固态红色硅烷化碳点及其制备方法与流程
本发明属于荧光纳米材料合成技术领域,具体涉及一种固态红色硅烷化碳点(CDs)及其制备方法。
背景技术:
荧光碳纳米材料具有较好的荧光性能与低毒性,在信息安全、光催化药物传输、细胞成像、传感、光电器件以及疾病诊断等领域具有良好的应用前景。作为荧光碳纳米材料之一的碳点因其独特的光学性质和环境友好性,从而受到人们的广泛关注。
但目前固态碳量子点难以制得,因为当碳点处于聚集态时,极易引起荧光猝灭。到目前为止,本领域的绝大部分工作都是关于碳点水溶液的荧光性能与应用,很少有报道涉及到碳点杂化固相材料和碳点团聚体的固态发光性能及相关应用。
本领域关于碳点发光性能的研究,绝大部分限于短波长的荧光(蓝色、绿色),而长波长的荧光(橙光、红光)报道很少。而长波长的荧光在特殊应用中有着短波长荧光无法企及的优良特性,所以对于红色荧光碳点的研究显得尤为重要。虽然目前已经有关于红光碳点的报道,但均是表现为液态荧光,对于固态红色荧光碳点的研究显得尤为重要。
为了克服现有技术的缺点与不足,本发明提供了一种新型固态红色碳点及其制备方法。本发明的制备方法操作工艺简单、快捷、原料廉价易得、产物稳定性好(如图9、10),不潮解,对于碳点的制备过程简单,后处理容易,粒子大小均匀(如图3所示),荧光效果显著,所述碳点不具备典型的荧光激发依赖性质(如图8所示),不同于绝大多数文献所报道的具有的激发依赖性质的碳点。
技术实现要素:
本发明公开了一种固态红色硅烷化碳点及其制备方法与应用,所述方法为回流法。本发明通过简易快捷的回流反应,无需后续强酸或表面钝化剂的处理,即可一步快速获得固态碳点,原料简单易得,价格低廉,反应条件温和。解决了现有固态红色碳点因制备工艺和原料的限制而无法规模化生产的问题,制备得到的碳点稳定性好(如图9、10),不潮解,粒子大小均匀(如图3所示),荧光效果显著,所述碳点不具备典型的荧光激发依赖性质(如图8所示),不同于绝大多数文献所报道的具有的激发依赖性质的碳点。
一方面,本发明提供了一种新型固态红色碳点的制备方法。本发明的制备方法操作工艺简单、快捷、原料廉价易得、产物稳定性好。
本发明提供的本发明所述碳点的制备方法,所述方法对于碳点的制备过程简单,后处理容易,粒子大小均匀(如图3所示),稳定性好(如图9、10),荧光效果显著,所述碳点不具备典型的荧光激发依赖性质(如图8所示),激发改变,发射峰位不变,其不同于绝大多数文献所报道的具有的激发依赖性质的碳点。
一方面,本发明涉及一种固态红色硅烷化碳点的制备方法,其中,所述方法为回流法,包括以下步骤:将柠檬酸溶解于丙酮中得到混匀的溶液,然后将所述混匀的溶液加至硅烷偶联剂中,加热反应,得到粘稠态的碳点,再将所得粘稠态的碳点放置于烘箱中烘干得到固态碳点。
在一些实施方案,本发明所述的方法,其中,将所得粘稠态的碳点放置于60℃烘箱中24h得到固态碳点。
在一些实施方案,本发明所述的方法,其中,所述加热反应是在150-240℃下进行的。
在一些实施方案,本发明所述的方法,其中,所述加热反应的反应时间为5-60min。
