一种GPR120小分子荧光探针及其应用的制作方法
本发明属于药物技术领域,具体涉及一种GPR120小分子荧光探针及其应用。
背景技术:
GPR120作为G蛋白偶联受体家族的重要成员,是长链不饱和游离脂肪酸(FFAs)的受体(Hirasawa et al.2005)。GPR120的mRNA在多种组织都有表达,其中小肠、肺、结肠、脂肪等组织部位GPR120的表达水平较高(Hirasawa et al.2005)。在细胞水平上,GPR120的mRNA在味蕾细胞(Matsumura et al.2007)、脂肪细胞(Gotoh et al.2007)、巨噬细胞(Oh,D et al.2010)以及肠内分泌细胞(Hirasawa et al.2005)等多种细胞系中表达丰富。这说明GPR120在组织和细胞上的表达及分布水平的高低可能与许多生理疾病息息相关,例如肥胖、饮食、糖尿病等。
研究表明,长链不饱和游离脂肪酸(FFAs)刺激GPR120会促进GLP-1的分泌、增加胰岛素敏感性、以及抗炎等一系列的生理过程(Hirasawa et al.2005)。其中,GLP-1是一种肠道分泌的荷尔蒙,具有调节肠道动力、抑制胃酸及胰高血糖素分泌等多种生理功能。因此,GPR120作为治疗糖尿病、肥胖症的潜在治疗靶标,值得我们进一步研究。
到目前为止,大量的G蛋白偶联受体的小分子荧光探针被相继报道出来,而且已经广泛地应用于细胞水平的受体可视化以及药理学研究(Ma,Z et al.2014)。小分子荧光探针具有快速、灵敏、高通量和易于自动化等特点,已经广泛应用于蛋白质、核酸等重要生物分子的生物学和药理学检测中,对疾病机制探讨、临床诊断及药物筛选等领域的发展具有重要意义。小分子荧光探针主要有三部分组成:药效基团、连接基团、荧光基团。药效基团与靶标蛋白成高亲和力的结合,连接基团即为药效基团和荧光基团之间的连接链,而荧光基团可通过外界激发从而发射荧光以标记靶标蛋白。
然而,对于GPR120目前还未有其小分子荧光探针的相关报道。如果我们能找到一个理想的GPR120探针分子作为荧光底物进行竞争性药理实验,那么就会为GPR120受体的拮抗剂或激动剂的活性研究提供一个快速药理筛选平台。因此研究建立针对GPR120的小分子荧光探针高通量筛选与标记的方法势必能推动GPR120的研究发展。
技术实现要素:
本发明针对现有技术不足,提供一种GPR120小分子荧光探针及其制备方法、光学性质、生物活性以及GPR120细胞成像研究中的应用。该小分子荧光探针对GPR120具有高选择性和高灵敏度,可以作为工具药去研究GPR120的生物学和生理学特征。
为实现上述目的,本发明采用下述技术方案:
本发明的第一个方面,提供一种GPR120的小分子荧光探针,具有下述结构通式(Ⅰ):
式中,R为各种荧光团;n=1-6。
优选的,所述R为香豆素类、萘二酰亚胺类荧光团;n=1-5。
进一步优选的,具有如下结构式的化合物,其中,n=1-5。
进一步优选的,n=1,2,3,5;R为含羧基的香豆素类荧光团。
进一步优选的,n=1,2,4;R为萘二酰亚胺类荧光团。
所述香豆素类荧光团的结构式如Ⅱ所示:
所述萘二酰亚胺类荧光团的结构式如Ⅳ所示,其中R1和R2本领域技术人员可以常规得到,在本发明的某些具体实施例中,R1可为氢,R2可为二甲胺基。
更优选的,是下述化合物:
本发明所述化合物可用作GPR120的小分子荧光探针。
本发明的第二个方面,提供一种所述GPR120的小分子荧光探针的制备方法,包括以下步骤:
(1)药效基团的制备:首先由对羟基苯丙酸和甲醇反应制备得到中间体1,然后将由中间体1与4-硝基卤苄反应制备得到中间体2,在将中间体2中的硝基还原为氨基得到中间体3,即得到药效基团;其中,所述中间体1的结构式如1所示,中间体2的结构式如2所示,中间体3的结构式如3所示:
(2)连接基团连接荧光基团得到中间体5,所述连接基团的结构式如(Ⅳ)所示,所述中间体5的结构式如5所示,其中,n=0~5,R为各种荧光团:
(3)荧光探针的制备:将所述中间体3与所述中间体5进行酰胺化反应制备得到中间体6,然后中间体6中的酯基水解为羧基,得到荧光探针,如(Ⅰ)所示;其中,所述中间体6的结构式如6所示,其中,n=0~5,R为各种荧光团:
步骤(1)中,所述4-硝基卤苄为4-硝基氯苄、4-硝基溴苄或4-硝基碘苄,优选4-硝基溴苄。
步骤(1)中,所述中间体1的具体制备方法为:对羟基苯丙酸和无水甲醇搅拌溶解后加入催化量的浓硫酸,在55~65℃(优选60℃)下搅拌反应5~7h,得到含有中间体1的反应液;中间体1的纯化方法为:向含有中间体1的反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得无色透明油状物,即为中间体1。
所述中间体2的具体制备方法为:将中间体1溶于乙腈中,加入碳酸铯和4-硝基卤苄,室温反应3~5h,得到含有中间体2的反应液;中间体2的纯化方法为:向含有中间体2的反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得白色固体,即为中间体2。
所述中间体3的具体制备方法为:将中间体2溶于甲醇中,加入饱和氯化铵溶液和锌粉,室温反应0.5~1.5h,得到含有中间体3的反应液;中间体3的纯化方法为:将所述含有中间体3的反应液用硅藻土过滤,甲醇洗涤,浓缩大部分滤液,加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥,过滤、浓缩,得白色固体,即为中间体3。
步骤(2)中,荧光团的制备为本领域的常规技术,可以根据现有文献的报道,制备各种荧光团。
当所述荧光基团为萘二酰亚胺类荧光团时,本发明中的一个具体实施例为:连接基团连接荧光基团得到中间体5的制备方法为:4-卤代-1,8-萘二甲酸酐与二甲胺反应制备得到中间体4,所述中间体4与连接基团反应制备得到中间体5ˊ。
所述中间体4的结构式如4所示,所述中间体5ˊ的结构式如5ˊ所示,n=0-5:
