一种光电材料及其制备方法和用途与流程
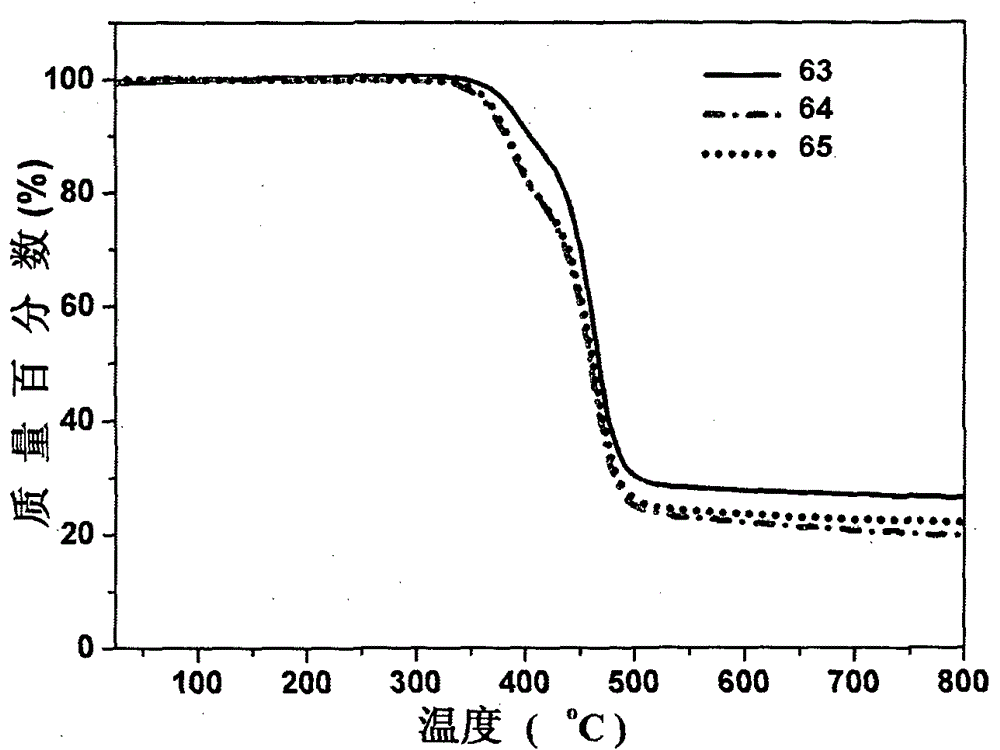
一种光电材料及其制备方法和用途领域本申请涉及材料化学领域。更具体地,本申请涉及光电材料领域背景太阳能是人类取之不尽、用之不竭、清洁无污染的可再生能源。与无机太阳能电池相比,有机太阳能电池具有质轻、价廉、可溶液处理、高的机械柔性、可制成柔性大面积器件等优点。概述一方面,本申请涉及下列通式的给受体型寡聚噻吩化合物:其中,x为0至50的整数,y为1至50的整数,R1和R2分别独立地选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,其中R1和R2可以相同也可以不同,但是R1和R2不能同时为H,L1、L2和L3分别独立地选自桥连的共轭电子给体单元或桥连的共轭电子受体单元,以及A2为端基受体单元。另一方面,本申请涉及选自通式(1)至通式(6)的给受体型寡聚噻吩化合物:其中,n为1至50的整数,R1和R2分别独立地选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,其中R1和R2可以相同也可以不同,但是R1和R2不能同时为H,D和D1分别独立地为桥连的共轭电子给体单元,A和A1分别独立地为桥连的共轭电子受体单元,以及A2为端基受体单元。另一方面,本申请涉及制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法,其中,给体桥连的含受体端基的寡聚噻吩通过双醛基给体桥连的寡聚噻吩与受体端基单体,在溶剂和催化剂的存在下,进行克内费纳格尔(Knoevenagel)缩合反应,得到所述化合物。再一方面,本申请涉及制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法,其中,给体桥连的含小分子染料端基的寡聚噻吩通过双醛基给体桥连的寡聚噻吩与有机小分子染料单体,在溶剂和催化剂的存在下,进行克内费纳格尔(Knoevenagel)缩合反应,得到所述化合物。又一方面,本申请涉及通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物在制备场效应晶体管中的用途。另一方面,本申请涉及通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物在制备光伏器件中的用途。再一方面,本申请涉及包含具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的活性层的三极管器件。又一方面,本申请涉及包含具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的活性层的光伏器件。另一方面,本申请涉及制备场效应晶体管的方法,其包括提供具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物。再一方面,本申请涉及制备光伏器件的方法,其包括提供具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物。其他方面,本申请涉及选自如下的化合物:以及附图说明图1为本申请实施例2和3中化合物的热重分析(TGA)曲线。图2为本申请实施例4、5和6中化合物的热重分析(TGA)曲线。图3为本申请实施例4、5和6中化合物的循环伏安曲线。图4为本申请实施例4、5和6中化合物的电流密度-电压曲线。图5示出了本申请实施例14中化合物的溶液与薄膜的紫外可见吸收光谱。图6示出了本申请实施例14中化合物的溶液与薄膜的循环伏安曲线。图7示出了本申请实施例14中化合物在不同给受体比例下的电流密度-电压曲线。图8示出了本申请实施例25中化合物在不同给受体比例下的电流密度-电压曲线。图9示出了本申请实施例25中化合物与C71PCBM在1∶0.8重量比下添加PDMS的电流密度-电压曲线。图10示出了本申请实施例13中化合物与C61PCBM在1∶0.8重量比下电流密度-电压曲线。图11示出了本申请实施例17中化合物与C61PCBM在1∶0.8重量比下电流密度-电压曲线。图12示出了本申请实施例21中化合物与C61PCBM在1∶0.5重量比下电流密度-电压曲线。图13示出了本申请实施例23中化合物与C61PCBM在1∶0.8重量比下电流密度-电压曲线。图14示出了本申请实施例24中化合物与C61PCBM在不同重量比下电流密度-电压曲线。图15示出了本申请实施例27中化合物与C61PCBM在不同重量比下电流密度-电压曲线。图16示出了本申请实施例28中化合物与C61PCBM在1∶0.5重量比下电流密度-电压曲线。图17示出了本申请实施例29中化合物与C61PCBM在1∶0.8重量比下电流密度-电压曲线。图18示出了本申请实施例30中化合物与C61PCBM在1∶0.5重量比下电流密度-电压曲线。图19示出了本申请实施例34中化合物与C61PCBM在1∶0.5重量比下电流密度-电压曲线。图20示出了本申请实施例35中化合物与C61PCBM在1∶0.5重量比下电流密度-电压曲线。详述在以下的说明中,包括某些具体的细节以对各个公开的实施方案提供全面的理解。然而,相关领域的技术人员会认识到,不采用一个或多个这些具体的细节,而采用其它方法、部件、材料等的情况下可实现实施方案。除非本申请中另外要求,在整个说明书和其后的权利要求书中,词语“包括”和“包含”应解释为开放式的、含括式的意义,即“包括但不限于”。在整个本说明书中提到的“一实施方案”或“实施方案”或“在另一实施方案中”或“在某些实施方案中”意指在至少一实施方案中包括与该实施方案所述的相关的具体参考要素、结构或特征。因此,在整个说明书中不同位置出现的短语“在一实施方案中”或“在实施方案中”或“在另一实施方案中”或“在某些实施方案中”不必全部指同一实施方案。此外,具体要素、结构或特征可以任何适当的方式在一个或多个实施方案中结合。定义由表明在所示化学基团中找到的碳原子总数的简化符号在前面标示本文中命名的某些化学基团。例如,C7-C12烷基描述具有总数为7至12个碳原子的如下定义的烷基,并且C4-C12环烷基烷基描述具有总数为4至12个碳原子的如下定义的环烷基烷基。简化符号中碳原子总数并不包含可能存在于所述基团的取代基中的碳。因此,非另有相反的说明,否则说明书及所附权利要求中所用的下列术语具有以下的意思:在本申请中,术语“烷基”系指由碳和氢原子组成的,不含不饱和键的,具有1至30个碳原子的,尤其是具有1至12个碳原子或1至8个碳原子的,且由单键与分子的其余部分相连的直链或支链烃链基团,例如甲基、乙基、正丙基、1-甲基乙基(异丙基)、正丁基、正戊基、1,1-二甲基乙基(叔丁基)、辛基等。在某些实施方案中,烷基是C1-C30烷基。在某些实施方案中,烷基是C1-C12烷基。在某些实施方案中,烷基是C1-C8烷基。烷基基团可以是任意取代的,亦即取代或未取代的。当被取代时,取代基团是单独地并且独立地选自下列的一个或多个基团:环烷基、芳基、杂芳基、杂脂环基、羟基、烷氧基、芳氧基、巯基、烷硫基、芳硫基、氰基、卤代、羰基、硫代羰基、O-氨基甲酰基、N-氨基甲酰基、O-硫代氨基甲酰基、N-硫代氨基甲酰基、C-酰氨基、N-酰氨基、S-亚磺酰氨基、N-亚磺酰氨基、C-羧基、O-羧基、异氰酸根合、氰硫基、异硫氰酸根合、硝基、甲硅烷基、三卤代甲烷磺酰基、-NR’R”或包括单-和二-取代的氨基基团在内的氨基,及其被保护的衍生物。在某些实施方案中,C1-C30烷基被卤素取代。在本申请中,术语“环烷基”指仅由碳和氢原子组成的,具有三至十五个碳原子的,尤其是具有3至30个碳原子的,并且其为饱和的,并且通过单键与分子的其余部分相连的稳定的非芳香族单环或双环烃基团,例如环丙基、环丁基、环戊基、环己基、环癸基等。