用于通过不饱和醛的气相氧化生产不饱和羧酸的催化剂的制作方法
然而,这些催化剂存在缺点。当被用作将丙烯醛非均相催化部分气相氧化为丙烯酸的催化剂时,形成丙烯酸的选择性不完全令人满意。发生的特别的副反应是过氧化为CO和CO2(以下统称为COX)。
当使用现有技术的催化剂时,通常仅在那种使形成丙烯酸的选择性不令人满意的条件下达到足够高的丙烯醛转化率。由此,在达到丙烯醛的足够高的转化率的温度下,通常存在过氧化并因此使形成丙烯酸的选择性降低。
WO 2011/134932公开了一种由空心圆柱状载体和涂覆到载体的外表面的催化活性氧化物组合物的壳所组成的蛋壳型催化剂,以及一种通过在包括所述蛋壳型催化剂的固定催化剂床上,在气相中催化氧化丙烯醛而制备丙烯酸的方法。在工作实施例中,操作100小时后,实现形成丙烯酸的选择性最高达97.5%。
所要解决的问题是提供一种催化剂,其伴随着丙烯醛的稳定的高转化率,可以减少过氧化为COX并可增加形成丙烯酸的选择性。
该问题通过一种用于通过气相氧化α,β-不饱和醛而制备α,β-不饱和羧酸的催化剂来解决,所述催化剂包括其上涂覆有活性组合物的成型载体,其中活性组合物覆盖度q
至多为0.3mg/mm2,其中Q是以重量%计的催化剂的活性组合物含量且Sm是以mg/mm2计的成型载体的比几何表面积。
优选地,活性组合物覆盖度q至多为0.26mg/mm2,优选至多0.22mg/mm2。通常,活性组合物覆盖度q至少为0.10mg/mm2,优选至少0.15mg/mm2。
成型载体优选具有限定的几何形状。
优选的成型载体是环状、球状、片状、穿孔的片状、三叶状、穿孔的三叶状、星形挤出状、星形片状、车轮状、挤出状、丸状、圆柱状和空心圆柱状。有利地,所述成型载体的最长尺寸(即连接成型体表面上两点的最长的直接直线)为1mm至10mm。
特别优选的成型载体是空心圆柱状。所述空心圆柱状载体优选具有2mm至5mm的高度和4mm至8mm的外径,外径和内径的中值差(median difference)是1mm至2mm。所述外径和内径的中值差对应于壁厚。特别优选的是具有7mm的外径、3mm的高度和4mm的内径的几何形状。
所述成型载体优选由惰性材料组成。“惰性”是指所述成型载体的材料在气相氧化的条件下不显著地改变并且相比于针对气相氧化而涂覆的活性组合物,其至多具有(如果有的话)可忽略的催化活性。有用的惰性材料特别包括氧化铝、二氧化硅、碳化硅、二氧化锆、二氧化钍、硅酸盐(如粘土、高岭土、滑石、浮石、硅酸铝和硅酸镁)以及它们的混合物。优选滑石。特别优选C220型滑石。非常特别优选的是购自CeramTec的C 220型滑石。
优选地,中空体具有不同的表面粗糙度(例如具有砂层的空心圆柱体)。有利地,空心圆柱状载体的表面是粗糙的,因为提高的表面粗糙度通常导致涂覆到空心圆柱状成型载体表面的活性组合物壳和/或前体组合物壳的粘结强度增加。表面粗糙度Rz优选为30μm至60μm,更优选40μm至50μm(根据DIN 4768表1用购自Hommelwerke的“用于DIN-ISO表面参数的Hommel Tester”来测定)。
惰性材料可以是多孔的或无孔的。优选地,惰性材料是基本上无孔的(总孔体积小于1%,基于载体的体积计)。惰性材料的几何密度通常在0.5g/cm3至8.0g/cm3的范围内,优选为1.0g/cm3至7.0g/cm3,进一步优选为1.5g/cm3至6.0g/cm3,更优选为2.0g/cm3至5.0g/cm3。化学惰性材料的几何密度用成型载体的质量除以其几何体积来计算。
几何体积可以由对理想基础几何形式的相应测量来计算。例如,空心圆柱体的几何体积可以基于圆柱体的高H、外径ED和内孔直径ID来计算。
催化剂的活性组合物含量Q(以重量%计)是基于活性组合物和成型载体质量的总和的活性组合物的质量。为了确定活性组合物的质量,可以由通过称重测定的催化剂的质量(在热处理以除去粘合剂以后;见下文)中减去已知的成型载体的质量。为了增加测量精度,可以测定大量催化剂或成型载体的质量并求平均值。例如,确定数目的催化剂的活性组合物的质量可以通过测定催化剂的总质量并减去成型载体重量来确定,所述成型载体重量是用成型载体重量乘以成型载体的数目算出的。活性组合物含量Q的测定也可以通过将活性组合物从成型载体上洗掉的方式进行。为了该目的,例如,可将经过涂覆的催化剂用氨水溶液反复煮沸并轻轻倒出得到的液体。随后可将余留的载体干燥。活性组合物含量由催化剂质量(洗掉活性组合物以前测定的)与载体质量(洗掉活性组合物并干燥以后测定的)之差,基于催化剂质量来计算。
相应地得出以重量%计的催化剂的载体材料含量(100-Q)。
成型载体的比几何表面积Sm是基于成型载体的质量的成型载体的几何表面积。
几何表面积可以由对理想基础几何形式的相应测量来计算。几何表面积是理想化的参数且并不考虑由成型体的孔隙度或表面粗糙度所引起的表面积的增加。
就球形成型载体而言,几何表面积是
4πr2
其中r是球形成型载体的半径。就空心圆柱状成型载体而言,几何表面积是
其中H为高度,ED为外径且ID为空心圆柱状成型载体的内径。
优选地,涂覆到成型载体上的活性组合物的平均厚度为50μm至400μm,优选75μm至350μm,更优选100μm至300μm且最优选100μm至200μm。
优选地,涂覆到成型载体上的活性组合物的厚度具有最大的均匀性。在各种成型载体之间,涂覆的活性组合物的厚度同样具有最大的均匀性。
用于通过气相氧化α,β-不饱和醛制备α,β-不饱和羧酸的活性组合物,其本身是已知的。例如,包含元素Mo和V的催化活性多元素氧化物组分是合适的,其中在催化活性多元素氧化物组分中除氧以外的所有元素的总量中,元素Mo的摩尔比是20mol%至80mol%,存在于催化活性多元素氧化物组分中的Mo相对于存在于催化活性多元素氧化物组分中的V的摩尔比,Mo/V,为15:1至1:1。优选地,多元素氧化物还包含元素Nb和W的至少一种;相应的摩尔比Mo/(W和Nb的总量)优选为80:1至1:4。通常,所述多元素氧化物组分还包含Cu,相应的摩尔比Mo/Cu为30:1至1:3。
前述多元素氧化物组分,除元素Mo、V和任选的Nb和/或W或Cu以外,还可以包括,例如,元素Ta、Cr、Ce、Ni、Co、Fe、Mn、Zn、Sb、Bi、碱金属(Li、Na、K、Rb、Cs)、H、碱土金属(Mg、Ca、Sr、Ba)、Si、Al、Ti和Zr。当然,多元素氧化物活性组合物也可以仅由元素Mo、V、O以及Cu和任选的W和/或Nb组成。它们特别适合作为用于丙烯醛至丙烯酸的非均相催化部分气相氧化的催化剂的活性组合物。
非常特别适合作为用于丙烯醛至丙烯酸的非均相催化部分气相氧化的催化剂的活性组合物,其组成包含以下通式(I)的多元素氧化物组分:
Mo12VaX1bX2cX3dX4eX5fOn(I)
其中
X1是W、Nb、Ta、Cr和/或Ce,
X2是Cu、Ni、Co、Fe、Mn和/或Zn,
X3是Sb和/或Bi,
X4是一种或更多种碱金属和/或碱土金属和/或N,
X5是Si、Al、Ti和/或Zr,
a是1至6范围内的数字,
b是0.2至4范围内的数字,
c是0至18范围内的数字,优选0.5至18,
d是0至40范围内的数字,
e是0至4范围内的数字,
f是0至40范围内的数字,且
n是元素氧的化学计量系数,其由(I)中除氧以外的元素的化学计量系数和它们的化合价来确定。
优选地,变量应在附带条件所指定的范围内进行选择,所述附带条件为在多元素氧化物组分(I)中除氧以外的所有元素的总量中,元素Mo的摩尔比是20mol%至80mol%。
优选地,多元素氧化物组分符合通式(II):
Mo12VaWbCucX4eX5fOn (II)
其中
X4是一种或更多种碱金属和/或碱土金属,
