一种中药组合物的指纹图谱检测方法与流程
本发明公开了一种中药组合物的指纹图谱检测方法,具体公开了一种由丁香、肉桂为原料药制成的中药组合物的指纹图谱检测方法,属于药物分析领域。
背景技术:
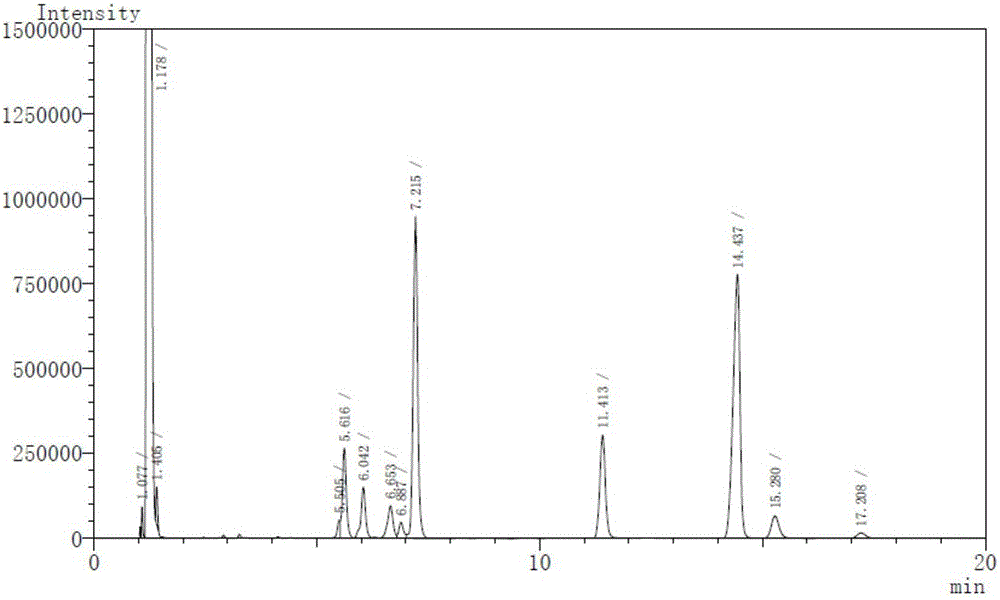
:本发明涉及由丁香、肉桂两味药按照一定比例组合后,提取其挥发油制备而成的中药制剂,其主要成分为丁香酚和桂皮醛。该制剂具有温散寒邪、行气止痛的作用,适用于肠易激综合症出现腹痛,遇寒加重,腹泻,便秘,脘腹胀痛,嗳气,畏寒,排便急迫或胀坠感,便后痛减,舌质淡,舌苔白,脉弦或紧等症状由于中药及其制剂均为多组分复杂体系,因此评价其质量应采用与之相适应的,能提供丰富鉴别信息的检测方法,但现行的显微鉴别、理化鉴别和含量测定等方法都不足以解决这一问题。中药指纹图谱是指某些中药材或中药制剂经适当处理后,采用一定的分析手段,得到的能够标示其化学特征的色谱图或光谱图。中药指纹图谱是一种综合的,可量化的鉴定手段,能较为全面地反映中药及其制剂中所含化学成分的种类与数量,进而对药品质量进行整体描述和评价。所以,中药指纹图谱已经成为目前国际上较为先进的中药质量控制手段。技术实现要素:本发明的目的是提供一种中药组合物的气相色谱指纹图谱检测方法。本发明的目的是通过如下技术方案实现的:一种中药组合物的气相色谱指纹图谱检测方法,包括以下步骤:气相色谱条件:选用毛细柱为色谱柱,氢焰离子化检测器,载气为氮气,分流比为5~20:1;柱流量为0.5~1ml/min,检测器温度(FID1)250℃-270℃,进样口温度(SPL1)230℃-250℃,起始柱温100-140℃,经升温后温度为200~240℃;制备供试品溶液:取中药组合物,以有机溶剂溶解,得供试品溶液;制备对照品溶液:取桂皮醛对照品适量,加有机溶剂制成桂皮醛对照溶液;进样检测:分别吸取对照品溶液和供试品溶液,注入气相色谱仪,测定。优选的,分流比为10:1,柱流量为0.85ml/min,检测器温度270℃,进样口温度250℃,升温程序为:起始柱温为100-120℃,保持0-15min,以2.5-10℃/min速度升温至220℃。进一步优选的,升温程序为:起始柱温为100℃,升温程序为:先以2.5℃/min速度升至135℃,再以4℃/min速度升至150℃,最后以10℃/min速度升至220℃;或,起始柱温为105℃,升温程序为:先以起始柱温保持15min,再以5℃/min速度升至150℃,最后以10℃/min速度升至220℃;或,起始柱温为120℃,升温程序为:先以起始柱温保持15min,再以10℃/min速度升至220℃。最优选的,起始柱温为120℃,升温程序:先以起始柱温保持15min,再以10℃/min速度升至220℃。优选的,制备供试品溶液步骤中所述有机溶剂包括正己烷、石油醚、乙醚、乙酸乙酯、无水乙醇、甲醇、丙酮中的任意一种或几种;进一步优选为乙酸乙酯;更进一步,所述供试品溶液制备方法为取中药组合物,加入乙酸乙酯制备成16mg/ml的供试品溶液。优选的,制备对照品溶液步骤中所述有机溶剂包括正己烷、石油醚、乙醚、乙酸乙酯、无水乙醇、甲醇、丙酮中的任意一种或几种;进一步优选为乙酸乙酯;更进一步,所述对照品溶液制备方法为取桂皮醛,加入乙酸乙酯制备成3.0mg/ml的桂皮醛对照品溶液。