一种消毒散口服液HPLC指纹图谱的建立方法与流程
本发明属于中药技术领域,具体涉及一种消毒散口服液指纹图谱的建立方法。
背景技术
中药指纹图谱作为一项质量控制技术,能全面、综合地控制药品的质量,具有特征明显、专属性强、重现性好等特点。随着中医药的推广应用,指纹图谱作为中草药及其提取物质量控制方法,目前已成为国内外广泛接受的一种对中药质量进行控制和评价的技术。
消毒散口服液是有由牛蒡子、荆芥穗、甘草3味中药组成,处方源自元代曾世荣的《活幼心书》,具有“解急惊风毒,赤紫丹瘤,壮热狂躁,睡卧不安,胸膈满闷,咽喉肿痛,九道有血妄行,及遍身疮疥”。药理研究表明,其具有镇咳、抗炎、镇痛等功效。目前还没查到有关消毒散口服液的质量研究和标准提升相关的文献资料及明确的检测方法,而对其中药味(牛蒡子、荆芥穗、甘草),多参考中国药典给出的方法,即单独对其中各药味进行薄层鉴别及对某些药味的主要成分进行含量测定,不能对其中多味药材的多个主要化学成分进行全面的、系统的检测和控制。
目前,以指纹图谱控制消毒散口服液的质量,国内外尚未见专利公开及文献报道。本发明中我们对消毒散口服液进行了指纹图谱试验研究,建立了指纹图谱测定方法,并对建立的方法进行了精密度、重复性等方法学验证试验,结果表明方法稳定可行。
技术实现要素:
本发明的目的是提供一种消毒散口服液hplc指纹图谱的建立方法,用于对消毒散口服液质量进行控制和评价。
本发明的hplc指纹图谱的建立方法包括:
1供试品溶液的制备:精密吸取消毒散口服液适量,置量瓶中,加入甲醇至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
2对照品溶液的制备:分别取甘草苷、牛蒡苷、牛蒡子苷元,甘草酸铵,胡薄荷酮对照品适量,加入甲醇制成每1ml分别约含甘草苷5~15μg、牛蒡苷230~270μg、牛蒡子苷元5~15μg、甘草酸铵230~270μg、胡薄荷酮40~70μg的混合对照品溶液;
3hplc色谱条件和系统适应性:用十八烷基硅烷键合硅胶为填充剂,以甲醇—乙腈—0.05%~0.1%磷酸溶液为流动相,梯度洗脱,流速为:0.5ml/min~1.5ml/min;检测波长:240nm~260nm,270nm~290nm;柱温:25℃~35℃;
4测定:分别精密吸取对照品溶液和供试品溶液各5μl~20μl,注入液相色谱仪,测定,即得。
优选的,本发明的hplc指纹图谱的最佳建立方法如下:
1供试品溶液的制备:取本品,精密吸取2ml,置25ml量瓶中,加入甲醇稀释至刻度,摇匀,用0.22μm微孔滤膜过滤,取续滤液,备用。
2对照品溶液的制备:分别精密称取甘草苷、牛蒡苷、牛蒡子苷元、甘草酸铵、胡薄荷酮对照品适量,加甲醇制成每1ml分别约含甘草苷10μg、牛蒡苷250μg、牛蒡子苷元10μg、甘草酸铵250μg、胡薄荷酮60μg的混合对照品。
3hplc色谱条件和系统适应性:用十八烷基硅烷键合硅胶为填充剂,以甲醇-乙腈-0.05%磷酸溶液为流动相,梯度洗脱,梯度洗脱表见表1,检测波长为250nm,280nm;流速:1.0ml·min-1;柱温:30℃;进样量10μl;理论踏板数按牛蒡苷计不得低于10000.