X5是Si、Al、Ti和Zr中的一种或更多种元素,
a是2至4范围内的数字,优选2.5至3.5范围内的数字,
b是0至3范围内的数字,优选0.2至3范围内的数字,优选0.5至2范围内的数字,更优选0.75至1.5范围内的数字,
c是0.5至3范围内的数字,优选0.7至2.7范围内的数字,优选0.9至2.4范围内的数字,更优选1至1.5范围内的数字,
e是0至4范围内的数字,优选0至2范围内的数字,优选0至1范围内的数字,更优选0至0.2范围内的数字,
f是0至40范围内的数字,优选0至15范围内的数字,优选0至8范围内的数字,更优选0,和
n是元素氧的化学计量系数,其由在(II)中除了氧以外的元素的化学计量系数和它们的化合价来确定。
元素X4和X5不是通式(II)的活性组合物的必需部分。它们通常在活性组合物中充当惰性稀释剂。将它们掺入活性组合物中可将比体积的催化剂活性调节至所需水平。
在一个实施方案中,如在DE 10 2007 010 422中所述的,活性组合物可以是以下物质的细碎混合物的形式:如式I或II的包含元素Mo和V的多元素氧化物组合物和钼氧化物源。钼氧化物源适当地选自钼的氧化物和在高温和分子氧的作用下形成钼的氧化物和钼的化合物。这些钼氧化物源包括氧化钼如MoO3、Mo18O52、Mo8O23和Mo4O11,或化合物如钼酸铵[(NH4)2MoO4]和多钼酸铵(如七钼酸铵四水合物[(NH4)6Mo7O244H2O])。替代实施例是水合氧化钼(MoO3xH2O)。氧化钼是优选的钼氧化物源。
根据本发明有利地,细碎的钼源的粒度(粒径或粒径分布)与细碎的包含元素Mo和V的多元素氧化物的粒度是相同的(这确保了与细碎的多元素氧化物特别均匀的混合)。当细碎的钼氧化物源是氧化钼(特别地MoO3)时尤其是这样。
钼氧化物源的额外使用可以预防性地对抗催化剂在将丙烯醛非均相催化部分气相氧化为丙烯酸的过程中的失活,或延迟失活的开始。
通常,催化剂是多孔的。催化剂优选具有特定平均直径的孔的特定分布。催化剂中的大孔的体积比例pvol优选至少是0.35,其中pvol由以下公式确定:
其中,
V0.26-2是平均直径为0.26μm至2μm的孔的体积,和
V0.02-6.5是平均直径为0.02μm至6.5μm的孔的体积。
平均直径在纳米和微米范围内的孔的体积可以通过汞孔隙率法(mercury porosimetry)(例如根据DIN No.66133)来测定。汞表现为相对于大多数固体的非润湿性液体。因此,汞不自发地被多孔材料吸收而只是在外部压力下才渗入到固体样品的孔中。该压力的程度取决于孔的尺寸。将这种性能利用到汞孔隙率测定法中,以便通过在外部施压下的压入而得到以容积计算的孔径。
这是通过将已预先排气(以将存在于多孔结构中的任意液体排气)的多孔系统(待分析的样品)浸入到汞浴中实现的,所述汞浴的压力可以改变。
由于汞不润湿样品材料,所以不得不将汞压入样品的孔中(在相应压力下等待平衡的建立)。汞向具有更大表面积的孔中的渗入在相对更低的压力下进行,而汞向更窄的孔中的渗入需要相对更高的压力。假定存在圆柱状的孔,那么借助于Washburn方程,迫使液体汞对抗汞的表面张力而进入适当直径的孔中(压入;压汞)所需的外部压力可与所述直径相关。汞孔隙率法分析过程中采用的压力范围与所覆盖的孔径范围相关联。
在25℃下实验确定的压汞曲线随后可被用于,通过计算,提取所覆盖的孔径的范围、孔的直径分布、孔的总内表面和孔的总内体积(总侵入体积;总孔体积)(参见Georges Reber(1991)在巴塞尔大学哲学和自然科学院的就职论文“Eigenschaften andvon Aerogelfenstern im Vergleich mit konventionellen sowie evakuierten Fenstern”[Properties and Possible Uses of Aerogel Windows Compared to Conventional and Evacuated Windows])。下文描述的Micromeritics Auto Pore IV 9520测量仪包括适于这些目的的标准计算程序。
本发明的催化剂通常通过将粉状活性组合物涂覆到成型载体上而获得,优选地通过下文描述的制备方法而获得。
粉状活性组合物可以以不同的方式制备。在一个实施方案中,活性组合物通过使用活性组合物元素组分的源制备均匀干燥的混合物而制备,所述均匀干燥的混合物是在350℃至600℃的温度下焙烧并然后转化成粉末形式的。
优选的活性组合物元素组分的源是活性组合物中存在的金属的氧化物。有用的活性组合物元素组分的源还包括那些至少在氧气的存在下通过加热可被转化为氧化物的化合物;特别是活性组合物中存在的金属的卤化物、硝酸盐、甲酸盐、草酸盐、柠檬酸盐、乙酸盐、碳酸盐、胺络合物、铵盐和/或氢氧化物。
优选地,均匀干燥的混合物通过均匀混合所述源来制备。均匀混合可以以干形式或湿形式进行。如果以干形式进行,那么源适当地以细碎的粉末的形式使用。特别地,当起始原料完全是以溶解形式存在的源时,是在混合的过程中获得均匀干燥的混合物。因此,源的均匀混合优选以湿形式进行。优选地,将源彼此之间以溶液和/或悬浮液的形式混合并随后将得到的湿的混合物干燥,以得到均匀干燥的混合物。所用的溶剂和/或悬浮液介质优选为水或水溶液。优选通过出口温度为100℃至150℃的喷雾干燥而对湿的混合物进行干燥。干燥气体料流优选为空气或分子氮。
在焙烧之前,干的混合物(例如通过喷雾干燥获得的干的混合物)可通过混合而进行材料处理操作。术语“混合”应理解为是指干混、捏合和搅拌,任选地添加液体。混合产生具有较窄的粒度分布的均质组合物。
特别有利地,混合操作在添加液体(例如水、乙酸等)以后以捏合操作的形式进行,由此获得延性或塑性材料。作用在其中的剪切力粉碎附聚物。延性材料适于挤出并产生可被干燥的稳定的挤出物。有利地,经干燥的挤出物尤其适于在旋转管式炉中进行焙烧。
焙烧可以在惰性气体下或在氧化气氛下,亦或在还原气氛下进行。优选地,焙烧在氧化气氛下进行。有用的惰性气体特别是氮气、水蒸气、稀有气体和它们的混合物。氧化气氛优选包含氧气,特别是空气。还原气氛优选包含H2、NH3、CO、甲烷和/或丙烯醛。活性组合物的催化活性通常呈现根据焙烧气氛的氧含量的最优。优选地,焙烧气氛的氧含量为0.5体积%至10体积%,更优选1体积%至5体积%。氧气含量高于或低于前述限度通常会降低所得的催化活性。焙烧时间可以是几分钟到几小时且通常随着焙烧温度的水平而降低。具有良好适应性的焙烧方法记载在,例如,WO 95/11081中。
焙烧干的混合物得到活性组合物。转化为粉末形式优选通过研磨实现。
在制备催化剂的另一方法中,首先将细碎的前体组合物涂覆到成型载体的表面,然后实施焙烧,在成型载体表面上将前体组合物焙烧成活性组合物。细碎的前体组合物优选包含活性组合物元素组分的源。活性组合物优选为通式(I)或(II)的活性组合物。
在根据本发明的用于制备催化剂的方法中,成型载体通过以下方式用活性组合物涂覆:在容器中混合大量的成型载体、粉状活性组合物和液体粘合剂,不使粉状活性组合物被液体粘合剂饱和,涂覆操作的持续时间小于30分钟。粉状活性组合物被液体粘合剂饱和是通过以下方式避免:选择液体粘合剂的量与粉状活性组合物的量的比例,使得粘合剂的量保持在粉状活性组合物的液体吸收能力以下。
例如,粉末的液体吸收能力可以通过以下方式测定:在搅拌器中搅动粉末并将液体施加到搅拌后的粉末中并测量搅拌器电机相对于时间的扭矩。最高达最大扭矩时已施于粉末的液体的量可用于计算粉末的液体吸收能力。
优选地,粉状活性组合物的最长尺寸在50μm以上的颗粒的比例为小于1%。