利用本发明提供的气相色谱指纹图谱检测方法对中药组合物进行检测,其检测结果为指纹图谱有10个共有特征峰,以桂皮醛的色谱峰为参照(S),各共有特征峰的相对保留时间为:0.5261±0.0263、0.5463±0.0273、0.5840±0.0292、0.6023±0.0301、0.6759±0.0338、0.7023±0.0351、0.7586±0.0379、1.0000(S)、1.0321±0.0516、1.0815±0.0541;在本发明的一些具体实施例中,指纹图谱有10个共有特征峰,以桂皮醛的色谱峰为参照(S),各共有特征峰的相对保留时间为:0.5256~0.5267、0.5457~0.5471、0.5833~0.5845、0.6018~0.6030、0.675~0.6766、0.7016~0.7034、0.7580~0.7592、1.0000(S)、1.0317~1.0323、1.0810~1.0819;在本发明的一些具体实施例中,指纹图谱10个共有特征峰的相对保留时间为0.5261、0.5463、0.5840、0.6023、0.6759、0.7023、0.7586、1.0000(S)、1.0321、1.0815。优选的,所述中药组合物的原料药包括:丁香1~10重量份、肉桂按1~10重量份;进一步优选的,所述中药组合物为原料药丁香1~10重量份、肉桂按1~10重量份按常规提取方法制备的提取物或进一步按常规制剂工艺制备的常规剂型;所述常规提取方法包括浸泡提取、超声提取、渗漉提取、回流提取或水蒸汽蒸馏提取;所述常规剂型包括片剂、硬胶囊剂、软胶囊剂、颗粒剂、散剂、口服液、丸剂、贴剂、膏剂、喷雾剂、注射剂等。更进一步优选的,所述中药组合物制备方法为:取丁香、肉桂,隔水蒸馏8~12小时,收集挥发油,冷藏24小时,除去水分,即得。经方法学考察,本发明气相色谱指纹图谱检测方法系统适应性良好、专属性强、精密度高、稳定性好、可重复性强,符合构建指纹图谱的要求,应用于中药组合物或其制剂的质量控制,可靠性高,简捷易行。附图说明图11#色谱条件下检测供试品溶液的色谱图。图22#色谱条件下检测供试品溶液的色谱图。图33#色谱条件下检测供试品溶液的色谱图。图44#色谱条件下检测供试品溶液的色谱图。图55#色谱条件下检测供试品溶液的色谱图。图66#色谱条件下检测供试品溶液的色谱图。图76#色谱条件下检测桂皮醛对照品溶液的色谱图。图86#色谱条件下检测丁香酚对照品溶液的色谱图。图9实施例3系统适应性试验中检测对照品溶液的色谱图。图10实施例3系统适应性试验中检测供试品溶液的色谱图。图11实施例3专属性试验中检测对照品溶液的色谱图。图12实施例3专属性试验中检测供试品溶液的色谱图。图13实施例3专属性试验中检测空白溶剂对照溶液的色谱图。图1410批中药组合物软胶囊剂的气相色谱图。图15实施例4的对照色谱图。图16实施例5待测品的色谱图。具体实施方式以下通过具体实施方式,对本发明的上述内容做进一步的详细说明。但不应该将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。实施例1中药组合物软胶囊的制备取丁香375g、肉桂375g,加8倍量水,隔水蒸馏8小时,收集挥发油,冷藏24小时,除去水分,共制36ml挥发油。将36ml挥发油与364ml大豆油混匀,备用。取明胶150g、甘油60g、单糖浆10g,加去离子水150g混匀,加热到60-70℃,保温0.5小时,加入对羟基苯甲酸甲酯0.15g、对羟基苯甲酸乙酯0.075g,搅拌,抽真空,70℃保温20分钟,制成囊材。取挥发油与大豆油的混合物,用囊材采用压制法制成软胶囊,检验,分装,即得软胶囊1000粒。实施例2色谱条件考察1仪器与试药岛津ShimadzuGC-2010Plus气相色谱仪,FID检测器;电子天平;Mili-Q纯水仪;KQ-500DE型超声波清洗仪。乙醇、甲醇、乙酸乙酯、正己烷、乙醚为分析纯;桂皮醛(98%,批号:PS151228-05)、丁香酚(98%,批号:PS0262-0500)均购自成都普思生物科技股份有限公司,中药组合物为按实施例1制备的软胶囊剂。