在某些实施方案中,环烷基是C3-C30环烷基。在某些实施方案中,环烷基是C3-C12环烷基。在某些实施方案中,环烷基是C3-C8环烷基。环烷基基团可以是任意取代的,亦即取代或未取代的。当被取代时,取代基团是单独地并且独立地选自下列的一个或多个基团:环烷基、芳基、杂芳基、杂脂环基、羟基、烷氧基、芳氧基、巯基、烷硫基、芳硫基、氰基、卤代、羰基、硫代羰基、O-氨基甲酰基、N-氨基甲酰基、O-硫代氨基甲酰基、N-硫代氨基甲酰基、C-酰氨基、N-酰氨基、S-亚磺酰氨基、N-亚磺酰氨基、C-羧基、O-羧基、异氰酸裉合、氰硫基、异硫氰酸根合、硝基、甲硅烷基、三卤代甲烷磺酰基、-NR’R”或包括单-和二-取代的氨基基团在内的氨基,及其被保护的衍生物。在某些实施方案中,C3-C30环烷基被卤素取代。在本申请中,术语“烷氧基”是指通式-OR,其中R是上文所定义的烷基,如甲氧基、乙氧基、正丙氧基、1-甲基乙氧基(异丙氧基)、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基、戊氧基、叔戊氧基等。烷氧基基团的烷基部分可以如对上述烷基基团定义的那样被任意地取代。在某些实施方案中,烷氧基是C1-C30烷氧基。在某些实施方案中,烷氧基是C1-C12烷氧基。在某些实施方案中,烷氧基是C1-C8烷氧基。在某些实施方案中,C1-C30烷氧基被卤素取代。在本申请中,术语“羧酸酯基”是指通式RC(=O)OR’-,其中R是烃基或氢,而R’为烃基。在某些实施方案中,羧酸酯基是C1-C30羧酸酯基。在某些实施方案中,羧酸酯基是C1-C12羧酸酯基。在某些实施方案中,羧酸酯基是C1-C8羧酸酯基。在本申请中,术语“卤素”系指溴、氯、氟或碘。在本申请中,术语“受体”系指具有电子接受能力的分子。在本申请中,术语“共轭电子给体”系指具有电子给予能力的共轭分子。在本申请中,术语“共轭电子受体”系指具有电子接受能力的共轭分子。在本申请中,术语“有机小分子染料”系指能将纤维或其他物质染色的在可见光区有较强吸收的有机小分子化合物。具体实施方式一方面,本申请涉及下列通式的给受体型寡聚噻吩化合物:其中,x为0至50的整数,y为1至50的整数,R1和R2分别独立地选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,其中R1和R2可以相同也可以不同,但是R1和R2不能同时为H,L1、L2和L3分别独立地选自桥连的共轭电子给体单元或桥连的共轭电子受体单元,以及A2为端基受体单元。另一方面,本申请涉及选自通式(1)至通式(6)的给受体型寡聚噻吩化合物:其中,n为1至50的整数,R1和R2分别独立地选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,其中R1和R2可以相同也可以不同,但是R1和R2不能同时为H,D和D1分别独立地为桥连的共轭电子给体单元,A和A1分别独立地为桥连的共轭电子受体单元,以及A2为端基受体单元。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中D和D1分别独立地选自基团7至基团20:其中R3选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中A和A1分别独立地选自基团21至基团30:其中R4选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的合受体端基的给受体型寡聚噻吩化合物中A2为有机小分子染料基团。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中A2选自基团31至基团60:其中R5和R6分别独立地选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基或其卤素取代的衍生物,以及X-为能够使A2形成中性基团的阴离子,当A2选自基团55时,通式(1)中n≥4。在某些实施方案中,当A2选自基团55时,通式(1)中n不为3。在某些实施方案中,所述化合物的结构选自:以及其中,n为1至50的整数,R1和R2分别独立地选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,其中R1和R2可以相同也可以不同,但是R1和R2不能同时为H,R3选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基或其卤素取代的衍生物,以及R5选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中,D和D1分别独立地选自基团7、基团10、基团15、基团16或基团20,其中R3选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中,A和A1分别独立地选自基团21或基团23,其中R4选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中,A2选自基团31、基团35、基团36、基团40、基团43、基团44、基团47或基团55,其中R5和R6分别独立地选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中,D和D1分别独立地选自基团7、基团10、基团15、基团16或基团20,A和A1分别独立地选自基团21或基团23,并且A2选自基团31、基团35、基团36、基团40、基团43、基团44、基团47或基团55,其中R3选自H、C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物,R4选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基、C1-C30羧酸酯基或其卤素取代的衍生物并且R5和R6分别独立地选自C1-C30烷基、C3-C30环烷基、C1-C30烷氧基或其卤素取代的衍生物。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中n为1至30的整数。在某些实施方案中,所述通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物中n为1至10的整数。在某些实施方案中,所述通式(1)至通式(6)的给受体型寡聚噻吩化合物中X-选自卤素离子、BF4-、PF6-、SO3-或者CF3SO3-。在某些实施方案中,所述通式(1)至通式(6)的给受体型寡聚噻吩化合物选自:以及另一方面,本申请涉及制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法,其中,给体桥连的含受体端基的寡聚噻吩通过双醛基给体桥连的寡聚噻吩与受体端基单体,在溶剂和催化剂的存在下,进行克内费纳格尔(Knoevenagel)缩合反应,得到所述化含物。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用的溶剂为极性溶剂。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用的溶剂为三氯甲烷。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用的催化剂为碱性化合物。能够用于本申请的示例性的碱性化合物包括但不限于碳酸钠、氢化钠、碳酸钾、叔丁基醇钾、三乙胺、N,N-二甲基吡啶、氢化钠和乙基二异丙基胺。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用的催化剂为烷基胺。能够用于本申请的示例性的烷基胺包括但不限于三乙胺、N,N-二甲基吡啶和乙基二异丙基胺。