术语“粘合剂”应理解为是指永久或暂时地提高活性组合物粉末颗粒彼此之间和/或其与成型载体材料之间的附着力的物质。优选地,粘合剂基本上在随后的干燥过程中蒸发或升华。在根据本发明的方法中,所使用的粘合剂,例如,可以是多元醇,例如乙二醇、丙二醇、丁二醇、甘油;或酰胺类,例如甲酰胺、N,N-二甲基甲酰胺、N,N-二乙基甲酰胺、N,N-二丁基甲酰胺、乙酰胺、吡咯烷酮或N-甲基吡咯烷酮。液体粘合剂优选选自水、甘油和甘油水溶液。优选的液体粘合剂是包含20重量%至99重量%的水的甘油水溶液。特别优选的液体粘合剂是包含75重量%的水的甘油水溶液。
优选地,首先将成型载体装入到容器中,然后在涂覆持续期间将粉状活性组合物和液体粘合剂分别加入到容器中。因此,粉状活性组合物和液体粘合剂仅在容器中彼此接触。优选地,粉状活性组合物和液体粘合剂仅在首先装入到容器中的成型载体的表面结合。这通过将液体粘合剂喷雾到容器中并向容器的液体粘合剂喷射锥角以外的区域中引入粉状活性组合物来实现。由此,避免了粉末颗粒的液体局部过载。在处理的持续期间内将粉状活性组合物和液体粘合剂引入到容器中,例如,可通过随着时间的推移连续地加入或不连续地加入各部分而进行。
混合优选通过容器的连续运动来实现。运动优选为转动。
DE-A 2909671(还参见EP-A 714 700和DE 10 2005 010 645)中公开的使用各个所需的液体粘合剂的方法原理,其特别适于用于实施上述制备催化剂的方法。
换言之,将待被涂覆的成型载体(优选空心圆柱状成型载体)引入到优选倾斜的(倾斜角通常为30°至90°)旋转容器中(例如旋转盘或涂覆罐(coating tank)或涂覆鼓(coating drum))。用于该目的的用途的合适的旋转容器特别是购自Freund Industrial Co.,Ltd,Tokyo(JP)的Hi-Coater HCF-100和购自GebrüderMaschinenbau GmbH,Paderborn,Germany的Hi-Coater LH 100。
旋转容器在两个以有利间隔连续排列的计量装置的下方处理成型载体,优选空心圆柱状成型载体。两个计量装置中的第一个适当地对应于喷嘴,通过所述喷嘴用液体粘合剂将在旋转盘(Hi-Coater)中滚动的成型载体以可控的方式润湿。就应用而言适当地,第二个计量装置在喷入的液体粘合剂的喷射锥角以外,并用于供应粉状活性组合物(例如经由振动槽)。成型载体附着活性组合物,原因是活性组合物通过滚动运动紧密结合成成型载体表面上的连贯外壳。
如果需要,那么在随后的旋转过程中使该经基础涂覆的成型载体,优选空心圆柱状载体再次经过喷嘴,被以可控的方式润湿(任选使用另一种液体粘合剂),以便能够在进一步的运动等的过程中附着一层(任选地另一层)粉状活性组合物(通常不需要中间干燥)。至少部分去除所使用的液体粘合剂,例如,可以遵从EP-A 714 700的教导或DE-A 10 2005 010 645的教导,通过最后的给热来实现,例如通过热气体(例如N2或空气)的作用来实现(所述热气体通过在旋转盘或涂覆罐或涂覆鼓(通常为旋转容器)中的像栅一样地配置的空间分离壁元件而进入和移出)。
对于所述涂覆方法的实施方案而言重要的是,以可控的方式进行待涂覆的成型载体的表面的润湿。简而言之,这意味着将载体表面适当地润湿,使得其吸收液体粘合剂但是在载体表面上并不会明显地观察到所述液体粘合剂。如果成型载体的表面太潮湿,那么细碎的活性组合物和/或前体组合物附聚产生单独的附聚物,而不是附着到所述表面上。关于这方面的更详细的信息可以在DE-A 2909671、EP-A 714 700和DE 10 2005 010 645中找到。所述方法的一个优势在于,所使用的液体粘合剂的去除可以以相对可控的方式进行,例如通过蒸发和/或升华。在最简单的情况下,正如已经解释过的,这可以在适当温度(通常为50至150℃)下通过热气体的作用来实现。热气体的这种作用通常导致预干燥。
粘合剂的去除可以在任何种类的干燥装置中(例如,在带式干燥机中)进行和/或直到处于壳管式反应器的固定催化剂床中时才进行,例如按照DE-A 10 2005 010 645所推荐的。优选地,本发明的催化剂通过在150℃至400℃,优选250℃至350℃的温度下进行干燥而去除经涂覆的成型载体中的液体粘合剂而得到。干燥优选在空气流中进行。优选地,干燥的持续时间为0.5小时至8小时,优选1小时至4小时。
本发明还提供一种通过越过固定催化剂床用分子氧气相氧化α,β-不饱和醛而制备α,β-不饱和羧酸的方法,其中所述固定催化剂床包括本发明的催化剂的床。优选地,通过引导分子氧和α,β-不饱和醛通过固定催化剂床而使分子氧和α,β-不饱和醛与固定催化剂床接触。优选地,将包含分子氧和α,β-不饱和醛的反应气体引导通过固定催化剂床,由此将反应气体转化为产物气体。
α,β-不饱和醛优选地选自包含3个至6个(即,3个、4个、5个或6个)碳原子的α,β-不饱和醛,特别选自丙烯醛和甲基丙烯醛。更优选地,α,β-不饱和醛是丙烯醛。所述方法特别适合于制备α,β-不饱和羧酸,尤其适合于将丙烯醛氧化为丙烯酸和将甲基丙烯醛氧化为甲基丙烯酸。优选地,其为通过丙烯醛的气相氧化而制备丙烯酸的一种方法。
分子氧优选以空气的形式供给到所述方法中。
反应气体中存在的α,β-不饱和醛的比例通常将为3体积%至15体积%,优选4体积%至10体积%,更优选5体积%至8体积%,各自基于反应气体计。
优选地,除水蒸汽以外,反应气体还包含至少一种惰性稀释剂气体。这应理解为是指那些在气相氧化过程中至少95mol%的程度,优选至少98mol%的程度的保持化学不变的气体。惰性稀释气体的实例是N2、CO2和稀有气体(例如氩气)。所使用的惰性稀释气体优选为分子氮。所述惰性稀释气体可以包含至少20体积%的分子氮气,优选至少40体积%,进一步优选至少60体积%,更优选至少80体积%,最优选至少95体积%。
反应气体还可包含水蒸汽。
反应气体还可包括循环气。循环气体应理解为意指当α,β-不饱和羧酸基本上选择性地从气相氧化的产物气体中分离出时所剩余的残余气体。
优选地,本发明用于制备α,β-不饱和羧酸的方法形成了烯烃到α,β-不饱和羧酸的两阶段气相氧化的第二阶段。在这样的两阶段气相氧化过程中,优选将第一阶段的产物气体供给到第二阶段。在被供给到第二阶段之前,可以将来自第一阶段的产物气体,例如,冷却和/或可以加入氧气(氧气的第二次加入,优选加入空气)。优选地,将循环气引导进入两阶段的第一阶段。
在反应气体中,O2:α,β-不饱和醛的摩尔比优选是1至3,优选1至2,更优选1至1.5。
优选地,反应气体所包含的α,β-不饱和醛:氧气:水蒸气:除了水蒸汽以外的惰性稀释气体的体积比为1:(1至3):(0至20):(3至30),优选1:(1至3):(0.5至10):(7至10)。
优选地,α,β-不饱和醛在床上的空速不多于600l(STP)/(lh),优选不多于300l(STP)/(lh),更优选不多于250l(STP)/(lh),最优选不多于200l(STP)/(lh)。优选地,α,β-不饱和醛在床上的空速不小于30l(STP)/(lh),优选不小于70l(STP)/(lh),更优选不小于90l(STP)/(lh),最优选不小于120l(STP)/(lh)。以l(STP)/(lh)表示的α,β-不饱和醛在床上的空速应理解为是指每升床每小时的以标准升计的α,β-不饱和醛的量,所述α,β-不饱和醛是作为反应气体组成被引导通过固定催化剂床的。一个标准升l(STP)是对应于α,β-不饱和醛的摩尔量的摩尔量的理想气体在标准条件下(即在25℃和1bar下)所占的以升计的体积。
通常,在反应气体中存在0.5bar至100bar的总压力,优选1bar至5bar,特别地1bar至3bar。在本文中所有的压力数值指的是绝对压力。