表1梯度洗脱条件
4测定:分别精密吸取供试品溶液和对照品溶液各10μl注入液相色谱仪,照高效液相色谱法测定,得到一种消毒散口服液的指纹图谱。将数据导入《中药色谱指纹图谱相似度评价系统2012版》进行评价,得到对照图谱。
其中所述一种消毒散口服液hplc指纹图谱,共有14个特征峰,归属于牛蒡子的特征峰为:3号峰,4号峰,5号峰,7号峰,8号峰,9号峰;归属于荆芥穗的特征峰为:6号峰,14号峰;归属于甘草的特征峰为:1号峰,2号峰,10号峰,11号峰,12号峰,13号峰。
其中所述一种消毒散口服液hplc指纹图谱的建立方法,在250nm,280nm波长检测下利用各对照品对主要峰进行比较确定:2号峰为甘草苷,5号峰为牛蒡苷,9号峰为牛蒡子苷元,12号峰为甘草酸,14号峰为胡薄荷酮。
其中所述一种消毒散口服液hplc指纹图谱,以牛蒡苷为参照峰s,计算各特征峰与s峰的相对保留时间,所述的相对保留时间峰1-3在第一规定值的±10%之内,峰4-13在第一规定值的±5%之内。所述的第一规定值为0.254(峰1),0.275(峰2),0.338(峰3),0.549(峰4),1.000(峰5,s),1.402(峰6),1.424(峰7),1.447(峰8),1.457(峰9),1.475(峰10),1.492(峰11),1.522(峰12),1.573(峰13),1.617(峰14)。
其中所述一种消毒散口服液hplc指纹图谱,采用《中药色谱指纹图谱相似度评价系统2012版》进行评价,供试品批与批间的相似度均大于0.90。
本发明提供的消毒散口服液hplc指纹图谱建立方法经精密度、重复性和稳定性测试,测试结果表明,该方法的精密度、稳定性和重复性良好,可对消毒散口服液的质量进行全面地评价,有效地保证了成品的质量,可克服现有技术检测指标单一,不能反映内在质量的特点。
附图说明
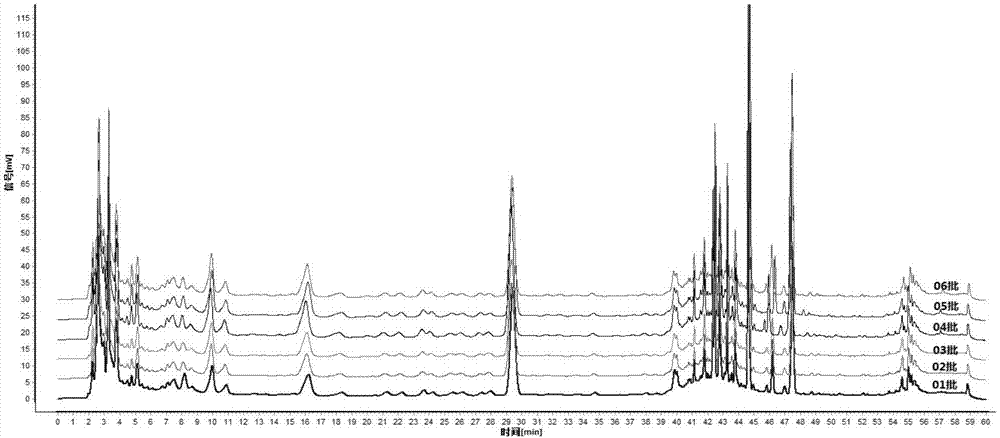
图1是一种消毒散口服液6批样品的hplc指纹图谱(250nm)。
图2是一种消毒散口服液6批样品的hplc指纹图谱(280nm)。
图3是一种消毒散口服液的hplc对照图谱(250nm)。
图4是一种消毒散口服液的hplc对照图谱(280nm)。
图5是一种消毒散口服液的hplc对照品图谱。
图6是一种消毒散口服液的hplc甘草苷对照品。
图7是一种消毒散口服液的hplc牛蒡苷对照品。
图8是一种消毒散口服液的hplc牛蒡子苷元对照品。
图9是一种消毒散口服液的hplc甘草酸对照品。
图10是一种消毒散口服液的hplc胡薄荷酮对照品。
图11是一种消毒散口服液的hplc特征峰归属图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
本发明提供了一种消毒口服液的hplc指纹图谱的建立方法,包括以下步骤:
(1)取消毒口服液适量,与溶剂混合后,得到供试品溶液;
(2)取甘草苷、牛蒡苷、牛蒡子苷元,甘草酸铵,胡薄荷酮对照品适量,用溶剂溶解,混合,得对照品溶液;
(3)分别将对照品溶液与供试品溶液注入高效液相色谱仪中进行检测,记录色谱,用指纹图谱软件对图谱进行处理,得到消毒散口服液指纹图谱。
在本发明中,供试品溶液按照以下方法配制:精密吸取消毒散口服液适量,置量瓶中,加入溶剂稀释,混匀,过滤,得到供试品溶液。
在本发明提供的上述配制供试品溶液的方法中,所精密吸取消毒散口服液最优量为2ml;所述的容量瓶为25ml;所述的溶剂为甲醇;所述过滤的滤膜孔径为0.1~0.45μm,优选0.22μm。