在一些实施方案,本发明所述的方法,其中,所述柠檬酸、丙酮和硅烷偶联剂的用量比为1~4g:5~10mL:10~20mL。
在一些实施方案,本发明所述的方法,其中,所述柠檬酸、丙酮和硅烷偶联剂的用量比为1g:5mL:10mL。
在一些实施方案,本发明所述的方法,其中,所述硅烷偶联剂包括含有2-3个烷氧硅基的氨基硅烷偶联剂、亚氨基硅烷偶联剂、环氧基硅烷偶联剂和巯基硅烷偶联剂中的至少一种。
在一些实施方案,本发明所述的方法,其中,所述硅烷偶联剂包括N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷、N-氨乙基-γ-氨丙基-三甲氧基硅烷、3-氨丙基三乙氧基硅烷、甲基三乙氧基硅烷和3-疏醇基丙基三甲氧基硅烷中的至少一种。
在一些实施方案,本发明所述的方法,其中,所述硅烷偶联剂为N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷。
另一方面,本发明涉及一种本发明所述的方法制备得到的固态红色硅烷化碳点,其中所述碳点粒径在5-7nm,其透射电镜图基本上如图3所示。
在一些实施方案,本发明所述的碳点具有一定程度的石墨化结构,其高分辨透射电镜图基本上如图4所示。
在一些实施方案,本发明所述的碳点含有Si元素,且C元素含量达到约63.42%,其XPS谱图基本上如图5所示。
在一些实施方案,本发明所述的碳点的XRD谱图基本上如图6所示。
本发明的目的通过下述技术方案实现:硅烷化红色固态碳点,包括最优制备条件和后处理方法。
一种固态红色硅烷化碳点的制备方法,包括以下步骤:
所述碳点的制备方法为回流法,包括以下步骤:
(1)将柠檬酸溶解于丙酮中,超声分散使其完全溶解,得到溶液;
(2)将上述步骤(1)中得到的溶液加入到硅烷偶联剂中,进行回流反应得到流动粘稠态的红色碳点;
(3)将步骤(2)中得到的流动粘稠态碳点置于60摄氏度烘箱中静置24小时,得到固态红色碳点;
所述的柠檬酸、丙酮和硅烷偶联剂的用量比为1g:5mL:10mL。
所述的回流反应优选为在150-240℃下反应5-60min,更优选为在150℃下回流5min。
本发明中所述的硅烷偶联剂包括但不限于含有2-3个烷氧硅基的氨基硅烷偶联剂、亚氨基硅烷偶联剂、环氧基硅烷偶联剂和巯基硅烷偶联剂中的至少一种,如N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷(AEAPMS),N-氨乙基-γ-氨丙基-三甲氧基硅烷(AEATMS),3-氨丙基三乙氧基硅烷(APTES),甲基三乙氧基硅烷(METS),3-疏醇基丙基三甲氧基硅烷(MPTES)等。更优选为N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷(AEAPMS)。
(4)步骤(3)得到的碳点稳定性好,在紫外光下可以发出明亮的红光,可与多种物质进行复合,得到性能优异的复合材料。