优选地,制备α,β-不饱和羧酸的方法在壳管式反应器中进行,所述壳管式反应器的反应管已经用固定催化剂床装填。
例如,床可以仅由本发明的催化剂组成。也可以是本发明的催化剂与成型的稀释剂体的基本均匀的混合物,所述成型的稀释剂体对于将出现在床中的气相氧化基本上是惰性的。用于成型稀释剂体的有用的材料包括,例如,多孔或无孔氧化铝、二氧化硅、二氧化锆、碳化硅、硅酸盐(如硅酸镁或硅酸铝和/或滑石(例如,购自德国CeramTec的C220型))。
原则上,成型的稀释剂体的几何形状可以是如期望的。换言之,它们可以是,例如,环状、球状、片状、穿孔的片状、三叶状、穿孔的三叶状、星形挤出状、星形片状、车轮状、挤出状、丸状、圆柱状和空心圆柱状
空心圆柱壳管式反应器优选为两区域壳管式反应器。优选的两区域壳管式反应器由DE-C 28 30 765公开。但是在DE-C 25 13 405、US-A 3147084、DE-A 22 01 528、EP-A 383224和DE-A 29 03 582中公开的两区域壳管式反应器也适合。
在两区域壳管式反应器中,优选使两个基本空间上分离的温度控制介质围绕反应管传导。温度控制介质优选为盐熔体。优选将温度控制介质的输入温度设定为230℃至300℃,优选240℃至290℃,更优选250℃至285℃。温度控制介质可以以与反应气体混合物并流或逆流的形式传导通过各个温度控制区。在温度控制区域内,温度控制介质优选以曲折方式传导。优选地选择各个温度控制区域内的温度控制介质的流率,使得热交换介质的温度从到温度区域的入口位置到离开温度区域的出口位置上升0℃至15℃,通常1℃至10℃,或2℃至8℃,或3℃至6℃。
在优选的实施方案中,固定催化剂床包括至少两个连续的反应区域,在这种情况下,床至少在最接近反应器入口的反应区域中包含本发明的催化剂。
在另一个优选的实施方案中,固定催化剂床包括至少两个连续的反应区域,在这种情况下,床至少在其中发生最高局部温度的反应区域中包含本发明的催化剂。
各个反应区域在选自下列的至少一种性质方面彼此不同:惰性成型稀释剂体的含量、催化剂的形状、催化剂的空间填充程度、催化剂的活性组合物含量和活性物质的化学组成。
因为不同的性质,所以一个反应区域的比体积催化剂活性可以与另一个反应区域的比体积催化剂活性不同。优选地,从反应器入口到反应器出口,比体积催化剂活性由一个反应区域到下一个增大。
在其他恒定的条件下,(相对)比体积催化剂活性可以测定为基于催化剂床体积的反应速率。
通过用成型的稀释剂体稀释催化剂可以使比体积催化剂活性变化。替代性地或额外地,可以通过改变活性组合物含量而对比体积催化剂活性进行调节。
优选地,空间活性组合物密度,即,以升计的每单位反应区域空间体积的以g计的活性组合物的量,其在最接近反应器入口的反应区域比在最接近反应器出口的反应区域更低。
从某一操作时间,操作持续时间逐渐增长伴随着催化剂装料质量逐渐减少。在优选的实施方案中,为了恢复床的质量,不是将整个用过的床而是仅将床的发生最高局部温度的部分移除并且用新鲜的床代替。例如,将在其中发生最高局部温度的反应区中的床用新鲜的床代替,并使所述新鲜的床保持在反应区的朝着反应气体流动方向的下游。
通常,壳管式反应器还具有热管,以测定催化剂床中的气体温度。合适地,选择热管的内径和在其中的用于热电偶的容纳套筒的直径,使得在热管和反应管中,散发反应热量的体积与去热表面积的比率相同或仅稍有不同。
基于相同的GHSV,压力降在反应管和热管中应该相同。在热管中的任意压力降可以,例如,通过向催化剂中添加碎催化剂而平衡掉。合适地,这种平衡遍布整个热管长度是均匀的。除此以外,热管的装填可以如EP-A 873783中记载的那样进行配置。
在热管中测得的温度可用于确定固定催化剂床的最高局部温度及其在固定催化剂床中的位置。
附图说明
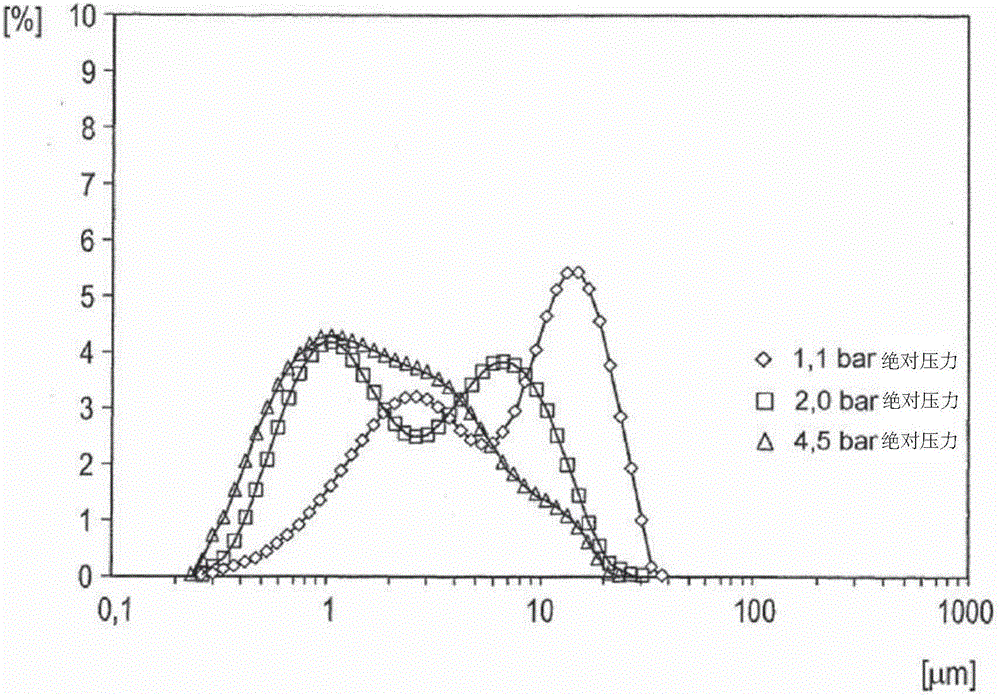
图1a示出了细碎的粉末P的累积粒度分布。横轴表示对数标度的以μm计的粒径。对应于特定粒径的分布曲线上的纵轴值表示由特定粒径或更小粒径的颗粒组成的总颗粒体积的百分比。
图1b示出了细碎的粉末P的微分粒度分布。横轴表示对数标度的以μm计的粒径。对应于特定粒径的分布曲线上的纵轴值表示由特定粒径的颗粒组成的总颗粒体积的百分比。
图2示出了细碎的粉末P的衍射图。
图3示出了C1的活性组合物外壳的孔的孔径分布。
图4示出了C2的活性组合物外壳的孔的孔径分布。
图5示出了C3的活性组合物外壳的孔的孔径分布。
图6示出了C4的活性组合物外壳的孔的孔径分布。
图7示出了C5的活性组合物外壳的孔的孔径分布。
图8示出了C6的活性组合物外壳的孔的孔径分布。
图9示出了C7的活性组合物外壳的孔的孔径分布。
图10示出了C8的活性组合物外壳的孔的孔径分布。
图11示出了C9的活性组合物外壳的孔的孔径分布。
图12示出了C10的活性组合物外壳的孔的孔径分布。
图13示出了C11的活性组合物外壳的孔的孔径分布。
在图3至13中,将以μm计的特定的孔径标绘在横坐标上(以10为底的对数坐标图)。以(ml/g的活性组合物)计的标绘在纵坐标上的是各孔径对特定的总孔体积的各自贡献的积分(对特定的总孔体积的累积贡献)(□曲线)。结束点是基于活性组合物(总侵入体积)的(特定的)总孔体积。
实施例
催化剂的制备
A)前体组合物的制备
在具有梁式搅拌器(beam stirrer)的水加热的1.75m3夹套的不锈钢容器中,在搅拌(速度:70转/分钟)的同时将8.2kg的乙酸铜水合物(含量:32.0重量%的铜,加入速率50kg/h,购自Goldschmidt)在室温下(25℃)溶解于274l的水中。获得溶液1。将所述溶液1再搅拌30分钟。
与此空间分开地,首先将具有梁式搅拌器的水加热的1.75m3夹套的不锈钢容器(速度:70转/分钟)装入614l的水并加热至40℃,然后在保持40℃的同时将73kg的七钼酸铵四水合物(81.5重量%的MoO3,加入速率300kg/h,购自H.C.Starck GmbH)搅拌加入。然后在搅拌的同时将容器内容物在30分钟内加热至90℃,同时保持该温度,接连按照提及的顺序加入以下物质:12.1kg的偏钒酸铵(77.6%的V2O5,加入速率150kg/h,在加入后进一步搅拌40分钟时间)和10.7kg的仲钨酸铵七水合物(89.6重量%的WO3,加入速率50kg/h,在加入后进一步搅拌30分钟时间)。获得溶液2。
将溶液2冷却到80℃,然后在梁式搅拌器的70转/分钟的搅拌器速度下将溶液1迅速转移到溶液2中,并继续搅拌。将所得混合物与133l的25重量%的NH3水溶液在25℃的温度下进行混合。在搅拌的同时,形成透明溶液,其暂时具有65℃的温度和8.5的pH。使所述透明溶液流到另一个具有梁式搅拌器的水加热的1.75m3夹套额不锈钢容器中。将容器内容物加热到80℃,在40转/分钟的搅拌器速度下搅拌并流通。通过自动加入25重量%的NH3水溶液的方式使容器内容物的pH保持在8.5的值。将容器内容物泵入到购自Niro(丹麦)的FS 15型转盘喷雾塔中并在350±10℃下的并流热空气入口温度、15 000rpm的盘速和2300m3(STP/h)的废空气(combustion air)体积流率下进行干燥,同时保持在喷雾塔中压力降为1mbar。调节计量进入喷雾塔中的液体体积流率,使得达到110±5℃的气体出口温度。得到的喷雾塔具有2μm至50μm的粒径和21±2重量%的灼烧损失(ignition loss)。灼烧损失通过在空气下在瓷坩埚中加热(在600℃下3小时)来测定。瓷坩埚已经预先在900℃下焙烧至恒重。使喷雾粉末分配进入具有塑料入口的专用容器或专用桶(200l)中。为了去除所有团块,使用了筛子衬垫。