在本发明中,对照品溶液按照以下方法配制:取甘草苷、牛蒡苷、牛蒡子苷元、甘草酸铵、胡薄荷酮对照品适量,用溶剂溶解,混合,得对照品溶液;所述溶剂优选为甲醇;所述的对照品溶液中,甘草苷、牛蒡苷、牛蒡子苷元,甘草酸铵,胡薄荷酮的含量分别为甘草苷5~15μg/ml、牛蒡苷230~270μg/ml、牛蒡子苷元5~15μg/ml、甘草酸铵230~270μg/ml、胡薄荷酮40~70μg/ml的混合对照品溶液,优选为甘草苷10μg/ml、牛蒡苷250μg/ml、牛蒡子苷元10μg/ml、甘草酸铵250μg/ml、胡薄荷酮60μg/ml。
配制好供试品溶液和对照品溶液后,分别将对照品溶液与供试品溶液注入高效液相色谱仪中进行检测,检测过程中,使用的色谱柱优选为c18柱,更优选为依利特-c18色谱柱;检测波长240nm-260nm,270nm-290nm,优选为250nm,280nm;色谱柱的柱温优选为25℃~35℃,更优选为30℃;流动相的流速优选为0.5ml/min~1.5ml/min,更优选为1.0ml/min;理论踏板数按牛蒡苷计不得低于10000。
检测过程中,优选以甲醇为流动相的b相,以乙腈为流动相的c相,以0.05%的磷酸为流动相的d相。检测过程中,洗脱方式优选为梯度洗脱,洗脱的程序设置为:0~15min,b:10%,c:15%,d:75%;15~35min,b:10%,c:15%~22%,d:75%~68%;35~40min,b:10%,c:22%~44%,d:68%~46%;40~45min,b:10%~7%,c:44%~46%,d:46%~47%;45~50min,b:7%~2%,c:46%~57%,d:47%~41%;50~51min,b:2%~10%,c:57%-90%,d:41%~0%;51~55min,b:10%,c:90%,d:0%;55~56min,b:10%,c:90%~15%,d:0%~75%;56~60min,b:10%,c:15%,d:75%。
检测结束后,得到色谱图,所述色谱图即可作为消毒散口服液hplc指纹图谱。在本发明提供的一个实施例中,也采用指纹图谱软件对所述色谱图进行处理,得到软件处理的消毒散口服液hplc指纹图谱。所述指纹图谱软件优选为国家药典颁布的《中药色谱指纹图谱相似度评价系统》。
本发明提供的一个实施例中,采用以下方法建立指纹图谱,该过程具体为:
(1)将多批次的消毒散口服液,与溶剂混合后,得到多个供试品溶液;
(2)取甘草苷、牛蒡苷、牛蒡子苷元,甘草酸铵,胡薄荷酮对照品适量,用溶剂溶解,混合,得对照品溶液;
(3)分别将对照品溶液与多个供试品溶液注入高效液相色谱仪中进行检测,得到多张消毒散口服液的色谱图;
在本发明上述实施例提供的方法中,所述共有峰的个数优选为10~16个,更优选为14个;所述指纹图谱软件优选为国家药典颁布的《中药色谱指纹图谱相似度评价系统》;所述处理方法优选为中位数法。
在本发明提供的一个实施例中,以牛蒡苷为参照峰s,计算各特征峰与s峰的相对保留时间,所述的指纹图谱相对保留时间峰1-3在第一规定值的±10%之内,峰4-13在第一规定值的±5%之内。所述的第一规定值为0.254(峰1),0.275(峰2),0.338(峰3),0.549(峰4),1.000(峰5,s),1.402(峰6),1.424(峰7),1.447(峰8),1.457(峰9),1.475(峰10),1.492(峰11),1.522(峰12),1.573(峰13),1.617(峰14)。
在本发明提供的一个指纹图谱共有峰的相对保留时间为上述数值的实施例中,所述指纹图谱2号峰为甘草苷,5号峰为牛蒡苷,9号峰为牛蒡子苷元,12号峰为甘草酸,14号峰为胡薄荷酮。