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用的催化剂为三乙胺。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法中使用催化剂的用量为0.1-20mol%。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法在保护气体下进行。在某些实施方案中,所述制备通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法在氩气保护下进行。在某些实施方案中,制备通式(1)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,其中,步骤①无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤②先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,催化剂用量0.1-20mol%,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤③先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,催化剂用量0.1-20mol%,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤④先将POCl3与DMF在冰浴下发应制成vilsmeier试剂,将其滴入到寡聚噻吩的1,2-二氯乙烷中,Vilsmeier试剂过量,加热回流1-7天;以及步骤⑤室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体A*过量,反应1-7天。在某些实施方案中,制备通式(2)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,其中,步骤①无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,与2-(三甲基锡)-3-烷基噻吩在甲苯中回流1-7天;步骤②先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20moi%,与2-(三甲基锡)-3-烷基噻吩在甲苯中回流1-7天;步骤④先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,与2-(三甲基锡)-3-烷基噻吩在甲苯中回流1-7天;步骤④先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到受体桥连的寡聚噻吩的1,2-二氯乙烷中,Vilsmeier试剂过量,加热回流1-7天;以及步骤⑤室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体A*过量,反应1-7天。在某些实施方案中,制备通式(3)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,式中,D和D1可以相同也可以不同,其中,步骤①先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到给体桥连的寡聚噻吩的1,2--二氯乙烷中,寡聚噻吩与Vilsmeier试剂的物质的量比为1∶0.5,加热回流1-7天;步骤②体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶1;步骤③无水,无氧,氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,溴代物与D的双锡化单体的物质的量的比为1∶0.5,加热回流反应1-7天;步骤④氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2moL/L的K2CO3水溶液,溴代物与D的双频哪醇硼酸酯的物质的量的比为1∶0,5,加热回流反应1-7天;以及步骤⑤室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体过量,反应1-7天。在某些实施方案中,制备通式(4)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,其中,步骤①先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到受体桥连的寡聚噻吩的1,2-二氯乙烷溶液中,受体桥连的寡聚噻吩与Vilsmeier试剂的物质的量比为1∶0.5,加热回流1-7天;步骤②体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶1;步骤③无水,无氧,氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20moi%,溴代物与D的双锡化单体的物质的量的比为1∶0.5,加热回流反应1-7天;步骤④氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与D的双频哪醇硼酸酯的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤⑤室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体过量,反应1-7天。在某些实施方案中,制备通式(5)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,其中,步骤①氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与A的双频哪醇硼酸酯单体的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤②室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体过量,反应1-7天。在某些实施方案中,制备通式(6)的含受体端基的给受体型寡聚噻吩化合物的方法如下所示,式中,A和A1可以相同也可以不同。其中,步骤①氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与A的双频哪醇硼酸酯单体的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤②室温,氩气保护下,三氯甲烷为溶剂,三乙胺为催化剂,催化剂用量0.1-20mol%,受体端基单体过量,反应1-7天。再一方面,本申请涉及制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法,其中,给体桥连的含小分子染料端基的寡聚噻吩通过双醛基给体桥连的寡聚噻吩与有机小分子染料单体,在溶剂和催化剂的存在下,进行克内费纳格尔(Knoevenagel)缩合反应,得到所述化合物。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的催化剂为酸性催化剂。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的催化剂为弱酸性催化剂。能够用于本申请所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中的示例性的弱酸性催化剂的实例包括但不限于乙酸铵、丙酸铵以及丁酸铵。