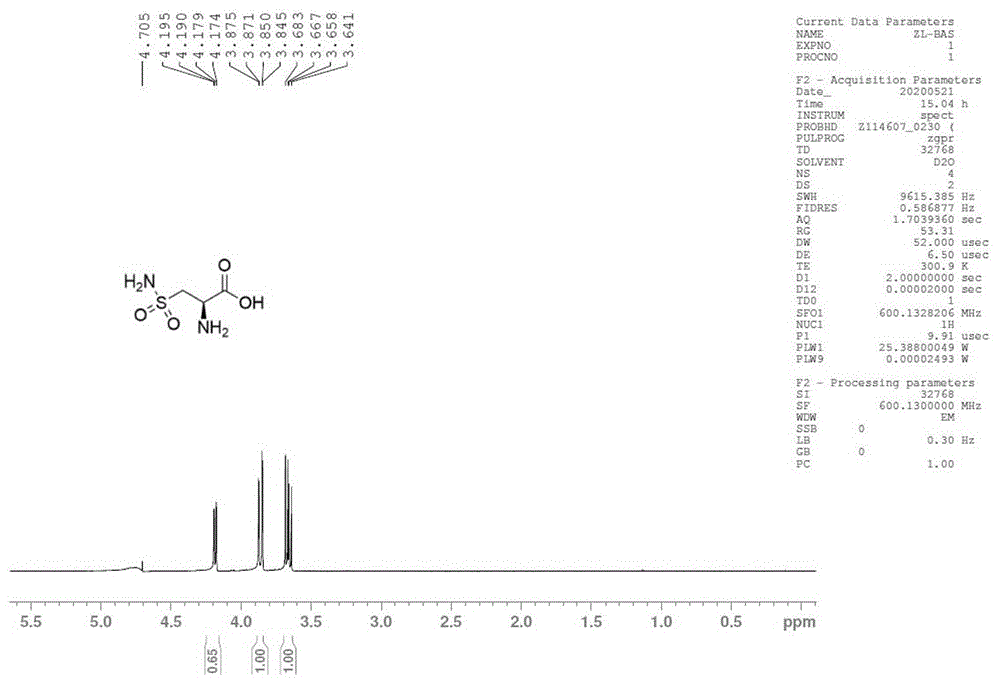
图1为化合物l-3-氨基磺酰基丙氨酸的核磁氢谱;
图2为化合物l-3-氨基磺酰基丙氨酸的核磁碳谱。
具体实施方式
以下结合具体实施例进一步解释和说明本发明,但不能用来限制本发明的范围。
实施例1:l-3-氨基磺酰基丙氨酸(l-i)的合成
室温下,向(l)-胱氨酸双(叔丁酯)二盐酸盐(5.00g,11.76mmol)的二氯甲烷溶液中(100ml),加入二碳酸二叔丁酯(6.40g,29.35mmol),三乙胺(8.18ml,58.80mmol),同温搅拌5小时,tcl检测反应完全(二氯甲烷:甲醇:氨水=10:1:0.005)。将该反应液倾入二氯甲烷(200ml)中,1n的盐酸水溶液洗涤(100ml),饱和碳酸氢钠水溶液洗涤(100ml),饱和食盐水洗(100ml),无水硫酸镁干燥,过滤,浓缩得化合物b(6.16g,收率95%)。
室温下,向化合物(l)-b(2.00g,3.62mmol)的甲醇(100ml)溶液中加入碳酸铵(0.69g,7.24mmol),二醋酸碘苯(8.16g,25.34mmol),同温搅拌过夜,tlc检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。残余物经硅胶柱层析(石油醚:乙酸乙酯=2:1)既得化合物(l)-c为白色固体(1.64g,收率70%)。
室温下,向化合物(l)-c(1g,3.08mmol)的二氯甲烷(20ml)溶液中加入三氟乙酸(5ml),同温搅拌2小时,tcl检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。向残余物中加入二氯甲烷(20ml),再次浓缩,随后油泵抽真空1小时,带走残余的三氟乙酸。向残余物中加入95%乙醇(20ml),加入吡啶2ml,静置有固体析出,过滤既得目标化合物l-3-氨基磺酰基丙氨酸为白色固体(420mg,收率81%),[a]25d=+11.87(c=0.32,h2o);1hnmr(500mhz,d2o)δ4.18(dd,j=2.75hz,9.89hz,1h),3.86(dd,j=15.37hz,2.75hz,1h),3.66(dd,j=15.37hz,9.89hz,1h)(见附图1);13cnmr(125mhz,cdcl3)δ170.58,54.22,50.29(见附图2);ms-esi(m/z):169(m+h)+。
实施例2:d-3-氨基磺酰基丙氨酸(d-i)的合成
室温下,向(d)-胱氨酸双(叔丁酯)二盐酸盐(5.00g,11.76mmol)的乙酸乙酯溶液中(100ml),加入二碳酸二叔丁酯(6.40g,29.35mmol),碳酸钾(8.11g,58.80mmol),同温搅拌5小时,tcl检测反应完全(二氯甲烷:甲醇:氨水=10:1:0.005)。将该反应液倾入乙酸乙酯(200ml)中,1n的盐酸水溶液洗涤(100ml),饱和碳酸氢钠水溶液洗涤(100ml),饱和食盐水洗(100ml),无水硫酸镁干燥,过滤,浓缩得化合物(d)-b(5.29g,收率84%)。