具体制备方法为:将4-卤代-1,8-萘二甲酸酐溶于二甲基甲酰胺(DMF)中,加入二甲胺和五水硫酸铜,在130~150℃下回流搅拌反应9~11h,得到含有中间体4的反应液,旋干反应液,加入无水乙醇,析出黄色固体,即为中间体4;将所述中间体4溶于DMF中,加入连接基团,在110~130℃反应9~11h,得到含有中间体5ˊ的反应液;所述中间体5ˊ的纯化方法为:向含有中间体5ˊ的反应液加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体,即为中间体5ˊ。
当所述荧光基团为含羧基的香豆素类荧光团时,本发明中的一个具体实施例为:连接基团连接荧光基团得到中间体5的制备方法为:将连接基团进行活化,得到中间体9;将所述中间体9与含羧基的香豆素荧光团反应制备得到中间体10;然后将中间体10的酯基水解得到中间体5”。
所述含羧基的香豆素荧光团的结构式如8ˊ所示,所述中间体9的结构式如9所示,所述中间体10的结构式如10所示,所述中间体11的结构式如5”所示,其中,n=1~6,R为甲基或乙基。
进一步的,所述中间体9的制备方法为:将连接基团溶于无水甲醇或乙醇中,在冰浴条件下滴加二甲亚砜,滴加完毕后,55~65℃(优选60℃)回流搅拌反应5~7h,旋干,析出白色固体,即为中间体9。
所述中间体10的制备方法为:向无水二氯甲烷加入所述含羧基的香豆素、N-甲基吗啉、O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯(TBTU),室温反应20~40min,最后加入中间体9,室温搅拌反应5~7h,得到含有中间体10的反应液;中间体10的纯化方法为:
向含有中间体10的反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体,即为中间体10。
所述中间体5”的制备方法为:向二氧六环中加入中间体10,加入盐酸,室温搅拌23~25h,得到含有中间体5”的反应液;中间体5”的纯化方法为:向含有中间体5”的反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体,即为中间体5”。
步骤(3)中,将中间体5ˊ和中间体3溶于DMF中,加入N-乙基二异丙胺(DIPEA)和2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU),室温反应12~18h,得到含有中间体6的反应液;所述中间体6的纯化方法为:向含有中间体6的反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体,即为中间体6;当中间体5为5ˊ所示的结构的化合物时,将所述中间体6溶于四氢呋喃(THF)中,加入LiOH的水溶液,室温反应10~14h,然后加入盐酸,得到含有荧光探针的反应液;当中间体5为5”所示的结构式的化合物时,向二氧六环中加入所述中间体6,加入盐酸,室温反应23~25h,得到含有荧光探针的反应液;
所述荧光探针的纯化方法为:向含有荧光探针的反应液中加入二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体,即为荧光探针。
本发明所述的室温为15~37℃。
本发明的第三个方面,提供以下应用:
(1)所述小分子荧光探针在制备用于检测GPR120高表达量的细胞或组织试剂中的应用;
探针分子可以标记GPR120高表达水平的细胞或组织,荧光成像后有较强的荧光。
(2)所述小分子探针在作为识别GPR120受体小分子荧光探针的应用,以及在GPR120生理、病理及其相关疾病研究中的应用;优选的,相关疾病为糖尿病或肥胖。
(3)所述小分子荧光探针在制备用于筛选GPR120相关疾病的药物的试剂中的应用;优选的,所述相关疾病为糖尿病或肥胖;优选的,所述GPR120相关疾病的药物为GPR120拮抗剂或GPR120激动剂。
上述技术方案中的一个技术方案具有如下有益效果:
本发明的GPR120小分子荧光探针对GPR120具有高选择性和高灵敏度,且该探针分子的荧光性质随着周围环境的改变而发生变化,尤其是其荧光会因周围环境的极性减小而显著增强;可以用于识别GPR120及其高表达的细胞或组织,以及其在生理、病理及相关疾病的研究中的应用;可用于GPR120激动剂或拮抗剂的筛选。此外,该类化合物制备方法成熟,反应条件温和,原料便宜易得,操作及后处理方便。
附图说明
图1.小分子荧光探针L3、L7在不同极性溶剂中的荧光发射光谱。
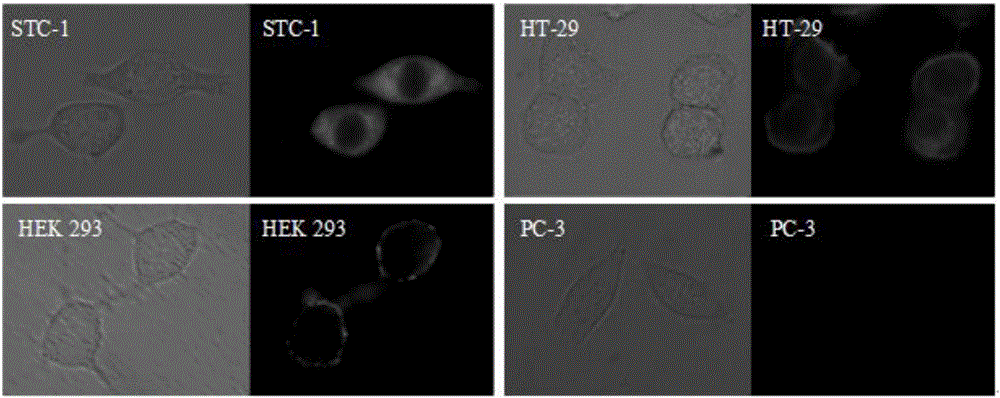
图2.小分子荧光探针L3对不同细胞的成像研究结果。
具体实施方式
下面的实施例可以使本专业技术人员更全面地理解本发明,但不以任何方式限制本发明。
实施例1:萘二酰亚胺类荧光团探针(L1-L3)的制备:
具体合成路线如下:
中间体1的制备
在100mL的圆底烧瓶中,加入对羟基苯丙酸(1.5g,9.03mmol)和无水甲醇(35mL)搅拌溶解后滴加催化量的浓硫酸(15滴),在60℃下回流搅拌6小时后,降至室温,将反应液旋出,加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得无色透明油状物1.54g,产率为95%。1H NMR(400MHz,DMSO):δ9.15(s,1H),6.99(d,J=8.4Hz,2H),6.65(d,J=8.5Hz,2H),3.57(s,3H),2.73(t,J=7.6Hz,2H),2.54(t,J=7.6Hz,2H);EI-MS:([M]+):180.1。
中间体2的制备