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的催化剂为乙酸铵。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的溶剂为酸性溶液。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的溶剂为弱酸性溶液。能够用于本申请所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中的示例性的弱酸性溶液的实例包括但不限于乙酸、丙酸以及丁酸。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中使用的溶剂为乙酸。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中所述催化剂的用量为过量。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中所述催化剂的用量为10-30mol%。在某些实施方案中,所述制备通式(1)至通式(6)的给受体型寡聚噻吩化合物的方法中所述催化剂的用量为20mol%。在某些实施方案中,制备通式(1)的给受体型寡聚噻吩化合物的方法如下所示,步骤①无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤②先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,催化剂用量0.1-20mol%,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤③先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Ni(dppp)Cl2催化剂,催化剂用量0.1-20mol%,与2-溴-3(和/或4)烷基噻吩的格氏试剂在乙醚中回流1-7天;步骤④先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到给体桥连的寡聚噻吩的1,2-二氯乙烷中,Vilsmeier试剂过量,加热回流1-7天;以及步骤⑤乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。在某些实施方案中,制备通式(2)的给受体型寡聚噻吩化合物的方法如下所示,步骤①无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,与2-(三甲基锡)-3(和/或4)-烷基噻在甲苯中回流1-7天;步骤②先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,与2-(三甲基锡)-3(和/或4)-烷基噻吩或2-(三丁基锡)-3(和/或4)烷基噻吩在甲苯中回流1-7天;步骤③先在体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶2,所得产物在无水,无氧,氩气保护下,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,与2-(三甲基锡)-3(和/或4)-烷基噻吩或2-(三丁基锡)-3(和/或4)烷基噻吩在甲苯中回流1-7天;步骤④先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到受体桥连的寡聚噻吩的1,2-二氯乙烷中,Vilsmeier试剂过量,加热回流1-7天;以及步骤⑤乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。在某些实施方案中,制备通式(3)的给受体型寡聚噻吩化合物的方法如下所示,式中,D和D1可以相同也可以不同,步骤①先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到给体桥连的寡聚噻吩的1,2-二氯乙烷中,寡聚噻吩与Vilsmeier试剂的物质的量比为1∶0.5,加热回流1-7天;步骤②体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,给体桥连的寡聚噻吩与NBS的物质的量比为1∶1;步骤③无水,无氧,氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,溴代物与D的双锡化单体的物质的量的比为1∶0.5,加热回流反应1-7天;步骤④氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与D的双频哪醇硼酸酯的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤⑤乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。在某些实施方案中,制备通式(4)的给受体型寡聚噻吩化合物的方法如下所示,步骤①先将POCl3与DMF在冰浴下发应制成Vilsmeier试剂,将其滴入到受体桥连的寡聚噻吩的1,2-二氯乙烷中,受体桥连的寡聚噻吩与Vilsmeier试剂的物质的量比为1∶0.5,加热回流1-7天;步骤②体积比为1∶1的三氯甲烷和冰乙酸中用NBS进行溴化,受体桥连的寡聚噻吩与NBS的物质的量比为1∶1;步骤③无水,无氧,氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,溴代物与D的双锡化单体的物质的量的比为1∶0.5,加热回流反应1-7天;步骤④氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与D的双频哪醇硼酸酯的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤⑤乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。在某些实施方案中,制备通式(5)的给受体型寡聚噻吩化合物的方法如下所示,步骤①氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与A1的双频哪醇硼酸酯单体的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤②乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。在某些实施方案中,制备通式(6)的给受体型寡聚噻吩化合物的方法如下所示,式中,A和A1可以相同也可以不同,步骤①氩气保护下,甲苯为溶剂,Pd(PPh3)4为催化剂,催化剂用量0.1-20mol%,加入适量2mol/L的K2CO3水溶液,溴代物与A的双频哪醇硼酸酯单体的物质的量的比为1∶0.5,加热回流反应1-7天;以及步骤②乙酸为溶剂,乙酸铵为催化剂,催化剂用量20mol%,受体端基单体A*过量,加热回流24小时。又一方面,本申请涉及通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物在制备场效应晶体管中的用途。另一方面,本申请涉及通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物在制备光伏器件中的用途。在某些实施方案中,通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物可以用于制备光伏器件是太阳能电池器件。在某些实施方案中,通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物可以用于制备光伏器件是太阳能电池器件中的光活性层。