2样品溶液的制备供试品溶液的制备方法:精密称取软胶囊内容物0.8g,置50mL容量瓶,分别以正己烷、乙醚、乙酸乙酯、无水乙醇、甲醇稀释至刻度,摇匀。结果表明,乙酸乙酯组澄清透明,溶解良好,其他组出现不同程度的分层。据此,选择乙酸乙酯组,经微孔滤膜(0.45μm)滤过,密封备用。对照品溶液的制备:分别精密称取丁香酚、桂皮醛对照品适量,各加乙酸乙酯制成浓度分别约为1.8、3.0mg/mL的对照品溶液,经微孔滤膜(0.45μm)滤过,密封,于4℃保存,备用。空白溶剂对照溶液:为乙酸乙酯试剂,微孔滤膜(0.45μm)滤过,即得。3色谱条件考察色谱条件:色谱柱:6%氰丙基苯基/94%二甲基聚硅氧烷为固定相的-624毛细柱(30m×0.25mm,1.4um);载气为氮气(99.99%);分流比为10:1;柱流量为0.85ml·min-1。对SPL1与FID1温度、升温程序进行考察。1#SPL1为230℃;FID1为250℃,起始柱温为120℃,以2℃·min-1升至150℃,再以4℃·min-1升至165℃,最后以10℃·min-1升至220℃。色谱图见图1。结果显示,8.995-10.787min的色谱峰分离度较差。2#SPL1为230℃;FID1为250℃,起始柱温为140℃,以1℃·min-1升至160℃,再以5℃·min-1升至200℃,最后以10℃·min-1升至220℃。色谱图见图2。结果显示,5.505-7.2115min的色谱峰分离度较差。3#SPL1为250℃;FID1为270℃,起始柱温为140℃,以1℃·min-1升至160℃,再以5℃·min-1升至200℃,最后以10℃·min-1升至220℃。色谱图见图3。结果显示,8.662-9.916min的色谱峰分离度较差。4#SPL1为250℃;FID1为270℃,起始柱温为100℃,以2.5℃·min-1升至135℃,再以4℃·min-1升至150℃,最后以10℃·min-1升至220℃。色谱图见图4。结果显示,各主要峰分离度得到改善。5#SPL1为250℃;FID1为270℃,起始柱温为105℃,保留15min,以5℃·min-1升至150℃,再以10℃·min-1升至220℃。色谱图见图5。结果显示,各主要峰分离度较4#无显著差异,但分析时间延长。6#SPL1为250℃;FID1为270℃,起始柱温为120℃,保留15min,以10℃·min-1升至220℃。色谱图见图6。结果显示,各主要峰分离度进一步改善,分离度良好,分析时间较短。根据试验结果,检测器温度270℃,进样口温度250℃;起始柱温为100-120℃,经升温后温度为220℃的色谱条件适宜分离、分析供试品溶液,并进一步构建相关指纹图谱,其中,4#、5#、6#为优选的色谱条件,6#为最优选的色谱条件。即最优色谱条件:色谱柱:6%氰丙基苯基/94%二甲基聚硅氧烷为固定相的-624毛细柱(30m×0.25mm,1.4um);载气为氮气(99.99%);SPL1为250℃;FID1为270℃,起始柱温为120℃,保留15min,以10℃·min-1升至220℃;分流比为10:1;柱流量为0.85ml·min-1,进样量为1uL。在最优选的6#色谱条件下,依次注入桂皮醛对照品溶液、丁香酚对照品溶液。色谱图见图7至图8。结果表明,桂皮醛色谱峰峰面积较大,保留时间适中,因此,选择桂皮醛色谱峰作为参照峰。实施例3方法学考察1样品溶液的制备供试品溶液的制备:精密称取软胶囊内容物0.8g,置50mL量瓶,加乙酸乙酯溶解、稀释至刻度,微孔滤膜(0.45μm)滤过,密封备用。