将中间体1(400mg,2.22mmol)溶于10mL乙腈中,加入碳酸铯(1.08g,3.33mmol),在搅拌下加入4-硝基溴苄(715.81mg,3.33mmol),室温反应4h,将反应液旋出,加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得白色固体580.62mg,产率为83%。1H NMR(400MHz,DMSO):δ8.26(d,J=8.8Hz,2H),7.71(d,J=8.8Hz,2H),7.15(d,J=8.6Hz,2H),6.94(d,J=8.6Hz,2H),5.25(s,2H),3.57(s,3H),2.78(t,J=7.6Hz,2H),2.58(t,J=7.6Hz,2H);EI-MS:([M+Na]+):338.1。
中间体3的制备
将中间体2(400mg,1.27mmol)溶于120mL甲醇中,加入饱和氯化铵溶液(15mL),搅拌下加入活化锌粉(1.6g,25.38mmol),室温反应1小时,反应液用硅藻土过滤,甲醇洗涤,浓缩大部分滤液,加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥,过滤、浓缩,无需进一步纯化,得白色固体278.8mg,产率为77%。
中间体4的制备
将4-溴-1,8-萘二甲酸酐(1.5g,5.41mmol)溶于30mL无水DMF中,加入质量分数为40%二甲胺水溶液(1.95g,43.28mmol)和五水硫酸铜(0.13g,0.54mmol),在150℃下回流搅拌反应10小时后,降至室温,将反应液旋出,加入无水乙醇,析出黄色固体,过滤、干燥,得黄色固体0.98g,产率为75%。1H NMR(400MHz,CDCl3):δ8.59(d,J=8.0Hz,1H),8.47(d,J=8.0Hz,1H),8.33(d,J=8.0Hz,1H),7.77(m,1H),7.20(d,J=8.0Hz,1H),3.17(s,6H);EI-MS:([M+H]+):242.1。
中间体5a的制备
将中间体4(0.20g,0.83mmol)溶于25mL无水DMF中,加入3-氨基丙酸(0.11g,1.24mmol),120℃下搅拌反应10小时后,降至室温,将大部分反应液旋出,向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体181mg,产率为70%。1H NMR(400MHz,DMSO):δ12.34(s,1H),8.51(d,J=8.5Hz,1H),8.45(d,J=7.3Hz,1H),8.33(d,J=8.3Hz,1H),7.75(t,J=7.9Hz,1H),7.21(d,J=8.3Hz,1H),4.28–4.20(m,2H),3.09(s,6H),2.60–2.53(m,2H);EI-MS:([M-H]-):311.1。
中间体5b的制备
中间体5b的合成以4-氨基丁酸(127.8mg,1.24mmol)为原料,按照中间体5a的合成方法,得黄色固体183mg,产率68%。1H NMR(400MHz,DMSO):δ12.02(s,1H),8.50(d,J=8.5Hz,1H),8.44(d,J=7.3Hz,1H),8.33(d,J=8.3Hz,1H),7.75(t,J=7.9Hz,1H),7.21(d,J=8.3Hz,1H),4.06(t,J=6.9Hz,2H),3.09(s,6H),2.29(t,J=7.4Hz,2H),1.87(p,J=7.2Hz,2H);EI-MS:([M-H]-):325.1。
中间体5c的制备
中间体5c的合成以6-氨基己酸(162.6mg,1.24mmol)为原料,按照中间体5a的合成方法,得黄色固体211mg,产率72%。1H NMR(400MHz,DMSO):δ11.98(s,1H),8.52(d,J=8.5Hz,1H),8.46(d,J=7.3Hz,1H),8.35(d,J=8.3Hz,1H),7.78–7.73(m,1H),7.22(d,J=8.3Hz,1H),4.05–3.98(m,2H),3.09(s,6H),2.21(t,J=7.3Hz,2H),1.58(m,4H),1.33(m,2H);EI-MS:([M-H]-):353.2。
中间体6a的制备
将中间体5a(200mg,0.64mmol)、中间体3(219mg,0.77mmol)溶于15mL无水DMF中,加入DIPEA(413mg,3.2mmol)和HATU(486.69mg,1.28mmol),室温反应过夜,向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体278mg,产率为75%。1H NMR(400MHz,DMSO):δ10.07(s,1H),8.52(d,J=8.5Hz,1H),8.47(d,J=6.6Hz,1H),8.35(d,J=8.3Hz,1H),7.79–7.74(m,1H),7.55(d,J=8.4Hz,2H),7.35(d,J=8.5Hz,2H),7.22(d,J=8.4Hz,1H),7.12(d,J=8.6Hz,2H),6.90(d,J=8.6Hz,2H),4.98(s,2H),4.34(t,J=7.4Hz,2H),3.57(s,3H),3.10(s,6H),2.77(t,J=7.6Hz,2H),2.69(t,J=7.5Hz,2H),2.58(t,J=7.6Hz,2H);EI-MS:([M+H]+):580.2。
中间体6b的制备
中间体6b的合成以5b(200mg,0.61mmol)为原料,按照中间体6a的合成方法,得黄色固体249mg,产率69%。1H NMR(400MHz,DMSO):δ9.89(s,1H),8.50(dd,J=8.5,1.0Hz,1H),8.46(dd,J=7.3,1.0Hz,1H),8.34(d,J=8.3Hz,1H),7.74(m,1H),7.49(d,J=8.5Hz,2H),7.28(d,J=8.5Hz,2H),7.21(d,J=8.4Hz,1H),7.12(d,J=8.6Hz,2H),6.89(d,J=8.6Hz,2H),4.95(s,2H),4.12(t,J=6.9Hz,2H),3.57(s,3H),3.09(s,6H),2.77(t,J=7.5Hz,2H),2.57(t,J=7.6Hz,2H),2.38(t,J=7.6Hz,2H),2.01–1.92(m,2H);EI-MS:([M+H]+):594.3。