再一方面,本申请涉及包含具有通式(1)至通式(6)的给受体型寡聚噻吩化合物的活性层的三极管器件。又一方面,本申请涉及包含具有通式(1)至通式(6)的给受体型寡聚噻吩化合物的活性层的光伏器件。在某些实施方案中,太阳能电池器件包含具有通式(1)至通式(6)的给受体型寡聚噻吩化合物的光活性层。另一方面,本申请涉及制备场效应晶体管的方法,其包括提供具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物。再一方面,本申请涉及制备光伏器件的方法,其包括提供具有通式(1)至通式(6)的含受体端基的给受体型寡聚噻吩化合物。其他方面,本申请涉及选自如下的化合物:在本申请的含受体端基的给受体型寡聚噻吩光电材料中,由于寡聚噻吩由于具有较高的空穴迁移率,所以本申请的给受体型寡聚噻吩光电材料也具有较高的空穴迁移率。本申请的含受体端基的给受体型寡聚噻吩光电材料结合了聚合物和普通共轭小分子的优点,与通常的聚合物相比具有精确的分子量、结构可控、易纯化等优点,与普通共轭小分子相比又具有较好的溶解度,可制成薄膜,有利于场效应晶体管和包括太阳能电池器件在内的光伏器件的制备。在本申请涉及的含有机小分子染料受体端基的给受体型寡聚噻吩化合物中,由于寡聚噻吩具有较高的空穴迁移率,而有机小分子染料具有高的吸电子性和摩尔吸光系数,所以本申请所涉及的这一类给受体型寡聚噻吩化合物也具有较高的空穴迁移率和摩尔吸光系数。本申请涉及的含有机小分子染料受体端基的给受体型寡聚噻吩化合物结合了聚合物和共轭小分子的优点,与常用的聚合物相比具有精确的分子量、可控的结构、简单的纯化过程,与普通的共轭小分子相比又具有较好的溶解度,使溶剂化过程成为可能,可制成薄膜,有利于制备高性能的有机场效应晶体管和包括有机太阳能电池器件在内的光伏器件。使用本申请涉及的含有机小分子染料受体端基的给受体型寡聚噻吩化合物制备的有机薄膜太阳能电池具有染料敏化电池材料高摩尔吸光系数的特点,同时有保留了有机太阳能电池可成柔性薄膜的特点。下文中,本发明将参照附图通过如下实施例进行详细解释以便更好地理解本发明的各个方面及其优点。然而,应当理解,以下的实施例是非限制性的而且仅用于说明本发明的某些实施方案。实施例实施例1寡聚噻吩前体的合成1)2-溴-3-辛基噻吩的合成在盛有3-辛基噻吩(10.00g,50.93mmol)的250mL双口瓶中加入60mLDMF。冰盐浴下,滴入NBS(9.26g,52.03mmol)的60mLDMF溶液。滴毕,缓慢升到室温,室温搅拌过夜。停止反应,倒入200mL水中,二氯甲烷(60mL×4)萃取。有机相依次用氢氧化钾水溶液(2M,100mL),饱和食盐水(100mL)和水(100mL×2)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得12.60g油状液体,产率为89%。其结构式如下所示:2)3,3”-二辛基-2,2’∶5’,2”-三噻吩(3T)的合成在盛有镁屑(704mg,28.96mmol)的100mL双口瓶中加入20mL乙醚,氩气保护下,缓慢滴入2-溴-3-辛基噻吩(4.00g,14.56mmol)、1,2-二溴乙烷(1.37g,7.28mmol)和20mL乙醚的混合液。滴毕,加热回流4小时,降到室温。将所得格氏试剂缓慢滴入到盛有Ni(dppp)Cl2(177mg,0.326mmol)、2,5-二溴噻吩(1.40g,5.56mmol)和25mL乙醚的混合液。滴毕,加热回流18小时。降到室温,加入20mL稀盐酸(2M),倒入200mL水中,二氯甲烷(100mL×3)萃取。有机相依次用碳酸钠水溶液(2M,100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得2.30g浅黄色油状液体,产率为84%。其结构式如下所示:3)5-三丁基锡-3,3”-二辛基-2,2’∶5’,2”-三噻吩(BS3T)的合成氩气保护下,在盛有三噻吩(3.65g,7.72mmol)的250mL三口瓶中,加入100mLTHF。降温至-78℃,滴加n-BuLi的正己烷溶液(3.3ml,2.4M,2.92mmol)后,升温至-40℃反应1h。再降温至-78℃,滴入三丁基氯化锡(3.02g,9.26mmol),室温搅拌过夜。将反应物倒入100mL水中,乙酸乙酯(30mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,得橙黄色油状液体4.83g,产率82%。其结构式如下所示:4)5,5”-二溴-3,3”-二辛基-2,2’∶5’,2”-三噻吩(3TBr2)的合成在盛有三噻吩1(1.20g,2.54mmol)的250mL双口瓶中,加入30mL氯仿和30mL冰乙酸,降温至0℃下,将NBS(0.96g,5.39mmol)分批加入,约20min加完。室温下搅拌3小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用碳酸钠水溶液(2M,100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得1.60g黄色油状液体,产率为100%。其结构式如下所示:5)3,3’,3’”,3””-四辛基-2,5’∶2’,5”∶2”,2’”∶5’”,2””-五噻吩(5T)的合成在盛有镁粉(0.36g,14.48mmol)的100mL双口瓶中加入20mL乙醚,氩气保护,室温下滴入2-溴-3-辛基噻吩(2.00g,7.28mmol),1,2-二溴乙烷(0.34g,1.82mmol)和20mL乙醚的混合液,滴毕,加热回流4小时。氩气保护下将所得格氏试剂滴入到盛有二溴三噻吩2(1.54g,2.44mmol),Ni(dppp)Cl2(90mg,0.17mmol)和20mL乙醚的混合液中,约半小时滴完。加热回流20小时,降到室温后,加入稀盐酸(20mL,1M),搅拌5分钟,将反应液倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得1.75g金黄色油状液体,产率为83%。其结构式如下所示:6)5,5””-二溴-3,3’,3’”,3””-四辛基-2,5’∶2’,5”∶2”,2’”∶5’”,2””-五噻吩(5TBr2)的合成在盛有五噻吩3(1.15g,1.33mmol)的250mL双口瓶中,加入30mL氯仿和30mL冰乙酸,降温至0℃下,将NBS(0.50g,2.81mmol)分批加入,约20min加完。室温下搅拌3小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用碳酸钠水溶液(2M,100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得1.22g黄色油状液体,产率为90%。其结构式如下所示:7)3,3’,3”,3’”,3””,3”””-六辛基-2,5’∶2’,5”∶2”,2’”∶5’”,2””∶5””,2’””∶5’””,2”””-七噻吩(7T)的合成在盛有镁粉(0.18g,7.24mmol)的100mL双口瓶中加入15mL乙醚,室温下滴入2-溴-3-辛基噻吩(1.00g,3.64mmol),1,2-二溴乙烷(0.17g,0.91mmol)和15mL乙醚的混合液,滴毕,加热回流4小时。氩气保护下将所得格氏试剂滴入到盛有二溴五噻吩4(1.24g,1.22mmol),Ni(dppp)Cl2(59mg,0.11mmol)和20mL乙醚的混合液中,约半小时滴完。加热回流20小时,降到室温后,加入稀盐酸(20mL,1M),搅拌5分钟,将反应液倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得1.09g金黄色油状液体,产率为72%。