本发明使用的术语“基本上如图所示”是指透射电镜图、XPS谱图或XRD谱图中至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少99%的特征或峰显示在其图中。
在本发明的上下文中,当使用或者无论是否使用“大约”或“约”等字眼时,表示在给定的值或范围的10%以内,适当地在5%以内,特别是在1%以内。或者,对于本领域普通技术人员而言,术语“大约”或“约”表示在平均值的可接受的标准误差范围内。每当公开一个具有N值的数字时,任何具有N+/-1%,N+/-2%,N+/-3%,N+/-5%,N+/-7%,N+/-8%或N+/-10%值以内的数字会被明确地公开,其中“+/-”是指加或减。
本发明的原理:
本发明利用柠檬酸,丙酮和硅烷偶联剂回流反应,原料简单易得,价格低廉,反应条件温和,制备的碳点产量高,荧光性能稳定,可以很好的应用于光电装置,光催化,生物成像及农用等领域。
本发明相对于现有技术,具有如下的优点及效果:
(1)本发明使用柠檬酸,丙酮和硅烷偶联剂回流反应,无需后续强酸或表面钝化剂处理,即可简单快速获得固态碳点。
(2)本发明原料广泛易得,价格低廉,制备条件温和,低能耗仅需150℃即可反应,且产率高,可以制得8mL液态粘稠态碳点,有望大规模工业化生产。
(3)本发明制得的碳点稳定性好,不潮解,且光稳定好(如图9、10),粒子大小均匀(如图3所示),所述碳点不具备典型的荧光激发依赖性质(如图8所示),激发改变,发射峰位不变,其不同于绝大多数文献所报道的具有的激发依赖性质的碳点,可应用于光电、光催化、气体传感以及农用等领域。
附图说明
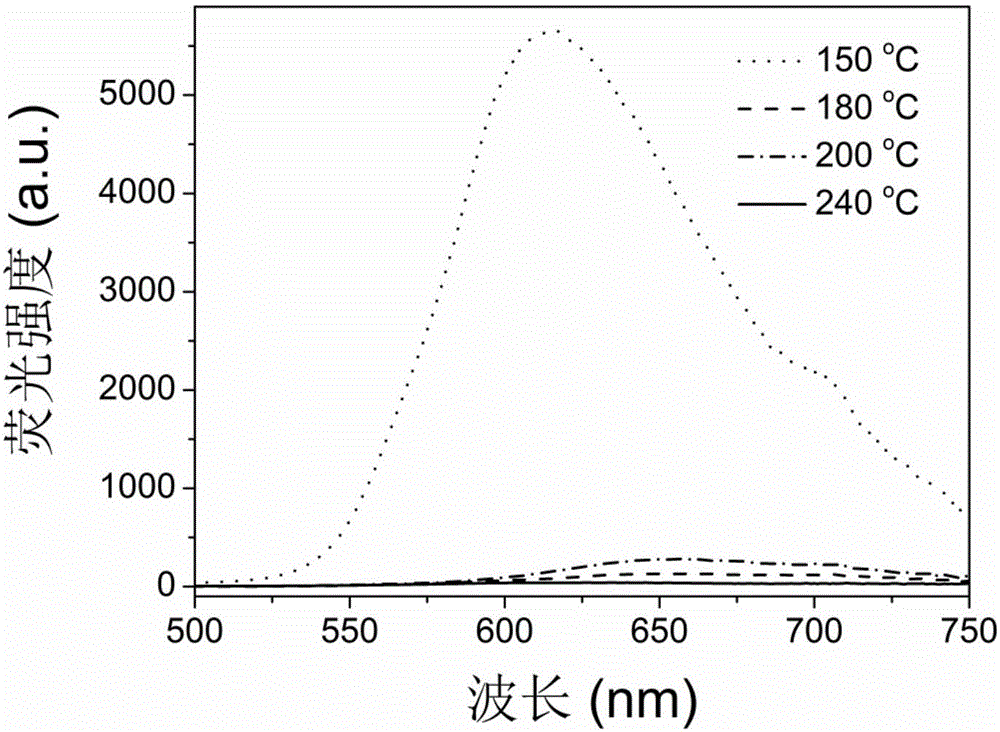
图1是实施例1在不同反应温度下制得的碳点的发射光谱图(λex=365nm)。
图2是实施例2在不同反应时间下制得的碳点的发射光谱图(λex=365nm)。
图3是实施例3在最优反应温度(150℃)与最优反应时间(5min)下制得的碳点的透射电镜图。
图4是实施例3在最优反应温度(150℃)与最优反应时间(5min)下制得的碳点的高分辨透射电镜图。
图5是实施例3制得的碳点的XPS谱图。
图6是实施例3制得的碳点的XRD谱图。
图7是实施例3制得的碳点的红外谱图。
图8是实施例3制得的碳点在不同激发下的发射光谱图。
图9是碳点在紫外灯下的稳定性研究结果。
图10是碳点自然光下的稳定性研究。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
1g柠檬酸溶于5mL丙酮中,超声使其完全溶解,再将得到的溶液加入到10mL的硅烷偶联剂(AEAPMS,工业级别,购于广州龙凯化工有限责任公司)中,在150-240℃下进行回流反应10min。即可制得在不同反应温度下的碳点,其发射光谱图如图1所示(用天美(中国)科学仪器有限公司的荧光光谱仪F-7000进行测试得到)。
实施例2