中间体6c的制备
中间体6c的合成以5c(200mg,0.56mmol)为原料,按照中间体6a的合成方法,得黄色固体207.6mg,产率63%。1H NMR(400MHz,DMSO):δ9.88(s,1H),8.51(dd,J=8.5,1.0Hz,1H),8.45(dd,J=7.3,1.0Hz,1H),8.34(d,J=8.3Hz,1H),7.75(dd,J=8.4,7.4Hz,1H),7.56(d,J=8.5Hz,2H),7.32(d,J=8.5Hz,2H),7.21(d,J=8.4Hz,1H),7.12(d,J=8.6Hz,2H),6.90(t,J=5.7Hz,2H),4.97(s,2H),4.08–3.99(m,2H),3.57(s,3H),3.09(s,6H),2.77(t,J=7.6Hz,2H),2.58(t,J=7.6Hz,2H),2.31(t,J=7.4Hz,2H),1.65(dq,J=14.5,7.4Hz,4H),1.41–1.32(m,2H);EI-MS:([M+H]+):622.3。
化合物L1的制备
将中间体6a(100mg,0.17mmol)溶于10mL THF中,加入LiOH的水溶液(21.73mg,0.51mmol),室温反应12小时,向反应液中加入水,加入1M HCl调PH使溶液显酸性,加入二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体49mg,产率为51%。1H NMR(400MHz,DMSO):δ12.13(s,1H),10.08(s,1H),8.53(d,J=8.4Hz,1H),8.47(d,J=7.2Hz,1H),8.36(d,J=8.2Hz,1H),7.77(t,J=7.9Hz,1H),7.55(d,J=8.4Hz,2H),7.35(d,J=8.3Hz,2H),7.23(d,J=8.4Hz,1H),7.13(d,J=8.4Hz,2H),6.90(d,J=8.4Hz,2H),4.98(s,2H),4.34(t,J=7.4Hz,2H),3.10(s,6H),2.71(m,4H),2.46(m,2H);13C-NMR(100MHz,DMSO):δppm:174.33,169.49,164.06,163.38,157.15,157.07,139.17,133.43,132.74,132.25,132.04,131.03,130.14,129.65,128.77,125.48,124.70,122.83,119.66,115.09,113.81,113.47,69.44,44.86,36.60,36.08,35.29,30.01;ESI-HRMS:[M-H]-calcd for C33H31N3O6:564.2213,found:564.2149。
化合物L2的制备
化合物L2的合成以中间体6b(100mg,0.16mmol)为原料,按照L1的合成方法,得黄色固体51mg,产率55%。1H NMR(400MHz,DMSO):δ12.08(s,1H),9.91(s,1H),8.50(d,J=8.5Hz,1H),8.45(d,J=6.8Hz,1H),8.34(d,J=8.3Hz,1H),7.78–7.70(m,1H),7.50(d,J=8.5Hz,2H),7.29(d,J=8.5Hz,2H),7.20(d,J=8.4Hz,1H),7.12(d,J=8.5Hz,2H),6.89(d,J=8.6Hz,2H),4.95(s,2H),4.12(t,J=6.9Hz,2H),3.09(s,6H),2.74(t,J=7.5Hz,2H),2.47(m,2H),2.38(t,J=7.5Hz,2H),2.03–1.92(m,2H);13C-NMR(100MHz,DMSO):δppm:174.29,171.04,164.23,163.58,157.15,157.00,139.33,133.40,132.74,131.94,131.91,131.02,130.13,129.64,128.66,125.44,124.70,122.85,119.33,115.09,113.90,113.45,69.44,44.86,39.45,36.04,34.55,29.99,24.14;ESI-HRMS:[M-H]-calcd for C34H33N3O6:578.2369,found:578.2284。
化合物L3的制备
化合物L3的合成以中间体6c(100mg,0.16mmol)为原料,按照L1的合成方法,得黄色固体47mg,产率49%。1H NMR(400MHz,DMSO):δ12.12(s,1H),9.92(s,1H),8.50(d,J=8.5Hz,1H),8.45(d,J=7.1Hz,1H),8.34(d,J=8.3Hz,1H),7.78–7.72(m,1H),7.57(d,J=8.5Hz,2H),7.33(d,J=8.5Hz,2H),7.21(d,J=8.4Hz,1H),7.13(d,J=8.5Hz,2H),6.89(d,J=8.6Hz,2H),4.97(s,2H),4.03(t,J=7.3Hz,2H),3.09(s,6H),2.74(t,J=7.5Hz,2H),2.47(m,2H),2.31(t,J=7.3Hz,2H),1.69–1.58(m,4H),1.42–1.32(m,2H);13C-NMR(100MHz,DMSO):δppm:174.34,171.62,164.09,163.44,157.15,157.01,139.38,133.42,132.74,131.99,131.97,131.03,130.06,129.64,128.74,125.47,124.69,122.79,119.40,115.10,113.80,113.47,69.45,44.85,36.69,36.07,30.01,27.93,26.67,25.31;ESI-HRMS:[M-H]-calcd for C36H37N3O6:606.2682,found:606.2610。