对照品溶液的制备:精密称取桂皮醛对照品适量,加乙酸乙酯稀释、定容至100mL容量瓶,制成浓度约为3.0mg/mL的对照品溶液,微孔滤膜(0.45μm)滤过,密封,于4℃保存,备用。2色谱条件色谱条件:色谱柱:6%氰丙基苯基/94%二甲基聚硅氧烷为固定相的-624毛细柱(30m×0.25mm,1.4um);载气为氮气(99.99%);SPL1为250℃;FID1为270℃,起始柱温为120℃,保留15min,以10℃·min-1升至220℃;分流比为10:1;柱流量为0.85ml·min-1,进样量为1uL。系统适应性试验:在上述色谱条件下,取对照品溶液重复进样5次,记录保留时间与峰面积,并计算RSD(%),结果见表1,色谱图见图9;取供试品溶液重复进样2次,记录色谱图,色谱图见图10。表1系统适应性试验结果结果表明:对照品溶液的重复性RSD小于5%,供试品溶液的分离度均大于1.5,系统适应性良好。专属性试验:在上述色谱图条件下,取上述对照品溶液、供试品溶液、空白溶剂对照溶液各进一针,记录色谱图,色谱图分别见图11至图13。结果表明:空白溶剂对照无干扰,方法专属性良好。精密度试验:在上述色谱条件下,取供试品溶液连续进样6次,以桂皮醛色谱峰为参照(S),计算各主要峰(A1-A9)的相对保留时间与相对峰面积,并计算RSD(%)。结果见表2和表3。表2精密度试验结果(相对峰面积)表3精密度试验结果(相对保留时间)试验结果表明,色谱图各共有峰(S、A1-A9)的相对保留时间、相对峰面积的RSD均小于5.0%,符合指纹图谱制订的相关要求。稳定性试验:取同一批样品,按上述供试品溶液的制备方法制备供试品溶液,室温下放置分别于0、2、4、8、12、24h进样,以桂皮醛色谱峰(S)为参照,计算各主要峰(A1-A9)的相对保留时间与相对峰面积,并计算RSD(%)。结果见表4和表5。表4稳定性试验结果(相对峰面积)表5稳定性试验结果(相对保留时间)试验结果表明,色谱图各共有峰的相对保留时间、相对峰面积的RSD均小于5.0%,符合指纹图谱制订的相关要求。重复性试验:取同一批样品,按上述供试品溶液制备方法制备6份样品溶液,依次进样,以桂皮醛色谱峰(S)为参照,计算各主要峰(A1-A9)的相对保留时间与相对峰面积,计算RSD(%)。结果见表6和表7。表6重复性试验结果(相对峰面积)表7重复性试验结果(相对保留时间)试验结果表明,色谱图各共有峰的相对保留时间、相对峰面积的RSD均小于5.0%,符合指纹图谱制订的相关要求。实施例4指纹图谱的建立及相似度评价将10批(S1-S10)按实施例1方法制备的中药组合物软胶囊按实施例2的方法制备供试品溶液,并根据最优选的6#色谱条件测试。将获得的10批样品色谱图,见图14,各色谱峰的相对峰面积和相对保留时间见表8-9。导入“中药色谱指纹图谱相似度评价系统”软件进行数据处理,生成对照色谱图,见图15。根据10个批次样品中各成分相对保留时间的一致性,从中选择了10个共有峰作为指纹图谱的特征峰,以桂皮醛色谱峰为参照(S),(t=19.955min),对照色谱图中各共有峰相对保留时间为:0.5261、0.5463、0.5840、0.6023、0.6759、0.7023、0.7586、1.0000(S)、1.0321、1.0815,RSD(%)均小于0.1%。相似度计算见表10,结果显示,各批次样品的相似度为0.933-0.981,均大于0.9,其指纹图谱具有较高的相似度。上述10批次中药组合物的指纹图谱共有特征峰,以各批次的桂皮醛色谱峰为参照(S),各共有峰的相对保留时间为0.5261±0.0263、0.5463±0.0273、0.5840±0.0292、0.6023±0.0301、0.6759±0.0338、0.7023±0.0351、0.7586±0.0379、1.0000(S)、1.0321±0.0516、1.0815±0.0541。