采用本发明提供的方法可建立消毒散口服液的指纹图谱,用于对消毒散口服液的质量进行控制和评价。对本发明提供的方法进行精密度、稳定性和重复性测试,试验结果表明,本发明提供的方法精密度和重复性良好,供试品溶液48小时内成分稳定。
为更清楚地阐述该方法,下面通过实施例进行详细说明。
本发明实施例中涉及的仪器与试药如下:
仪器:天平(mettlertoledoxpe205);agilent1260高效液相色谱仪(dad-检测器,自动进样器);色谱柱:依利特公司hypersilods2色谱柱5μm(4.6mm×250mm),批号:14025
试药:牛蒡子(辽宁清原县,批号:20161101);荆芥穗(河北新乐县,批号:20161101);甘草(新疆温宿县,批号:20161201);甘草苷(中国食品药品检定研究院,批号:111610-201106,93.1%);牛蒡苷(中国食品药品检定研究院,批号:110819-201611,纯度95.9%);甘草酸铵(中国食品药品检定研究院,批号:110731-201619,纯度93.0%);胡薄荷酮(中国食品药品检定研究院,批号:11706-201205,纯度99.8%);牛蒡子苷元(成都瑞芬思生物科技有限公司,批号:n-013-160406,纯度>98%);乙腈、甲醇为色谱纯,磷酸为分析纯,水为纯化水。6批次消毒散口服液(参照中国专利cn107243020a所述方法制备)
实施例1指纹图谱的建立
1供试品溶液和对照品溶液的制备
1.1供试品溶液:取消毒散口服液2ml,置25ml容量瓶中,加入甲醇稀释至刻度,摇匀,用0.22μm滤膜过滤,取续滤液,即得供试品溶液。
1.2对照品溶液:取甘草苷、牛蒡苷、牛蒡子苷元、甘草酸铵、胡薄荷酮对照品适量,加甲醇制成每1ml分别含10μg、250μg、10μg、250μg、60μg的混合对照品。
2色谱条件
色谱柱:依利特-c18(4.6mm×250mm,5μm);流动相:甲醇-乙腈-0.05%磷酸水溶液,梯度洗脱,见表2;柱温:30℃;流速:1.0ml·min-1;检测波长:250nm、280nm;色谱图记录时间:60min。进样量10μl。
表2梯度洗脱程序
3建立对照指纹图谱取6批消毒散口服液按“1.1”项下方法制备供试品溶液,再按“2”项下方法进行测定,记录色谱图。将6批图谱数据导入指纹图谱相似度评价软件(中药色谱指纹图谱相似度评价系统,2012.1版),以01批次样品图谱为参照图谱,匹配色谱峰,以中位数法生成对照指纹图谱。
4相似度计算采用相似度评价软件(中药色谱指纹图谱相似度评价系统,2012.1版),以对照指纹图谱作为参照图谱,计算6个批次的消毒散口服液指纹图谱的相似度,结果见表3,表4:
表36批消毒散口服液相识度计算结果(250nm)
表46批消毒散口服液相识度计算结果(280nm)
结果表明6个批次的样品在280nm检测条件下得到的指纹图谱,相似度均大于0.99,250nm检测条件下得到的指纹图谱,相似度均大于0.95,均具有较高相似度。
5特征峰的确定根据色谱峰保留时间稳定、峰面积相对较高及检测率达100%原则,共选择了14个重复性较好且专属性较强的色谱峰作为特征峰,以保留时间适中、峰面积较大的有效成分5号牛蒡苷峰为参照峰,计算各特征峰的相对保留时间,结果见表5。
表56批消毒散口服液特征峰的相对保留时间