实施例2:香豆素类荧光团探针(L4-L7)的制备:
具体合成路线如下:
中间体7的制备
在100mL的圆底烧瓶中,加入4-(二乙氨基)水杨醛(1.5g,7.76mmol)和40mL无水乙醇,再加入丙二酸二乙酯(1.24g,7.76mmol),搅拌使其溶解,将吗啡啉(68mg,0.78mmol)和乙酸(20μL)溶于2mL无水乙醇中,得到混合液,然后将混合液加入上述反应液中,回流搅拌24h,冰温冷却反应液,将反应液旋干,得中间体7,未经纯化,直接用于下一步。
中间体8的制备
将中间体7(400mg,1.38mmol)溶于少量甲醇溶液,加入25mL的2M NaOH溶液,室温下搅拌24小时,用1M HCl调PH使溶液显酸性,析出大量黄色沉淀,过滤、干燥,得黄色固体288mg,产率为80%。1H NMR(400MHz,DMSO):δ12.54(s,1H),8.60(s,1H),7.65(d,J=9.1Hz,1H),6.81(dd,J=9.0,2.4Hz,1H),6.58(d,J=2.3Hz,1H),3.49(q,J=7.0Hz,4H),1.14(t,J=7.0Hz,6H);ESI-MS:([M+H]+):262.3。
中间体9a的制备
将氨基乙酸(500mg,6.66mmol)溶于20mL无水甲醇中,在冰浴条件下,滴加二氯亚砜(2.37g,19.98mmol),滴毕,在60℃下回流搅拌反应6小时后,降至室温,将反应液旋干,析出白色固体,无需进一步纯化,得白色固体791mg,产率为95%。1H NMR(400MHz,DMSO):δ8.53(s,3H),3.78(s,2H),3.73(s,3H);EI-MS:([M+H]+):90.1。
中间体9b的制备
将3-氨基丙酸(400mg,4.48mmol)溶于20mL无水乙醇中,在冰浴条件下,滴加二氯亚砜(1.6g,13.47mmol),滴毕,在80℃下回流搅拌反应6小时后,降至室温,将反应液旋干,析出白色固体,无需进一步纯化,得白色固体665mg,产率为97%。1H NMR(400MHz,DMSO):δ8.21(s,3H),4.10(q,J=7.1Hz,2H),2.99(t,J=7.2Hz,2H),2.71(t,J=7.2Hz,2H),1.20(t,J=7.1Hz,3H);EI-MS:([M+H]+):118.1。
中间体9c的制备
中间体9c的合成以4-氨基丁酸(450mg,4.36mmol)为原料,按照中间体9a的合成方法,得白色固体620mg,产率93%。1H NMR(400MHz,DMSO):δ8.16(s,3H),3.60(s,3H),2.78(s,2H),2.44(t,J=7.5Hz,2H),1.86–1.77(m,2H);EI-MS:([M+H]+):118.1。
中间体9d的制备
中间体9d的合成以6-氨基己酸(600mg,4.57mmol)为原料,按照中间体9a的合成方法,得白色固体753mg,产率91%。1H NMR(400MHz,DMSO):δ8.06(s,3H),3.59(s,3H),2.76–2.69(m,2H),2.31(t,J=7.4Hz,2H),1.61–1.48(m,4H),1.35–1.25(m,2H);EI-MS:([M+H]+):146.1。
中间体10a的制备
在25mL无水二氯甲烷中加入中间体8(200mg,0.76mmol),N-甲基吗啉(309.91mg,3.06mmol),TBTU(366.03mg,1.14mmol),室温搅拌30min,最后加入中间体9a(114.06mg,0.912mmol),室温搅拌6h反应基本完成。向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体184mg,产率为73%。1H NMR(400MHz,DMSO):δ9.00(t,J=5.7Hz,1H),8.67(s,1H),7.70(d,J=9.0Hz,1H),6.82(dd,J=9.0,2.3Hz,1H),6.63(d,J=2.1Hz,1H),4.11(d,J=5.7Hz,2H),3.66(s,3H),3.49(q,J=7.0Hz,4H),1.15(t,J=7.0Hz,6H);EI-MS:([M+H]+):333.1。
中间体10b的制备
中间体10b的合成以中间体9b(140.69mg,0.92mmol)为原料,按照中间体10a的合成方法,得黄色固体181mg,产率69%。1H NMR(400MHz,DMSO):δ8.84(t,J=5.9Hz,1H),8.66(s,1H),7.69(d,J=9.0Hz,1H),6.81(dd,J=9.0,2.3Hz,1H),6.61(d,J=2.1Hz,1H),4.08(q,J=7.1Hz,2H),3.57–3.43(m,6H),2.57(t,J=6.6Hz,2H),1.19(t,J=7.1Hz,3H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):361.2。
中间体10c的制备
中间体10c的合成以中间体9c(153.69mg,0.92mmol)为原料,按照中间体10a的合成方法,得黄色固体151mg,产率55%。1H NMR(400MHz,DMSO):δ8.68–8.63(m,2H),7.68(d,J=9.0Hz,1H),6.80(dd,J=9.0,2.4Hz,1H),6.62(d,J=2.2Hz,1H),3.59(s,3H),3.48(q,J=7.0Hz,4H),3.32(dd,J=12.9,6.7Hz,2H),2.35(t,J=7.4Hz,2H),1.77(p,J=7.1Hz,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):361.2。