将由此得到的75kg的喷雾粉末计量加入到15转/分钟的螺杆转速下的购自AMK(Aachener Misch-and Knetmaschinen Fabrik)的VM 160型(Sigma叶片)捏合机中。随后,在15转/分钟的螺杆转速下将6.5l的乙酸(100重量%,冰醋酸)和5.2l的水计量加入到所述捏合机中。在4分钟至5分钟(螺杆速度:20转/分钟)的捏合时间后,加入另外6.5l的水并继续捏合操作至30分钟过后(捏合温度约40℃至50℃)。在捏合的过程中,观察功率消耗。当超过25%的功率消耗时,按需将另外的约1l的水加入到捏合物料中。然后,将捏合物料排空到挤出机中且通过挤出机(购自Bonnot Company(Ohio),类型:G 103-10/D7A-572K(6”Extruder W Packer))装置成型,以得到挤出物(长度:1cm至10cm;直径6mm)。在三区域带式干燥机中,在10厘米/分钟的带速度和64分钟的总停留时间(resulting residence time)和155℃的气体输入温度下对挤出物进行干燥。气体温度的期望值为在区域中90℃至95℃,在区域2中约115℃且在区域3约125℃。
B)式Mo12V3W1.2Cu1.2On的活性组合物的制备
在DE 10360057A1中记载的旋转管式炉中,在下列条件下进行热处理:
热处理以306kg的物料量分批实施,所述物料已经按照在A中的描述而制备;
旋转管相对于水平的斜角为≈0°;
旋转管以1.5转/分钟右旋;
在整个热处理过程中,将205m3(STP)/h的气体料流引导通过旋转管,所述气体料流(在置换最初存在的空气后)具有以下的组分并在其由旋转管输出处用另外的25m3(STP)/h的载气氮气进行补充:80m3(STP)/h的由基载氮气和在旋转管中释放的气体组成的组分、25m3(STP)/h的载气氮气、30m3(STP)/h的空气和70m3(STP)/h的再循环循环气体。载气氮气在25℃的温度下提供。在每种情况下,在各物料在旋转管中所具有的温度下使来自加热器的其他气体料流的混合物引导进入旋转管中。
在10h内,将物料温度从25℃以基本线性的方式升高到300℃;然后在2h内将物料温度以基本线性的方式升高至360℃;随后,在7h内将物料温度以基本线性的方式降低至350℃;然后在2h内将物料温度以基本线性的方式升高到420℃并将该物料温度保持30分钟;然后通过相应地增加基载氮气而将引导通过旋转管的气体料流中的30m3(STP)/h的空气代替(这结束了实际的热处理操作),将旋转管的加热关闭并通过开启借由向内吸入环境空气进行的旋转管的快速冷却而在2h内将物料冷却到低于100℃的温度,且最终达到室温;此处,在25℃的温度下将气体料流进料到旋转管中;在整个热处理期间,气体料流在(紧接着)旋转管出口处的压力比外部压力低0.2mbar。
在热处理的所有阶段中,旋转管式炉中的气体气氛的氧含量为2.9体积%。在还原性热处理的整个持续时间内,以算术平均计,旋转炉中的气体气氛的氨浓度为4体积%。
通过双向横流分级磨(biplex crossflow classifying mill)(BQ 500)(购自Hosokawa-Alpine Augsburg)将得到的催化活性物料粉碎,以得到细碎的粉末P。此处,将24个长叶片安装到研磨轨道中。研磨速度为2500转/分钟。通风机节流阀完全打开。将剂量设定为2.5转/分钟。空气输出体积流率为1300m3/h,压力差10mbar至20mbar。研磨得到的细碎的粉末的粉末颗粒的50%通过了1mbar至10μm筛目的筛子。细碎的粉末中的最长尺寸高于50μm的颗粒的比例为小于1%。上述经粉碎的催化活性多元素氧化物组分粉末的颗粒尺寸分布作为用于干燥分散的压缩空气的分散压力的函数示于图1a和1b中(◇=1.1bar(绝对压力);□=2.0bar(绝对压力);Δ=4.5bar(绝对压力))。
构成图1a和1b的粒径分布的基础的测量方法是激光衍射法。这涉及引导多元素氧化物组分粉末通过分散通道进入到Sympatec RODOS干燥分散器(Sympatec GmbH,System-Partikel-Technik,Am Pulverhaus 1,D-38678Clausthal-Zellerfeld),用压缩空气(其具有1.1bar或2bar或4.5bar(绝对)的各自的分散压力)将其干燥分散在那里并将其在自由射流中吹到测量室。然后,根据ISO 13320,用Malvern Mastersizer S激光衍射分光计(Malvern Instruments,Worcestershire WR14 1AT,United Kingdom)测定在其中的基于体积的粒径分布(遮蔽3-7%)。
图2示出了细碎的粉末P的X-射线衍射图。横轴表示2Θ标度的衍射角[度]。标绘在纵坐标上的是绝对强度。
C)活性组合物的成形
C1(对比实施例)
将1600g的空心圆柱状载体(外径7mm,长度3mm,内径4mm,购自CeramTec的具有45μm的表面粗糙度Rz的C220滑石(砂砾层))用粉碎的细碎的粉末P涂覆。涂覆在HiCoater LHC 25/36(购自D-33102Paderborn)中实施。该HiCoater已被改进以确保连续的粉末计量。该HiCoater包括漏斗形粉末储存器,所述漏斗形粉末储存器经聚乙烯软管(内径:8mm,外径11.1mm;购自Saint-Gobain Performance,89120Charny,France)连接到HiCoater的滚筒。滚筒半径为18cm。滚筒的深度为20cm。滚筒绕着旋转的轴是水平对准的。关于涂覆,将750g的粉碎的催化活性氧化物组分粉末引入到粉末储存器中。通过连续的压力计量而计量加入粉末。时控脉冲阀设定为50ms且压力设定为高于环境压力(~1atm)0.7bar。在涂覆期间,对漏斗形粉末储存器中的粉末进行连续搅拌以确保均匀计量(搅拌器运行时间:2s,搅拌器中止时间:1s,改进的V-型锚式搅拌器,在BASF SE机构内部构建)。粘合剂是75重量%的水和25重量%的甘油的水溶液。将其经由液体计量装置单独地喷雾到滚筒中。将液体通过Watson-Marlow HPLC泵(323型)泵入到计量臂,所述计量臂在滚筒中(喷射压力3bar,成形压力2bar,流率:3g的甘油/水溶液(1:3)/分钟)。粉末计量装置和液体计量装置彼此平行排列。安装在计量臂上的购自Schlick(DE)的570/0S75型喷嘴和同样固定在计量臂下面的固体计量装置的出口孔,它们相距6cm平行对齐且,在角度测量仪的辅助下,它们与水平面呈40°角。粉末在喷嘴的喷射锥角外面计量加入。喷嘴孔和固体计量装置出口孔显示滚筒的旋转方向。在涂覆期间,滚筒以15rpm顺时针方向旋转。涂覆在25℃下进行50分钟时间。此后,将经涂覆的成型载体材料在130℃的空气入口温度和81℃的空气出口温度下干燥27分钟。此后,使它们在滚筒中经30分钟静止而冷却到25℃。在涂覆期间,所提供的粉末大部分附着在成型载体的表面。没有附着的部分被收集在滚筒下游的过滤器中。没有观察到细碎的氧化物组分的成对,以及附聚。
将经涂覆的成型载体在购自Memmert GmbH+Co.KG(UM 400型;内部体积=53l;空气流率=800l/h)的空气环流干燥箱中进行处理,以除去仍然存在于样品中的甘油。为此目的,将空气环流干燥箱在2h内加热到300℃(包括空气温度),然后在300℃下保持2h。在干燥过程中,将待干燥物料分多层放置在位于干燥箱中央的穿孔板上(均匀分布在穿孔板上的通孔的孔径=0.5cm;穿孔板的孔比率为60%;穿孔板的总横截面积为35cm×26cm=910cm2)(床高=2cm)。此后,将空气环流干燥箱在2h至3h之内冷却至40℃至50℃并移出样品。从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C1具有基于其总质量计为29.1重量%的氧化物蛋壳含量。