其结构式如下所示:8)5,5”-二甲醛-3,3”-二辛基-2,2’∶5’,2”-三噻吩(3T(CHO)2)的合成0℃下,将POCl3(0.71mL,7.74mmol)缓慢滴入到DMF(3.00mL,38.70mmol)中,搅拌10分钟,氩气保护下将所得液滴入到盛有3T(1.22g,2.58mmol)和30mL1,2-二氯乙烷的混合混合液中。加热到60℃反应12小时,冷至室温,倒入200mL冰水中,碳酸钠中和,二氯甲烷(100mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂,过柱分离,得1.13g珊瑚色固体,产率为83%。其结构式如下所示:9)5,5””-二甲醛-3,3’,3’”,3””-四辛基-2,5’∶2’,5”∶2”,2’”∶5’”,2””-五噻吩(5T(CHO)2)的合成方法同3T(CHO)2的合成。得深橙色固体,产率为85%。其结构式如下所示:10)5,5”””-二甲醛基-3,3’,3”,3””,3””,3”””-六辛基-2,5’∶2’,5”∶2”,2’”∶5’”,2””∶5””,2’””∶5’””,2”””-七噻吩(7T(CHO)2)的合成方法同3T(CHO)2的合成。得1.13g褐色固体,产率为81%。其结构式如下所示:11)化合物TBT的合成在250mL的双口瓶中,加入4,7-二溴苯并噻二唑(6.00g,20.4mmol),2-三丁基锡-4-辛基噻吩(55g,113.3mmol),以及Pd(PPh3)2Cl2(320mg,0.46mmol)。氩气保护下加入120mL无水新蒸四氢呋喃。加热回流24小时,停止反应。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为洗脱剂过柱分离,得8.8g红色固体,产率为82%。其结构式如下所示:12)化合物BrTBTBr的合成在100mL的双口瓶中,加入化合物TBT(0.96g,1.83mmol),60mL氯仿。冰盐浴下分批加入NBS(0.65g,3.66mmol)。保持次温度反应1小时,撤掉冰浴。室温反应过夜。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为洗脱剂过柱分离,得1.08g红色固体,产率为86%。其结构式如下所示:13)化合物2TB2T的合成在100mL的双口瓶中,加入化合物BrTBTBr(1.02g,1.49mmol),2-三丁基锡-4-辛基噻吩(2.18g,4.48mmol),以及Pd(PPh3)2Cl2(105mg,0.15mmol)。氩气保护下加入65mL无水新蒸四氢呋喃。加热回流40小时,停止反应。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为洗脱剂过柱分离,得1.26g紫色固体,产率为94%。其结构式如下所示:14)化合物Br2TB2TBr的合成在100mL的双口瓶中,加入化合物2TB2T(0.86g,0.94mmol),70mL氯仿。冰盐浴下分批加入NBS(0.29g,1.61mmol)。保持次温度反应1小时,撤掉冰浴。室温反应过夜。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为洗脱剂过柱分离,得0.79g红色固体,产率为46%。其结构式如下所示:15)化合物3TB3T的合成在250mL的双口瓶中,加入化合物Br2TB2TBr(130mg,0.12mmol),3-丁基锡-4-辛基噻吩(357mg,0.36mmol),以及Pd(PPh3)2Cl2(8.5mg,0.01mmol)。氩气保护下加入60mL无水新蒸四氢呋喃。加热回流40小时,停止反应。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为洗脱剂过柱分离,得139mg黑色固体,产率为89%。其结构式如下所示:16)5TCHO的合成0℃下,将POCl3(0.84mL,9.2mmol)缓慢滴入到DMF(4.24mL,55.0mmol)中,搅拌10分钟,氩气保护下取十分之一的所得液滴入到盛有5T(0.79g,0.92mmol)和30mL1,2-二氯乙烷的混合液中。加热到70℃反应24小时,冷至室温,倒入200mL冰水中,碳酸钠中和,二氯甲烷(100mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂,过柱分离,得0.46g红色固体,产率为56%。其结构式如下所示:17)Br5TCHO的合成在盛有5TCHO(0.32g,0.36mmol)的100mL双口瓶中,加入30mL氯仿和30mL冰乙酸,将NBS(64mg,0.36mmol)分批加入,约20min加完。室温下搅拌3小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用碳酸钠水溶液(2M,100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚为淋洗剂,过柱分离,得0.31g红色固体,产率为89%。其结构式如下所示:18)化合物3TB3T(CHO)的合成方法同5TCHO的合成。得1.08g褐色固体,产率为70%。其结构式如下所示:19)化合物Br3TB3T(CHO)的合成方法同Br5TCHO的合成。得0.82g褐色固体,产率为81%。其结构式如下所示:20)化合物D3TBT的合成氩气保护下,在盛有对二溴并噻吩(0.40g,1.34mmol)、单三丁基锡三噻吩(2.46g,3.23mmol)和40mL干燥甲苯的双口瓶中加入三苯基膦钯(0.078g,0.068mmol)110℃回流过夜。将反应液倒入100mL水中,二氯甲烷(30mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-石油醚为洗脱剂,过柱分离,得0.60g橙红色固体,产率为41%。其结构式如下所示:21)化合物3T(TT)3T(CHO)2的合成0℃下,将POCl3(0.76mL,8.32mmol)缓慢滴入到DMF(3.19mL,41.25mmol)中,搅拌10分钟,氩气保护下将所得液滴入到盛有D3TBT(0.60g,0.55mmol)和25mL1,2-二氯乙烷的混合混合液中。加热到60℃反应12小时,冷至室温,倒入100mL冰水中,碳酸钠中和,二氯甲烷(30mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为洗脱剂,过柱分离,得0.40g黑色固体,产率为63%。其结构式如下所示:实施例2氩气保护下,在盛有双醛基三噻吩3T(CHO)2(200mg,0.38mmol)和50mL氯仿的100mL双口瓶中加入三滴三乙胺和0.1mL氰基乙酸乙酯,氩气保护下,搅拌回流过夜。降到室温,倒入200mL水中,静置,抽滤,固体用乙醇洗涤,将所得固体以二氯甲烷为淋洗剂,过柱分离,得226mg褐色固体,产率为83%。1HNMR(400MHz,CHCl3):δ8.23(s,2H),7.61(s,2H),7.33(s,2H),4.34-4.40(q,J=7.0,4H),2.83(t,J=7.5Hz,4H),1.69(m,4H),1.40(t,J=7.0Hz,6H),1.27(m,20H),0.87(t,J=6.1Hz,6H).HRMS(MALDI-FTICR):C40H50N2O4S3[M]+,理论值,718.2933;实测值,718.2937.其结构式如下所示:实施例3方法同实施例2。得到墨绿色固体,产率为80%。