1g柠檬酸溶于5mL丙酮中,超声使其完全溶解,再将得到的溶液加入到10mL的硅烷偶联剂(AEAPMS,工业级别,购于广州龙凯化工有限责任公司)中,在150℃下进行回流反应5-60min。即可制得在不同反应时间下的碳点,其发射光谱图如图2所示(用天美(中国)科学仪器有限公司的荧光光谱仪F-7000进行测试得到)。
实施例3
1g柠檬酸溶于5mL丙酮中,超声使其完全溶解,再将得到的溶液加入到10mL的硅烷偶联剂(AEAPMS,工业级别,购于广州龙凯化工有限责任公司)中,在150℃下进行回流反应5min(即最优反应温度与反应时间),然后冷却至室温即可制得呈现粘稠态的碳点。对制得的碳点进行观察与测试,结果见图3-8。
参见图3,是本实施制备的碳点的透射电镜图,从图中可以明显看出碳点是均分分布的,分散性良好,粒径在5-7nm。
参见图4,是本实施制备的碳点的高分辨透射电镜图,从图中可以明显看出碳点是具有明显的晶格条纹,说明制得的碳点具有一定程度的石墨化结构。
参加图5,是本实施制得的碳点的XPS谱图及其元素含量,从图中可以明显看出材料中含有Si元素的存在。
参见图6,是本实施制得的碳点的XRD谱图及其元素含量,从图中可以明显看出碳点呈现宽峰,峰位在25度,这与碳点的石墨化结构的特征峰对应,对应为石墨的[002]晶面,这可以更进一步解释图4的石墨化的晶格条纹。这也进一步证实了该材料为石墨化了的碳点。
参见图7,是本实施制得的碳点的红外光谱图,从图中可以看出该碳点中含有Si-O-Si、Si-O-CH、Si-CH2键的吸收,表明了制得的产物是硅烷功能化的碳点。
参加图8,是本实施制得的碳点在不同激发波长下的发射光谱图,从图中可以看出,随着激发波长的持续增加500-580,荧光强度持续增加,峰位在640nm,表面所得碳点为明显的红光发射,所述碳点不具备典型的荧光激发依赖性质,不同于绝大多数文献所报道的具有的激发依赖性质的碳点。
上述实施例中的硅烷偶联剂除了AEAPMS外,还可以使用APTES、AEATMS或MPTES等其他硅烷偶联剂代替,其效果一致。
本发明还详细探讨了本发明所述碳点的稳定性:
1碳点在紫外灯下的稳定性研究
取2mL的粘稠态碳点于石英比色皿中,将光谱仪的双狭缝调为5,电压调为500V,激发波长与发射波长分别设为360nm和440nm,使其持续激发,研究紫外光下碳点荧光强度随着时间的变化如图9所示。(用天美(中国)科学仪器有限公司的荧光光谱仪F-7000进行测试得到)。
从图9中可以看出,在紫外光线(UV)持续激发下,荧光强度几乎没有变化,表明本发明所述碳点可以稳定存在于UV下。
2碳点在自然光下的稳定性研究
取2mL的粘稠态碳点于石英比色皿中,将比色皿至于白光下,每隔一天就用光谱仪记录下荧光强度。将光谱仪的双狭缝调为5,电压调为500V,激发波长与发射波长分别设为360nm和440nm,研究碳点荧光强度随着时间的变化如图10所示。(用天美(中国)科学仪器有限公司的荧光光谱仪F-7000进行测试得到)。
从图10可以看出,在自然光激发下,荧光强度几乎不变,表明本发明所述碳点可以稳定存在于自然光下。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!