6特征峰相对保留时间规定值的确定以6批消毒散口服液图谱中14个特征峰的相对保留时间的平均值作为规定值,并结合方法学考察中各变动因素对特征峰相对保留时间的影响确定其规定值的波动范围。最终规定:供试品特征图谱中应呈现14个特征峰,与牛蒡苷参照物相应色谱峰为s峰,计算各特征峰与峰(s)的相对保留时间,其中峰1-3相对保留时间应在规定值的±10%之内,峰4-14相对保留时间应在规定值的±5%之内。特征峰相对保留时间规定值为0.254(峰1),0.275(峰2),0.338(峰3),0.549(峰4),1.000(峰5,s),1.402(峰6),1.424(峰7),1.447(峰8),1.457(峰9),1.475(峰10),1.492(峰11),1.522(峰12),1.573(峰13),1.617(峰14)。
7特征峰峰指认采用甘草苷、牛蒡苷、牛蒡子苷元、甘草酸铵、胡薄荷酮对照品进行定位,其中峰2为甘草苷,峰5为牛蒡苷,峰9为牛蒡子苷元,峰12为甘草酸,峰14为胡薄荷酮。
实施例2
精密度试验
取消毒散口服液(批号20170324)样品1份,按实施例1“1.1”项下方法制备供试品,按“2”项下方法,连续进样6次,测定,记录色谱图,以牛蒡苷参照物相应色谱峰为参照峰,计算各特征峰的相对保留时间和相对峰面积,结果显示,各特征峰的相对保留时间和相对峰面积的rsd值均小于3.0%,直观观察图谱的全貌无明显变化,经中药指纹图谱相似度评价系统计算相似度大于0.999,表明仪器精密度良好。
实施例3重复性试验
取消毒散口服液(批号20170324)样品6份,按实施例1“1.1”项下方法制备供试品,按“2”项下方法测定,记录色谱图,以牛蒡苷参照物相应色谱峰为参照峰,计算各特征峰的相对保留时间和相对峰面积,结果显示,各特征峰的相对保留时间均小于2.0%,相对峰面积的rsd值均小于5.0%,直观观察图谱的全貌无明显变化,经中药指纹图谱相似度评价系统计算相似度大于0.999,表明该方法重复性良好。
实施例4稳定性试验
取消毒散口服液(批号20170324)样品1份,按实施例1“1.1”项下方法制备供试品,按“2”项下方法分别于0,1,2,6,4,8,12,24,48h测定,记录色谱图,以牛蒡苷参照物相应色谱峰为参照峰,计算各特征峰的相对保留时间和相对峰面积,各特征峰的相对保留时间rsd小于2.0%,除特征峰2,峰6相对峰面积rsd小于10.0%,其余相对峰面积rsd均小于5.0%,直观观察图谱的全貌无明显变化,经中药指纹图谱相似度评价系统计算相似度大于0.95,表明溶液在48h内基本稳定。
实施例5
指纹图谱中共有峰的归属
分别取处方中除某味药材的其他药材,按处方工艺制备成消毒散口服液阴性对照样品,取各阴性对照样品适量,按实施例1“1.1”项下方法制备成阴性对照溶液,共3份阴性对照溶液。分别取各单味处方药相应的饮片,按处方提取工艺制备成相应的单位药材供试品,取各供试品适量,按实施例1“1.1”项下方法制备成相应的单味药材供试品溶液,共制备3份单味药材供试品溶液。
分别吸取供试品溶液、各阴性对照溶液和单味药材供试品溶液各10μl,
实施例1“2”项下方法测定,记录色谱图;依据各色谱峰的紫外吸收光谱和相对保留时间,确定各特征峰的归属。结果显示,本文建立的消毒散口服液特征图谱中的14个特征峰均可在药材中找到归属,制剂与药材之间存在良好的相关性。其中,归属于牛蒡子的特征峰为:峰3、峰4、峰5、峰7、峰8、峰9;归属于荆芥穗的特征峰为:峰6,峰14;归属于甘草的特征峰为:峰1,峰2,峰10,峰11,峰12,峰13;处方中牛蒡子、荆芥穗、甘草3味药均能在消毒散口服液特征图谱中体现。
本发明根据处方中各药味有效成分的吸收特点,采用hplc所建立的消毒散口服液特征图谱,共有14个特征峰,专属性均较强,且对各特征峰归属做了较为全面的研究,分别将各峰归属到各味药材中,能较好地对处方中的牛蒡子、荆芥穗和甘草进行控制;指认了5个指标性成分,能较为全面地对处方中主要成分进行控制,使产品的质量更加稳定、可控。
- 还没有人留言评论。精彩留言会获得点赞!