中间体10d的制备
中间体10d的合成以中间体9d(179.45mg,0.92mmol)为原料,按照中间体10a的合成方法,得黄色固体209mg,产率71%。1H NMR(400MHz,DMSO):δ8.67–8.60(m,2H),7.68(d,J=9.0Hz,1H),6.80(dd,J=9.0,2.4Hz,1H),6.61(d,J=2.2Hz,1H),3.58(s,3H),3.48(q,J=7.0Hz,4H),3.29(dd,J=13.0,6.8Hz,2H),2.31(t,J=7.4Hz,2H),1.60–1.46(m,4H),1.35–1.25(m,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):389.2。
中间体11a的制备
在10mL二氧六环中加入中间体10a(100mg,0.3mmol),搅拌状态下滴加3M HCl(5mL),室温搅拌24小时。向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体43mg,产率为45%。1H NMR(400MHz,DMSO):δ12.75(s,1H),8.96(t,J=5.5Hz,1H),8.68(s,1H),7.70(d,J=9.0Hz,1H),6.82(dd,J=9.0,2.3Hz,1H),6.63(d,J=2.0Hz,1H),4.02(d,J=5.5Hz,2H),3.49(q,J=7.0Hz,4H),1.14(t,J=7.0Hz,6H);EI-MS:([M-H]-):317.1。
中间体11b的制备
中间体11b的合成以中间体10b(100mg,0.29mmol)为原料,按照中间体11a的合成方法,得黄色固体41mg,产率41%。1H NMR(400MHz,DMSO):δ12.32(s,1H),8.83(t,J=5.9Hz,1H),8.66(s,1H),7.68(d,J=9.1Hz,1H),6.81(dd,J=9.0,2.4Hz,1H),6.61(d,J=2.3Hz,1H),3.54–3.45(m,6H),2.48(m,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):333.1。
中间体11c的制备
中间体11c的合成以中间体10c(100mg,0.28mmol)为原料,按照中间体11a的合成方法,得黄色固体47mg,产率47%。1H NMR(400MHz,DMSO):δ12.09(s,1H),8.72–8.57(m,2H),7.68(d,J=9.0Hz,1H),6.80(dd,J=9.0,2.4Hz,1H),6.62(d,J=2.2Hz,1H),3.48(q,J=7.0Hz,4H),3.30(m,2H),2.26(t,J=7.4Hz,2H),1.74(p,J=7.2Hz,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M-H]-):345.1。
中间体11d的制备
中间体11d的合成以中间体10d(100mg,0.26mmol)为原料,按照中间体11a的合成方法,得黄色固体53mg,产率55%。1H NMR(400MHz,DMSO):δ11.99(s,1H),8.67–8.60(m,2H),7.68(d,J=9.0Hz,1H),6.80(dd,J=9.0,2.4Hz,1H),6.61(d,J=2.2Hz,1H),3.48(q,J=7.0Hz,4H),3.29(dd,J=13.0,6.8Hz,2H),2.21(t,J=7.4Hz,2H),1.57–1.47(m,4H),1.36–1.26(m,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M-H]-):373.2。
中间体12a的制备
将中间体11a(100mg,0.33mmol)、中间体3(112mg,0.39mmol)溶于15mL无水DMF中,加入HATU(251mg,0.66mmol)和DIPEA(213mg,1.65mmol),室温反应过夜,向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体127mg,产率为66%。1H NMR(400MHz,DMSO):δ10.16(s,1H),9.09(t,J=5.3Hz,1H),8.69(s,1H),7.70(d,J=9.0Hz,1H),7.61(d,J=8.5Hz,2H),7.38(d,J=8.5Hz,2H),7.13(d,J=8.6Hz,2H),6.90(d,J=8.6Hz,2H),6.82(dd,J=9.1,2.2Hz,1H),6.64(d,J=1.9Hz,1H),5.00(s,2H),4.18(d,J=5.3Hz,2H),3.57(s,3H),3.50(q,J=6.9Hz,4H),2.77(t,J=7.6Hz,2H),2.58(t,J=7.6Hz,2H),1.18–1.12(m,6H);EI-MS:([M+H]+):586.2。
中间体12b的制备
中间体12b的合成以中间体11b(100mg,0.31mmol)为原料,按照中间体12a的合成方法,得黄色固体130mg,产率70%。1H NMR(400MHz,DMSO):δ10.05(s,1H),8.87(t,J=5.8Hz,1H),8.66(s,1H),7.68(d,J=9.0Hz,1H),7.59(d,J=8.4Hz,2H),7.36(d,J=8.4Hz,2H),7.12(d,J=8.5Hz,2H),6.90(d,J=8.6Hz,2H),6.80(dd,J=9.0,2.2Hz,1H),6.60(d,J=1.9Hz,1H),4.98(s,2H),3.63–3.58(m,2H),3.57(s,3H),3.48(q,J=7.0Hz,4H),2.77(t,J=7.5Hz,2H),2.65–2.54(m,4H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):600.3。