C2(对比实施例)
将335kg的粉碎的细碎的粉末P与50kg的购自Climax的三氧化钼(66.6重量%的钼,MoO3满足DE 10 2007 010 422A1中所列所有要求)在购自Aachen(DE)的AMK的R645混合机中剧烈混合10分钟。其为具有切割叶片的倾斜式混合机(强力混合机)。混合臂以39转/分钟旋转。得到的粉末在下文中称为PMo粉末。
然后,如下地进行成形:将61kg的空心圆柱状载体(外径7mm,长度3mm,内径4mm,购自Ceram Tec的表面粗糙度Rz为45μm且基于载体体积计的总孔体积为≤1体积%的C220型滑石,参见DE-A-2135620)引入到容量200l的涂布罐(倾斜角90°,购自DE的Hicoater)中。随后,设置所述涂布罐以16rpm旋转。经由Schlick0.5mm,90°型,的喷嘴,将3.8至4.2升的75重量%的水和25重量%的甘油的水溶液在约1.8bar的液体供给压力下在40分钟内喷洒到载体上。同时,在相同的周期内,经由在雾化器喷嘴的喷射锥角外的振动槽将18.3kg的PMo粉末连续地计量加入。在涂覆期间,供应的粉末全部附着在载体的表面;没有观察到细碎的氧化物活性组合物的附聚或者成对。
在活性组合物粉末和水溶液的添加已经结束以后,将110℃(或者80℃至120℃)下的空气(约400m3/h)吹入到2rpm的旋转速度下的涂布罐中40分钟(或者15分钟至60分钟)。
获得约2kg的涂覆了活性组合物的粉末的样品。将仍存在于样品中的甘油在购自Memmert GmbH+Co.KG(UM 400型;容积=53l;空气流率=800l/h)的空气环流干燥箱中去除。热处理条件与实施例C1中的那些相同。从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C2具有基于其自身总质量计为22重量%的氧化物蛋壳含量。
C3(对比实施例)
然后,如下地进行成形:将70kg的空心圆柱状载体(外径7mm,长度3mm,内径4mm,购自Ceram Tec的表面粗糙度Rz为45μm的且基于载体的体积总孔体积为≤1体积%的C220型滑石,参见DE-A-2135620)引入到容量200l的涂布罐(倾斜角90°,购自DE的Hicoater)中。随后,设置涂布罐以16rpm旋转。经由Schlick 0.5mm,90°型,的喷嘴,将3.8升至4.2升的75重量%的水和25重量%的甘油的水溶液在约1.8bar的液体供给压力下在40分钟内喷洒到载体上。同时,在相同的周期内,经由雾化器喷嘴的喷射锥角外的振动槽将18.2kg的粉碎的细碎的粉末P(其比表面积为16m2/g)连续地计量加入。在涂覆期间,供应的粉末全部附着在载体的表面;没有观察到细碎的氧化物活性组合物的附聚或者成对。
在活性组合物粉末和水溶液的添加已经结束以后,将110℃(或者80℃至120℃)下的空气(约400m3/h)吹入到2rpm的旋转速度下的涂布罐中40分钟(或者15分钟至60分钟)。获得了空心圆柱状蛋壳型催化剂,其中氧化活性组合物基于总组分的比例为20重量%。
获得约2kg的涂覆了活性组合物的粉末的样品。将仍存在于样品中的甘油在购自Memmert GmbH+Co.KG(UM 400型;容积=53l;空气流速=800l/h)的空气环流干燥箱中去除。热处理条件与实施例C1中的那些相同。从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C3具有基于其自身总质量计为20重量%的氧化物蛋壳含量。
C4(对比实施例)
催化剂C4的形成如C1那样进行,不同之处在于,相比于C1,仅将600g的粉末引入到粉末储存器中且涂覆进行40分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地实施,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C4具有基于其自身总质量计为20.0重量%的氧化蛋壳含量。
C5(对比实施例)
涂覆在购自ERWEKA(DE)的具有造粒系统的旋转涂布滚筒(内径=25.5cm;36rpm)中进行。滚筒的旋转轴设定为相对于水平面呈51.6°角。涂覆滚筒填充800g的空心圆柱状载体(外径7mm,长度3mm,内径4mm,购自CeramTec的具有45μm的表面粗糙度Rz的C220滑石(砂砾层))。所用的粘合剂是75重量%的水和25重量%的甘油的水溶液。在45分钟的涂覆时间内,经由喷嘴(喷嘴直径=1mm)将约76g的液体粘合剂喷雾到成型载体上。同时,在相同的周期内,经由雾化器喷嘴的喷射锥角外的螺旋输送机装置而连续地计量加入200g的粉碎的细碎的粉末P。在涂覆期间,所供应的粉末全部附着在载体的表面。没有观察到细碎的氧化物活性组合物的附聚。重复所述涂覆操作。两次涂覆实验的总量结合成一个样品。将所述样品在购自Memmert GmbH+Co.KG(UM 400型;容积=53l;空气流率=800l/h)的空气环流干燥箱中进行处理,以去除仍存在于样品中的甘油。热处理条件与实施例C1中的那些相同。从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C5具有基于其自身总质量计为19.6重量%的氧化物蛋壳含量。
C6(实施例)
催化剂C6的形成如C1那样进行,不同之处在于,相比于C1,仅将451g的粉末引入到粉末储存器中且涂层进行30分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地实施,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C6具有基于其自身总质量计为18.0重量%的氧化物蛋壳含量。
C7(实施例)
催化剂C7的形成如C1那样进行,不同之处在于,相比于C1,仅将377.5g的粉末引入到粉末储存器中且涂层进行25分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地实施,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C7具有基于其自身总质量计为15.8重量%的氧化物蛋壳含量。
C8(实施例)
催化剂C8的形成如C5那样进行,不同之处在于,相比于C5,仅将约44g的液体粘合剂经由喷嘴(喷嘴直径=1mm)喷雾到载体上且涂覆在27.5分钟内实现。在空气环流干燥箱中的热处理后——如在C1中那样地进行,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C8具有基于其自身总质量计为15.5重量%的氧化物蛋壳含量。
C9(实施例)
催化剂C9的形成如C5那样进行,不同之处在于,相比于C5,仅将约29g的液体粘合剂经由喷嘴(喷嘴直径=1mm)喷雾到载体上且涂覆在16分钟内实现。在空气环流干燥箱中的热处理后——如在C1中那样地进行,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C9具有基于其自身总质量计为10.8重量%的氧化物蛋壳含量。
C10(实施例)
催化剂C10的形成如C1那样进行,不同之处在于,相比于C1,仅将300g的粉末引入到粉末储存器中且涂进行20分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地进行,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C10具有基于其自身总质量计为10.4重量%的氧化物蛋壳含量。