其中10个批次中药组合物的指纹图谱共有特征峰,以各批次的桂皮醛色谱峰为参照(S),各共有特征峰的相对保留时间为:0.5256~0.5267、0.5457~0.5471、0.5833~0.5845、0.6018~0.6030、0.675~0.6766、0.7016~0.7034、0.7580~0.7592、1.0000(S)、1.0317~1.0323、1.0810~1.0819。表810批样品相对峰面积计算结果序号SA1A2A3A4A5A6A7A8A9110.03050.16070.01280.09560.06330.03870.67070.97143.7558210.06160.31520.02190.18300.12800.06971.30790.69383.6566310.07440.37650.03610.22120.14610.08251.54420.52113.4829410.03930.20350.01720.10960.07020.04440.80490.74264.6011510.08340.42970.04120.25550.16050.08821.75460.44723.7860610.09480.48690.04680.28920.18700.10071.97050.31433.6420710.10600.55140.05140.32340.20850.11362.22620.18233.4152810.08470.41780.03850.24640.16400.08991.72430.46213.9375910.09390.48160.04080.28340.19040.10552.00290.30623.60751010.11350.57670.05030.34080.22300.12332.39440.05843.1979表910批样品相对保留时间计算结果序号SA1A2A3A4A5A6A7A8A9110.52670.54710.58360.60300.67630.70280.75921.03201.0817210.52650.54680.58400.60290.67660.70340.75921.03231.0815310.52630.54640.58430.60240.67620.70270.75881.03211.0814410.52580.54570.58420.60180.67500.70160.75831.03191.0819510.52560.54600.58410.60210.67530.70200.75811.03221.0815610.52560.54580.58330.60180.67550.70190.75801.03231.0817710.52580.54590.58370.60200.67570.70190.75841.03201.0813810.52660.54680.58450.60300.67610.70280.75921.03221.0815910.52610.54650.58420.60250.67640.70240.75921.03231.08161010.52660.54680.58440.60270.67660.70260.75901.03171.0810表10指纹图谱相似度计算结果序号相似度序号相似度S10.976S60.959S20.981S70.980S30.952S80.933S40.943S90.942S50.972S100.961注:表中S1-S10为10批次的编号。