中间体12c的制备
中间体12c的合成以中间体11c(100mg,0.30mmol)为原料,按照中间体12a的合成方法,得黄色固体134mg,产率73%。1H NMR(400MHz,DMSO):δ9.97(s,1H),8.68(t,J=5.7Hz,1H),8.63(s,1H),7.67(d,J=9.0Hz,1H),7.58(d,J=8.4Hz,2H),7.32(d,J=8.4Hz,2H),7.12(d,J=8.5Hz,2H),6.88(d,J=8.5Hz,2H),6.80(dd,J=9.0,2.2Hz,1H),6.61(d,J=1.9Hz,1H),4.94(s,2H),3.57(s,3H),3.48(q,J=6.9Hz,4H),3.39(m,2H),2.77(t,J=7.5Hz,2H),2.58(t,J=7.6Hz,2H),2.36(t,J=7.4Hz,2H),1.84(p,J=7.0Hz,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):614.3。
中间体12d的制备
中间体12d的合成以中间体11d(100mg,0.27mmol)为原料,按照中间体12a的合成方法,得黄色固体133mg,产率77%。1H NMR(400MHz,DMSO):δ9.91(s,1H),8.68–8.62(m,2H),7.68(d,J=9.1Hz,1H),7.58(d,J=8.5Hz,2H),7.32(d,J=8.5Hz,2H),7.12(d,J=8.6Hz,2H),6.89(d,J=8.6Hz,2H),6.80(dd,J=9.1,2.3Hz,1H),6.62(d,J=2.2Hz,1H),4.96(s,2H),3.57(s,3H),3.48(q,J=7.0Hz,4H),3.32–3.28(m,2H),2.77(t,J=7.6Hz,2H),2.58(t,J=7.6Hz,2H),2.31(t,J=7.4Hz,2H),1.68–1.50(m,4H),1.34(m,2H),1.14(t,J=7.0Hz,6H);EI-MS:([M+H]+):642.3。
化合物L4的制备
在10mL二氧六环中加入中间体12a(100mg,0.18mmol),搅拌状态下滴加2M HCl(5mL),室温搅拌24小时。向反应液中加入水和二氯甲烷萃取,合并有机层,饱和食盐水洗,无水MgSO4干燥过夜,过滤、浓缩、柱层析纯化,得黄色固体28mg,产率为28%。1HNMR(400MHz,DMSO):δ12.04(s,1H),10.16(s,1H),9.09(t,J=5.2Hz,1H),8.69(s,1H),7.70(d,J=9.0Hz,1H),7.61(d,J=8.4Hz,2H),7.38(d,J=8.4Hz,2H),7.13(d,J=8.5Hz,2H),6.90(d,J=8.5Hz,2H),6.82(dd,J=9.1,2.0Hz,1H),6.64(d,J=1.7Hz,1H),4.99(s,2H),4.18(d,J=5.2Hz,2H),3.49(dd,J=13.9,6.9Hz,5H),2.74(t,J=7.5Hz,2H),2.46(m,2H),1.15(t,J=7.0Hz,6H);13C-NMR(100MHz,DMSO):δppm:174.43,167.83,162.85,162.10,157.81,157.12,152.99,148.35,138.88,133.48,132.42,132.16,129.65,128.91,119.47,115.07,110.62,109.45,108.10,96.35,69.38,44.82,43.76,36.16,30.04,12.79;ESI-HRMS:[M-H]-calcd for C32H33N3O7:570.2319,found:570.2245。
化合物L5的制备
化合物L5的合成以中间体12b(100mg,0.17mmol)为原料,按照L4的合成方法,得黄色固体32mg,产率33%。1H NMR(400MHz,DMSO):δ12.19(s,1H),10.09(s,1H),8.87(t,J=5.7Hz,1H),8.67(s,1H),7.68(d,J=9.0Hz,1H),7.60(d,J=8.4Hz,2H),7.36(d,J=8.4Hz,2H),7.12(d,J=8.4Hz,2H),6.89(d,J=8.4Hz,2H),6.79(d,J=9.0Hz,1H),6.60(s,1H),4.98(s,2H),3.59(dd,J=12.0,6.0Hz,2H),3.47(dd,J=13.7,6.7Hz,4H),2.74(t,J=7.5Hz,2H),2.62(t,J=6.3Hz,2H),2.47(t,J=7.6Hz,2H),1.13(t,J=6.9Hz,6H);13C-NMR(100MHz,DMSO):δppm:170.16,162.64,162.14,157.70,157.12,152.89,148.21,139.17,133.48,132.21,132.08,129.65,128.83,119.48,115.07,110.57,109.69,108.10,96.30,69.41,44.79,36.61,36.19,35.65,30.05,12.78;ESI-HRMS:[M-H]-calcd for C33H35N3O7:584.2475,found:584.2403。
化合物L6的制备