1HNMR(400MHz,CHCl3):δ8.15(s,2H),7.49(s,2H),7.13(s,2H),7.09(s,2H),4.29(q,J=6.6Hz,4H),2.74(t,J=6.6Hz,8H),1.61(m,8H),1.32(t,J=6.5Hz,6H),1,21(m,40H),0.80(t,J=6.1Hz,12H).HRMS(MALDI-FTICR):C64H86N2O4S5[M]+,理论值,1106.5191;实测值,1106.5188,其结构式如下所示:实施例4方法同实施例2。得到180mg墨绿色固体,产率为75%。1HNMR(400MHz,CHCl3):δ8.14(s,2H),7.49(s,2H),7.12(s,2H),7.05(s,2H),6.96(s,2H),4.27-4.31(q,J=7.1Hz,4H),2.75(t,J=7.8Hz,12H),1.58-1.68(m,12H),1.32(t,J=7.1Hz,6H),1.21(m,60H),0.80(t,J=6.1Hz,18H).HRMS(MALDI-FTICR):C88H122N2O4S7[M]+,理论值,1494.7450;实测值,1494.7460。其结构式如下所示:实施例5方法同实施例2。用氰基乙酸辛酯代替氰基乙酸乙酯,产率为81%。1HNMR(400MHz,CHCl3):δ8.20(s,2H),7.56(s,2H),7.19(s,2H),7.12(s,2H),7.03(s,2H),4.29(t,J=6.7Hz,4H),2.83m,12H),1.71(m,16H),1.42-1.29(m,80H),0.88(t,J=5.9Hz,24H).MALDI-TOFMS(m/z):C100H146N2O4S7[M]+,理论值,1662.93;实测值,1662.93。其结构式如下所示:实施例6方法同实施例2。用氰基乙酸-2-乙基己酯代替氰基乙酸乙酯,产率为78%。1HNMR(400MHz,CHCl3):δ8.20(s,2H),7.57(s,2H),7.19(s,2H),7.12(s,2H),7.03(s,2H),4.22(d,J=Hz,4H),2.83(m,12H),1.71(m,14H),1.42(m,16H),1.31(m,60H),0.94(t,J=8.1Hz,12H),0.88(t,J=5.9Hz,18H).MALDI-TOFMS(m/z):C100H146N2O4S7[M]+,理论值,1662.93;实测值,1662.93。其结构式如下所示:实施例7在100mL的双口瓶中加入9(0.16g,0.12mmol),40mL氯仿。氩气保护下加入0.1mL三乙胺,滴入0.4mL丙二腈,室温反应过夜。倒入100mL水中,用二氯甲烷(100mL×3)萃取,有机相用水(100mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为洗脱剂过柱分离,得0.13g黑色固体,产率为76%。1HNMR(400MHz,CHCl3):δ8.40(s,2H),7.98(s,2H),7.84(s,2H),7.11(d,J=7.7Hz,2H),7.07(br,2H),4.37(q,J=7.0Hz,4H),2.83(m,12H),1.71(m,12H),1.40(t,J=7.0Hz,6H),1.29(m,60H),0.89(br,18H).MS(MALDI-TOF):calcdforC86H112N6S7[M]+,1452.70;foud,1452.67。其结构式如下所示:实施例8氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.26g,0.20mmol),1,3-二乙基-2-硫代巴比妥酸(0.20g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷为洗脱剂,过柱分离,得到0.23g黑色固体,产率为70%。MALDI-TOFMS(m/z):C95H134N4O4S8[M]+,理论值,1650.82;实测值,1650.83。其结构式如下所示:实施例9氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.26g,0.20mmol),丙二酸二乙酯(0.16g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷为洗脱剂,过柱分离,得到0.24g暗红色固体,产率为75%。MALDI-TOFMS(m/z):C92H132O8S7[M]+,理论值,1588.80;实测值,1588.81。其结构式如下所示:实施倒101)(3TB3T)T(3TB3T)(CHO)2的合成在100mL的双口瓶中加入50mL甲苯,Br3TB3T(CHO)(0.28g,0.20mmol),2,5-二(三甲基锡)噻吩(41mg,0.10mmol)。氩气保护下加入Pd(PPh3)4(20mg,0.017mmol),加热至90℃回流。24小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用饱和食盐水(100mL)和水(100mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂过柱分离,得到0.21g黑色固体,产率为72%。2)目标化合物的合成氩气保护下,在盛有(3TB3T)T(3TB3T)(CHO)2(0.22g,0.08mmol),氰基乙酸乙酯(0.3mL)和60mL干燥三氯甲烷的100mL双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷和石油醚(体积比为1∶1)为淋洗剂,过柱分离,得到0.19g黑色固体,产率为82%。MS(MALDI-TOF):C172H234N6O4S15[M]+,理论值,2927.41;实测值,2927.43。其结构式如下所示:实施例111)11T(CHO)2的合成在100mL的双口瓶中加入50mL甲苯,Br5TCHO(0.39g,0.40mmol),2,5-一(三甲基锡)噻吩(0.08g,0.20mmol)。氩气保护下加入Pd(PPh3)4(20mg,0.017mmol),加热至90℃回流。24小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用饱和食盐水(100mL)和水(100mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂过柱分离,得到0.30g褐色固体,产率为81%。2)目标化合物的合成氩气保护下,在盛有双醛基寡聚噻吩11T(CHO)2(0.28g,0.15mmol),氰基乙酸乙酯(0.3mL)和60mL干燥三氯甲烷的100mL双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷和石油醚(体积比为1∶1)为淋洗剂,过柱分离,得到0.26g黑色固体,产率为84%。MS(MALDI-TOF):C120H162N2O4S11[M]+,理论值,2046.95;实测值,2046.97。其结构式如下所示:实施例121)5T(BDT)5T(CHO)2的合成在100mL的双口瓶中加入60mL甲苯,Br5TCHO(194g,0.20mmol),2,6-二(三甲基锡)-4,8-二(2-乙己基)苯并二噻吩(0.08g,0.10mmol)。氩气保护下加入Pd(PPh3)4(20mg,0.017mmol),加热至90℃回流。24小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用饱和食盐水(100mL)和水(100mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂过柱分离,得到0.16g红褐色固体,产率为73%。