C11(实施例)
如在实施例C2中地,将细碎的粉末P与MoO3混合。然而,相比于实施例C2,仅将约1.9-2.1升的水溶液(甘油/水=1/3)在约20分钟内喷雾到载体上。同时,在相同的周期内,仅连续计量加入将约8kg的PMo粉末。涂覆后——如在实施例C2中那样地进行,在涂覆装置中实施热处理。
获得约2kg的涂覆了活性组合物的样品。将仍存在于样品中的甘油在购自Memmert GmbH+Co.KG(UM 400型;容积=53l;空气流率=800l/h)的空气环流干燥箱中去除。热处理条件与实施例C1中的那些相同。从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C11具有基于其自身总质量计为10重量%的氧化物蛋壳含量。
C12(实施例)
催化剂C12的形成如C1那样进行,不同之处在于,相比于C1,仅将345.5g的粉末引入到粉末储存器中且涂覆进行23分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地进行,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C12具有基于其自身总质量计为14重量%的氧化物蛋壳含量。本发明的蛋壳型催化剂C12的活性组合物覆盖度为0.23mg/mm2。
C13(实施例)
催化剂C13的形成如C1那样进行,不同之处在于,相比于C1,仅将360.7g的粉末引入到粉末储存器中且涂覆进行24分钟时间。在空气环流干燥箱中的热处理之后——如在C1中那样地进行,从空气环流干燥箱中移出的空心圆柱状蛋壳型催化剂C13具有基于其自身总质量计为13.4重量%的氧化物蛋壳含量。
根据实施例C1至C11的催化剂的性能示于表1中。在所有实施例中,成型载体的比几何表面积Sm为0.725mm2/mg。其是用成型载体的几何表面积(155.5mm2)除以其质量(214.4mg)来计算的。
表1:
*:非本发明的
1)催化剂的活性组合物含量
2)活性组合物覆盖度
3)平均直径为0.26μm至2μm的孔的体积
4)平均直径为0.02μm至6.5μm的孔的体积
5)平均直径为0.26μm至2μm的大孔的体积相对于0.02μm至6.5μm的总孔体积的比率
6)涂层操作的持续时间
在本文中,除非另外明确说明,所有涉及固体物质的孔特征的数值都是基于通过压汞法使用购自Norcross,Georgia 30093-1877,USA的Micromeritics的Auto Pore IV 9520仪器的的测定结果。关于粉末检测,引入到样品空间的样品的量在每种情况下为2.5g。关于蛋壳型催化剂研究,将各5片各个蛋壳型催化剂引入到样品空间(在所测试的案例中,与活性组合物外壳的孔的贡献相比,蛋壳型催化剂的几何形状载体的孔的贡献是可忽略不计的)中。
样品空间延续到细长的毛细管中,因此小的压力变化对应着伸入到毛细管中的汞线的长度上的显著变化。在所有情况下,所用的毛细管体积是25体积%至91体积%,基于总毛细管体积的。
在特定样品分析开始前,在每种情况下,将样品空间(在25℃下)抽降至内部压力为9.3×10-4bar,并且在该温度和该压力下将样品脱气20分钟。然后,迫使汞进入到样品空间中,压力随着时间推移而上升到最高达4137bar的最终压力。起始压力为0.04bar。这对应于覆盖0.003μm至360μm的范围的孔径。
本文的图3示出了C1的活性组合物外壳的孔的孔径分布。标绘在横坐标上的是以μm计的各个孔径(以10为底的对数坐标图)。在左侧的纵坐标([ml]/[g的活性组合物])上标绘的是各个孔径各自对比总孔体积的贡献的积分(对上述比总孔体积的累积贡献)(□曲线)。结束点是基于活性组合物计的(比)总孔体积(总侵入体积)。
丙烯醛至丙烯酸的气相氧化
反应管(V2A钢:30mm的外径;壁厚2mm;内径26mm;长度464cm)从顶部向下包括:
段1:长度79cm,空管;
段2:长度62cm,几何形状7mm×3mm×4mm的滑石环(外径×长×内径;购自CeramTec的C220滑石)的初级床;
段3:长度100cm,由均匀混合物组成的固定催化剂床,所述均匀混合物由20重量%的几何形状为7mm×3mm×4mm的滑石环(外径×长度×内径;购自CeramTec的C220块滑石)和80重量%的各个催化剂组成;
段4:长度200cm,固定催化剂床,其仅由用于段3中的相应的催化剂组成;
段5:长度10cm,与段2中相同的滑石环的下游床;
段6:长度14cm,由V2A钢制成的为调整固定催化剂床的催化剂基底。
将反应气体混合物引导通过各个反应管(从顶部向下流过反应管),所述反应气体混合物在进入反应管的入口处具有以下的内含物:
4.3体积%的丙烯醛,
0.2体积%的丙烯,
0.2体积%的丙烷,
0.3体积%的丙烯酸,
5.4体积%的O2,
7.0体积%的H2O,
0.4体积%的CO和CO2
余量为N2。
在每种情况下,丙烯醛在固定催化剂床上的空速为75l(STP)/(lH)。
在反应管的整个长度上(除段1的空管的最后10cm和段6的管的最后3cm以外),使搅拌且外部电加热的盐浴(53重量%的硝酸钾、40重量%的亚硝酸钠和7重量%的硝酸钠的混合物;50kg的盐熔体)围绕反应管流动(管内的流率为3m/s(在垂直于管的纵向轴线的平面上))。
盐浴温度TB对应于盐熔体被引导进入盐浴时的温度。在所有的情况下,其设定应使得产生99.3mol%的丙烯醛转化率CA,基于反应混合物单程通过固定催化剂床计。由于沿着反应管加热,所以不存在盐浴温度的变化(盐浴散发的热量比反应管向盐浴释放的热量更多)。在反应管的入口,使反应气体的温度对应于各自的盐浴温度TB。最高局部温度TH通过在反应管中进行的点测量来确定。使用各种催化剂所获得的结果总结于表2中。
在本文中,丙烯酸形成的选择性(SAA(mol%))应理解为是指:
(转化率数值各自基于反应混合物单程通过固定催化剂床计)。
下表2示出了因所使用的蛋壳型催化剂各自在操作100个小时以后的作用所产生的结果:
表2
*:非本发明的
1)丙烯酸形成的选择性
在催化剂具有不超过0.3mg/mm2的本发明的活性组合物覆盖度的情况下,SAA为97.6mol%或更多。在催化剂具有更高活性组合物覆盖度的情况下,SAA更低,其值为96.7mol%至97.2mol%。虽然具有281℃至299℃的更高的热点温度TH,但是本发明的活性组合物覆盖度的优势建立起来了。
高的大孔体积比(Pvol)——根据本发明优选的,其优势与具有基本相同的活性组合物覆盖度的本发明的催化剂已经达到的结果相比是明显的。在活性组合物覆盖度为0.15mg/mm2至0.17mg/mm2的催化剂组中,当Pvol=0.5时SAA特别高(97.9mol%,C10),而当Pvol=0.24时SAA较低(97.6mol%,C11)。在活性组合物覆盖度为0.25mg/mm2至0.26mg/mm2的两种催化剂之间,当Pvol=0.49时SAA是高的(97.7mol%,C7),而当Pvol=0.40时SAA较低(97.5mol%,C8)。
使用具有两个连续反应区域的固定催化剂床的丙烯醛至丙烯酸的气相氧化
反应管(不锈钢型1.4541(EU标准号EN10088-3);外径33.7mm;壁厚2mm;内径29.7mm;长度400cm,热电偶套管4mm)从底部向上如下装填:
段1:长70cm
几何形状7mm×3mm×4mm的滑石环(外径×长×内径;购自CeramTec的C220滑石)的上游床;
段2:长度100cm
各个蛋壳型催化剂的固定催化剂床;