实施例5指纹图谱在中药组合物质量控制中的应用制备供试品溶液:取10批次的中药组合物软胶囊供试品,分别精密称取内容物0.8g,置50mL量瓶,加乙酸乙酯溶解、稀释至刻度,经0.45μm微孔滤膜滤过,得供试品溶液。确定气相色谱条件:色谱柱的固定相为6%氰丙基苯基/94%二甲基聚硅氧烷,尺寸为30m×0.25mm,1.4um,优选-624毛细柱;氢焰离子化检测器,检测器温度为270℃;载气为氮气,分流比为10:1;柱流量为0.85ml·min-1,进样口温度为250℃;起始柱温为100-120℃,升温程序为:先以起始柱温保持0-15min,然后以2.5-10℃·min-1升至220℃,优选起始柱温为120℃,保持15min,再以10℃·min-1升至220℃;进样量为1uL;制定标准指纹图谱:将上述制得的10批供试品溶液注入气相色谱仪,以上述的色谱条件检测,测得供试品溶液气相色谱图,比较各色谱图,得到由共有特征峰构成的中药组合物制剂的标准指纹图谱;测试中药组合物软胶囊待测品:取待测品,按制备供试品溶液方法制得待测品溶液,注入气相色谱仪,以上述的色谱条件检测,测得待测品溶液气相色谱图,见图16;检测待测品质量:将待测品色谱图与上述标准指纹图谱进行比较,计算相似度,相似度大于0.9。实施例6中药组合物片剂的制备取实施例1制备的挥发油3ml,加微粉硅胶2.5mg,乳糖5.5mg,微晶纤维素5.3mg,糖粉3.0mg,用10%PVP‐k30水溶液制粒,外加低取代羟丙基纤维素0.5mg和硬酯酸镁0.2mg,压片,即得。实施例7中药组合物颗粒剂的制备取实施例1制备的挥发油5ml,并溶于少量乙醇中,备用。取40gβ‐环糊精,加入300ml蒸馏水,水浴加热溶解,温度降至35℃时加入挥发油乙醇溶液,混合均匀,超声包合30min,冷藏过夜,抽滤,沉淀于38℃低温干燥,得挥发油β‐环糊精包合物。将β‐环糊精包合物与适量糊精混合,采用90%乙醇为润湿剂制软材,过20目筛进行湿法制粒,65℃干燥,整粒,即得。实施例8中药组合物指纹图谱检测色谱条件:选用毛细柱为色谱柱,氢焰离子化检测器,载气为氮气,分流比为10:1;柱流量为1ml/min,检测器温度270℃,进样口温度250℃,升温程序为:起始柱温为100℃,升温程序为:先以2.5℃/min速度升至135℃,再以4℃/min速度升至150℃,最后以10℃/min速度升至220℃;制备供试品溶液:取实施例6或7的中药组合物制剂0.8g,置于50ml容量瓶,以乙酸乙酯溶解,并稀释至刻度,摇匀,经微孔滤膜(0.45μm)滤过,得供试品溶液;制备对照品溶液:取桂皮醛对照品适量,加乙酸乙酯制成3.0mg/ml的桂皮醛对照溶液,经微孔滤膜(0.45μm)滤过,备用;进样检测:分别吸取对照品溶液和供试品溶液,注入气相色谱仪,测定。实施例9中药组合物指纹图谱检测色谱条件:选用毛细柱为色谱柱,氢焰离子化检测器,载气为氮气,分流比为10:1;柱流量为1ml/min,检测器温度270℃,进样口温度250℃,起始柱温为105℃,升温程序为:先以起始柱温保持15min,再以5℃/min速度升至150℃,最后以10℃/min速度升至220℃;制备供试品溶液:取实施例6或7的中药组合物制剂0.8g,置于50ml容量瓶,以乙酸乙酯溶解,并稀释至刻度,摇匀,经微孔滤膜(0.45μm)滤过,得供试品溶液;制备对照品溶液:取桂皮醛对照品适量,加乙酸乙酯制成3.0mg/ml的桂皮醛对照溶液,经微孔滤膜(0.45μm)滤过,备用;进样检测:分别吸取对照品溶液和供试品溶液,注入气相色谱仪,测定。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1