室温下,向化合物(d)-b(2.00g,3.62mmol)的甲醇(100ml)溶液中加入碳酸铵(0.69g,7.24mmol),二醋酸碘苯(8.16g,25.34mmol),同温搅拌过夜,tlc检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。残余物经硅胶柱层析(石油醚:乙酸乙酯=2:1)既得化合物(d)-c为白色固体(1.55g,收率66%)。
室温下,向化合物(d)-c(1g,3.08mmol)的二氯甲烷(20ml)溶液中加入三氟乙酸(5ml),同温搅拌2小时,tcl检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。向残余物中加入二氯甲烷(20ml),再次浓缩,随后油泵抽真空1小时,带走残余的三氟乙酸。向残余物中加入95%乙醇(20ml),加入吡啶2ml,静置有固体析出,过滤得目标化合物(d)-3-氨基磺酰基丙氨酸为白色固体(383mg,收率74%),[a]25d=-10.65(c=0.45,h2o);ms-esi(m/z):169(m+h)+。
实施例3:(d,l)-3-氨基磺酰基丙氨酸(d,l)-i的合成
室温下,向(d,l)-胱氨酸双(叔丁酯)二盐酸盐(5.00g,11.76mmol)的二氯甲烷溶液中(100ml),加入二碳酸二叔丁酯(6.40g,29.35mmol),n,n-二异丙基乙胺(10.25ml,58.80mmol),同温搅拌5小时,tcl检测反应完全(二氯甲烷:甲醇:氨水=10:1:0.005)。将该反应液倾入二氯甲烷(200ml)中,1n的盐酸水溶液洗涤(100ml),饱和碳酸氢钠水溶液洗涤(100ml),饱和食盐水洗(100ml),无水硫酸镁干燥,过滤,浓缩得化合物(d,l)-b(5.91g,收率91%)。
室温下,向化合物b(2.00g,3.62mmol)的甲醇(100ml)溶液中加入碳酸铵(0.69g,7.24mmol),二醋酸碘苯(8.16g,25.34mmol),同温搅拌过夜,tlc检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。残余物经硅胶柱层析(石油醚:乙酸乙酯=2:1)既得化合物(d,l)-c为白色固体(1.81g,收率77%)。
室温下,向化合物(d,l)-c(1g,3.08mmol)的二氯甲烷(20ml)溶液中加入三氟乙酸(5ml),同温搅拌2小时,tcl检测反应完全(石油醚:乙酸乙酯=1:1),浓缩。向残余物中加入二氯甲烷(20ml),再次浓缩,随后油泵抽真空1小时,带走残余的三氟乙酸。向残余物中加入95%乙醇(20ml),加入吡啶2ml,静置有固体析出,过滤既得目标化合物(d,l)-3-氨基磺酰基丙氨酸为白色固体(339mg,收率65%),ms-esi(m/z):169(m+h)+。
实施例4:l-3-氨基磺酰基丙氨酸(l-i)的重结晶
向l-3-氨基磺酰基丙氨酸(500mg)加入95%乙醇(15ml),加热回流1小时至溶清,冷却至室温有固体析出,过滤得l-3-氨基磺酰基丙氨酸(l-i)纯品。
- 还没有人留言评论。精彩留言会获得点赞!