段3:长度200cm
各个蛋壳型催化剂的固定催化剂床;
段4:长度8cm
与段1中相同的滑石环的下游床;
段5:长度23cm
空管
将反应气体混合物引导通过如上所述装填的各个反应管(从顶部向下流过所述反应管),所述反应气体混合物具有以下的内含物:
4.5体积%的丙烯醛,
0.1体积%的丙烯,
0.07体积%的丙烷,
0.5体积%的丙烯酸,
5.4体积%的O2,
7体积%的H2O,
0.6体积%的CO和CO2,和
余量为N2。
在每种情况下,丙烯醛在固定催化剂床(如在DE-A 19927624中所定义的)上的空速为75l(STP)/(lH)。
在反应管的整个长度上,使搅拌且外部电加热的盐浴(53重量%的硝酸钾、40重量%的亚硝酸钠和7重量%的硝酸钠的混合物;50kg的盐熔体)围绕反应管流动(管内的流率为3m/s(在垂直于管的纵向轴线的平面上))。
在所有的情况下,调节盐浴温度TB(℃)(盐浴供应温度),使得丙烯醛转化率为98.3mol%,基于反应混合物单程通过固定催化剂床计。由于加热,所以沿着反应管不存在盐浴温度方面的变化(盐浴散发的热量比反应管向盐浴释放的热量更多)。将反应气体混合物的进料温度各自(在进入反应管的入口处)调节到各自的盐浴温度。
通过热电偶装置连续测量催化剂床中的温度,所述热电偶是已经安放在位于反应管内部的热电偶套管中的且已在提升装置(pulling machine)的辅助下使其在反应床中从底部向上移动。该测量中的最高温度对应于热点温度TH。
下表3示出了操作100小时后得到的结果,所述结果是在用不同的本发明的蛋壳型催化剂和非本发明的蛋壳型催化剂装填反应器段2和段3之后建立的。
表3
*:非本发明的
**:几何形状7mm×3mm×4mm的滑石环(购自CeramTec的C220滑石)
***:存在于段2中的热点与段1到段2的过渡之间的距离
表3中的实施例D2与对比实施例的比较表明,由D2比对比实施例(97mol%)具有更好的丙烯酸选择性(97.5mol%)得出,就丙烯酸形成的选择性而言,用本发明的蛋壳型催化剂装填具有最高温度(热点是在段2的D2和对比物中,见表3)的反应区域是有利的。
部分更换固定催化剂床的上游反应区域
DE-A-10232748记载了将固定催化剂床的一部分更换为新鲜催化剂负载。在下文中,测试了本发明的催化剂是否也有利于DE10232748中记载的固定催化剂床的部分更换。
反应管(不锈钢型1.4541(EU标准号EN10088-3);外径33.7mm;壁厚2mm;内径29.7mm;长度400cm,热电偶套管4mm)从底部向上如下装填:
段1:长75cm
几何形状7mm×3mm×4mm的滑石环(外径×长×内径;购自CeramTec的C220滑石)的上游床;
段2:长度110cm
各个蛋壳型催化剂的固定催化剂床;
段3:长度190cm
固定催化剂床,其由根据DE 103 60 057A1的工作实施例1制备的蛋壳型催化剂(71重量%)和29重量%的几何形状7mm×3mm×4mm的滑石环(外径×长×内径;购自CeramTec的C220滑石)组成;
段4:长度3cm
与段1中相同的滑石环的下游床;
段5:长度23cm
空管
将反应气体混合物引导通过如上所述装填的各个反应管(从顶部向下流过反应管),所述反应气体混合物具有以下的内含物:
4.5体积%的丙烯醛,
0.1体积%的丙烯,
0.07体积%的丙烷,
0.5体积%的丙烯酸,
5.4体积%的O2,
7体积%的H2O,
0.6体积%的CO和CO2,和
余量为N2。
在每种情况下,丙烯醛在固定催化剂床(如在DE-A 19927624中所定义的)上的空速为75l(STP)/(lH)。
在反应管的整个长度上,使搅拌且外部电加热的盐浴(53重量%的硝酸钾、40重量%的亚硝酸钠和7重量%的硝酸钠的混合物;50kg的盐熔体)围绕反应管流动(管内的流速为3m/s(在垂直于管的纵向轴线的平面上))。
在所有的情况下,调节盐浴温度TB(℃)(盐浴供应温度),使得丙烯醛转化率为99.3mol%,基于反应混合物单程通过固定催化剂床计。由于加热,所以沿着反应管不存在盐浴温度方面的变化(盐浴散发的热量比反应管向盐浴释放的热量更多)。在每种情况下,将反应气体混合物的进料温度各自(在进入反应管的入口处)调节到各自的盐浴温度。通过热电偶装置连续测量在催化剂床中的温度,所述热电偶是已经安放在位于反应管内部的热电偶套管中的且已在提升装置的辅助下使其在反应床中从底部向上移动。在该测量中的最高温度对应于热点温度TH。
下表4示出了操作100小时后得到的结果,所述结果是在用本发明的和非本发明的蛋壳型催化剂装填反应器段2之后建立的。
表4
*:非本发明的
**:几何形状7mm×3mm×4mm的滑石环(购自CeramTec的C220滑石)
***:存在于段2中的热点与段1到段2的过渡之间的距离
表4中的实施例E1与对比实施例的比较表明,由实施例E1(97.5mol%)比对比实施例(96.9mol%)具有更好丙烯酸选择性得出,在部分催化剂更换的情况下,就丙烯酸形成的选择性而言,用本发明的蛋壳型催化剂装填上游反应区域是有利的。
使用具有两个连续加热区的固定催化剂床的丙烯醛至丙烯酸气相氧化
反应管(不锈钢型1.4541(EU标准号EN10088-3);外径33.7mm;壁厚2mm;内径29.7mm;长度400cm,热电偶套管4mm)从底部向上如下装填:
段1:长70cm
几何形状7mm×3mm×4mm的滑石环(外径×长×内径;购自CeramTec的C220滑石)的上游床;
段2:长度100cm
蛋壳型催化剂C13的固定催化剂床;
段3:长度200cm
蛋壳型催化剂C3的固定催化剂床;
段4:长度8cm
与段1中相同的滑石环的下游床;
段5:长度23cm
空管
将反应气体混合物引导通过如上所述装填的各个反应管(从顶部向下流过反应管),所述反应气体混合物具有以下的内含物:
4.5体积%的丙烯醛,
0.1体积%的丙烯,
0.07体积%的丙烷,
0.5体积%的丙烯酸,
5.4体积%的O2,
7体积%的H2O,
0.6体积%的CO和CO2,和
余量为N2。
丙烯醛在固定催化剂床LACR(如在DE-A 19927624中所定义的)上的空速为75、100和145l丙烯醛(STP)/(lH)。
如在DE 2010-10048405中描述的,用两种不同的盐浴加热反应管。第一个190cm通过逆流抽吸的盐浴的方式进行恒温,该盐浴以温度TA提供。第二个210cm通过逆流抽吸的盐浴B的方式恒温,该盐浴以温度TB提供。两种盐浴都由53重量%的硝酸钾、40重量%的亚硝酸钠和7重量%的硝酸钠的混合物组成;50kg的盐熔体。管内的流率为3m/s(在垂直于管的纵向轴线的平面上)。
在所有的情况下,调节盐浴温度TA和TB(℃)(两种盐浴的供应温度),使得丙烯醛转化率在75和100l丙烯醛(STP)/(l H)的丙烯醛空速下为99.4mol%且在145l丙烯醛(STP)/(l H)的丙烯醛空速下为98.5mol%,基于反应混合物单程通过催化剂床计。由于加热,所以沿着反应管不存在盐浴温度方面的变化(盐浴散发的热量比反应管向盐浴释放的热量更多)。在每种情况下,将反应气体混合物的进料温度各自(在进入反应管的入口处)调节到各自的盐浴温度。
通过热电偶装置连续测量在催化剂床中的温度,所述热电偶是已经安放在位于反应管内部的热电偶套管中的且已在提升装置的辅助下使其在反应床中从底部向上移动。在该测量中的最高温度对应于热点温度TH1和TH2。
下表5示出了操作100小时后得到的结果,所述结果是在用本发明的蛋壳型催化剂在不同的空速下建立的。
表5
*:第一个热点与段1到段2的过渡之间的距离
**:第二个热点与段1到段2的过渡之间的距离。
- 还没有人留言评论。精彩留言会获得点赞!