化合物L6的合成以中间体12c(100mg,0.16mmol)为原料,按照L4的合成方法,得黄色固体23mg,产率25%。1H NMR(400MHz,DMSO):δ12.09(s,1H),9.97(s,1H),8.68(t,J=6.0Hz,1H),8.64(s,1H),7.68(d,J=9.0Hz,1H),7.58(d,J=8.5Hz,2H),7.32(d,J=8.5Hz,2H),7.12(d,J=8.6Hz,2H),6.89(d,J=8.6Hz,2H),6.80(dd,J=9.0,2.3Hz,1H),6.62(d,J=2.1Hz,1H),4.94(s,2H),3.48(q,J=7.0Hz,4H),3.40–3.35(m,2H),2.74(t,J=7.6Hz,2H),2.47(m,2H),2.36(t,J=7.3Hz,2H),1.83(dd,J=14.3,7.2Hz,2H),1.13(t,J=7.0Hz,6H);13C-NMR(100MHz,DMSO):δppm:174.33,171.28,162.75,162.18,157.66,157.13,152.83,148.11,139.39,133.41,132.03,131.96,129.64,128.74,119.33,115.06,110.54,109.91,108.10,96.29,69.40,44.78,38.97,36.08,34.39,30.00,25.73,12.78;ESI-HRMS:[M+H]+calcd for C34H37N3O7:600.2632,found:600.2703。
化合物L7的制备
化合物L7的合成以中间体12d(100mg,0.15mmol)为原料,按照L4的合成方法,得黄色固体32mg,产率35%。1H NMR(400MHz,DMSO):δ12.08(s,1H),9.92(s,1H),8.66(m,2H),7.68(d,J=9.0Hz,1H),7.58(d,J=8.4Hz,2H),7.32(d,J=8.4Hz,2H),7.12(d,J=8.5Hz,2H),6.88(d,J=8.5Hz,2H),6.80(dd,J=9.0,2.1Hz,1H),6.62(d,J=1.7Hz,1H),4.95(s,2H),3.48(dd,J=13.9,6.9Hz,4H),3.32–3.27(m,2H),2.74(t,J=7.5Hz,2H),2.47(m,2H),2.31(t,J=7.3Hz,2H),1.66–1.49(m,4H),1.39–1.29(m,2H),1.13(t,J=7.0Hz,6H);13C-NMR(100MHz,DMSO):δppm:174.35,171.65,162.53,162.26,157.66,157.15,152.85,148.08,139.39,133.45,132.00,129.63,128.74,119.42,115.09,110.57,109.98,108.14,96.33,69.44,44.79,39.20,36.75,36.11,30.03,29.42,26.57,25.30,12.78;ESI-HRMS:[M-H]-calcd for C36H41N3O7:626.2945,found:626.3052。
实验例一:光学活性测定
表一:探针分子的光学特性
注:以上所有光学性质均在PH=7.4的Tris-HCl缓冲液中测量。
实验例二:测定周围环境对配体分子荧光性质的影响
以探针分子L3、L7为研究对象,该类探针分子的荧光性质随着周围环境的改变而发生变化,尤其是其荧光会因周围环境的极性减小而显著增强(如图1所示),所采用溶剂为PBS缓冲液、甲醇(MeOH)、乙醇(EA)、二氯甲烷(DCM);说明该类荧光探针分子属于环境敏感型的荧光探针。
实验例三:生物活性的测定
采用测量BRET信号的方法确定荧光探针的EC50:通过瞬时转染的方法,将β-arrestin2-RLuc和GPR120-YFP两种质粒同时转入HEK 293细胞中,转染后36-48h,加入海肾荧光素酶(使其终浓度为5μM)孵育5-10min,用不同浓度梯度的荧光探针进一步刺激5min后用酶标仪(POLARstar Omega mocroplate reader)检测荧光强度比(460nm/520nm)变化测定化合物EC50。结果如表二所示。其中,GPR120激动剂TUG-891(中文名称为4-[(4-氟-4’-甲基-[1,1’-联苯]-2基)甲氧基]苯丙酸)为阳性对照。合成的探针分子L1、L2、L4、L5、L6、L7的活性较阳性TUG-891低,探针L3的活性与TUG-891的活性相当。
表二:BRET实验测定结果
注:EC50为荧光探针的BRET信号引起50%最大效应的浓度。
实验例四:探针分子在有关表达GPR120细胞成像中的应用
以探针分子L3为研究对象,考察其在细胞成像中的应用。对于细胞成像,以STC-1细胞,HT-29细胞,HEK 293细胞,PC-3细胞(GPR120相对低表达)为研究对象。
具体操作步骤为:HEK 293细胞用含有10%胎牛血清的高糖DMEM培养基;STC-1、HT-29、PC-3细胞用含有10%胎牛血清的1640培养基,在37℃以及5%CO2环境中进行培养。将细胞消化离心后接种于共聚焦小皿中,培养18-24h,待细胞形态贴壁完好后,将培养基吸去,将探针分子加入孵育5min后,然后吸出培养基,用不含血清的培养基洗涤2次,用Zeiss Axio Observer A1成像。
成像结果如图2所示,探针分子可以标记GPR120高表达水平的细胞(STC-1细胞,HT-29细胞,HEK 293细胞),而低表达的PC-3细胞表达很弱的荧光,当扣除相同的荧光背景值时,其荧光几乎不亮。这说明了该荧光探针在GPR120生理、病理及其相关疾病的研究中有广阔的应用前景。
另外,经过大量的实验验证,本发明的其他探针分子(L1、L2、L4、L5、L6以及其他探针分子)在GPR120高表达水平的细胞荧光成像中也具有良好的效果。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案基础上,本领域技术人员不许要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!