2)目标化合物的合成氩气保护下,在盛有双醛基寡聚噻吩5T(BDT)5T(CHO)2(0.11g,0.05mmol),氰基乙酸辛酯(0.2mL)和40mL干燥三氯甲烷的100mL双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷和石油醚(体积比为2∶1)为淋洗剂,过柱分离,得到0.10g黑色固体,产率为77%。1HNMR(400MHz,CHCl3):δ8.20(s,2H),7.56(s,2H),7.26(s,2H),7.20(s,2H),7.14(br,8H),4.28(t,J=6.5Hz,4H3,4.20(br,4H),2.82(br,16H),1.86(s,2H),1.71(m,24H),1.29(m,112H),1.07(t,J=7.0Hz,6H),1.00(br,6H),0.89(br,30H).MS(MALDI-TOF):[M]+,理论值,2577.36;实测值,2577.82。其结构式如下所示:实施例131)5TB5T(CHO)2的合成在100mL的双口瓶中加入50mL甲苯,脱气10分钟。加入Br5TCHO(0.29g,0.30mmol),4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼烷)苯并噻二唑(58mg,0.15mmol),2M的K2CO3水溶液8mL。氩气保护下加入Pd(PPh3)4(20mg,0.017mmol),加热至90℃回流。24小时后,将反应物倒入100mL水中,二氯甲烷(100mL×3)萃取。有机相依次用饱和食盐水(100mL)和水(100mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比1∶1)为淋洗剂过柱分离,得到0.21g暗红色固体,产率为72%。2)目标化合物的合成氩气保护下,在盛有5TB5T(CHO)2(0.38g,0.20mmol),氰基乙酸乙酯(0.3mL)和60mL干燥三氯甲烷的100mL双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗涤,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷为淋洗剂,过柱分离,得到0.37g黑色固体,产率为88%。1HNMR(400MHz,CHC13):δ8.18(s,2H),7.94(d,J=Hz,2H),7.75(d,J=Hz,2H),7.52(s,2H),7.17(s,2H),7.09(m,6H),7.92-7.97(m,1H),4.34(q,J=Hz,4H),2.80(br,16H),1.70(m,16H),1.29(m,86H),0.89(m,24H)。MS(MALDI-TOF):C122H162N4O4S11[M]+,理论值,2098.95;实测值,2099.02。其结构式如下所示:实施例14在50mL单口瓶中加入双醛基七噻吩7T(CHO)2(50mg,0.038mmol)和30mL乙酸,搅拌使分散均匀,再加入3-乙基洛丹宁(20mg,0.12mmol)和乙酸铵(20mg,0.12mmol),搅拌加热回流过夜。降到室温,倒入200mL水中,加入50mL二氯甲烷萃取,有机相加入50mL水洗(三次)。有机相无水硫酸镁干燥,过滤,旋干,以二氯甲烷和石油醚(1∶1)为淋洗剂,柱色谱分离,得到60mg褐色固体,产率为98.4%。1HNMR(400MHz,CHC13):67.764(s,2H)7.209(s,2H),7.100(s,4H),7.011(s,2H),4.21(q,4H,J=7.0Hz),2.74(t,12H,J=6.7Hz),1.709(m,12H),1.300(m,60H),0.882(t,18H,J=6.6Hz).HRMS(MALDI-FTICR):C88H122N2O2S11[M]+,理论值:1592.64;实测值:1591.10其结构式如下所示:实施倒15在50mL单口瓶中加入双醛基七噻吩(100mg,0.077mmol),六氟代乙酰丙酮(77mg,0.37mmol),再加入20mL乙酸,5mL氯仿,搅拌溶解。在搅拌下加热回流过夜。降到室温,倒入200mL水中,加入50mL二氯甲烷萃取,有机相加入50mL水洗(一次),50ml饱和碳酸氢钠溶液洗(一次),50mL水洗(一次)。有机相无水硫酸镁干燥,过滤,旋干,以二氯甲烷和石油醚(2∶1)为淋洗剂,柱色谱分离,得到120mg墨绿色固体,产率为93.0%。1HNMR(400MHz,CHC13):δ7.878(s,2H)7.396(s,2H),7.155(8,4H),6.976(s,2H),2.757(t,12H,J=6.7Hz),1.630(m,12H),1.215(m,60H),0.810(t,18H,J=6.6Hz).HRMS(MALDI-FTICR):C88H112F12O4S7[M]+,理论值:1686.26;实测值:1685.32其结构式如下所示:实施例16在25mL双口瓶中加入双醛基七噻吩(50mg,0.038mmol),正丁基胺(44mg,0.6mmol),加入10mL二氯甲烷搅拌溶解,再加入无水硫酸钠(1g,7ml),室温搅拌24小时。将反应体系旋干,加入15mL苯溶解,再加入三氟乙酰乙酸乙酯(73mg,0.4mmol),乙酸酐(0.1g,0.98mmol),加热回流4小时后,降至室温,旋干溶剂,加入50mL二氯甲烷重新溶解,50mL水洗(三次),有机相无水硫酸镁干燥,柱色谱分离,得墨绿色固体32mg,产率52.6%。1HNMR(400MHz,CHC13):δ7.853(s,2H)7.409(s,1H),7.342(s,1H)7.134(s,4H),7.021(s,2H),4.32(dd,4H,J=6.9Hz,J=32.3Hz)2.803(t,12H,J=6.7Hz),1.685(m,18H),1.259(m,60H),0.878(t,18H,J=6.6Hz).HRMS(MALDI-FTICR):C90H122F6O6S7[M]+,理论值:1638.37;实测值:1637.72其结构式如下所示:实施例17氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0,13g,0.10mmol),1,3-二甲基-巴比妥酸(0.156g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴哌啶,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-乙酸乙酯为洗脱剂,过柱分离,得0.12g蓝黑色固体,产率为79%。MALDI-TOFMS(m/z):C90H124N4O6S7[M]+,理论值:1580.76;实测值:1580.71其结构式如下所示:实施倒18氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.13g,0.10mmol),2,2,2-三氟乙基2-氰基乙酸酯(0.167g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-石油醚为洗脱剂,过柱分离,得0.11g墨绿色固体,产率为70%。MALDI-TOFMS(m/z):C88H116F6N2O4s7[M]+,理论值:1603.69;实测值:1603.71其结构式如下所示:实施例19氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.13g,0.10mmol),2,2,3,3,3-五氟丙基2-氰基乙酸酯(0.217g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-石油醚为洗脱剂,过柱分离,得0.12g墨绿色固体,产率为70%。MALDI-TOPMS(m/z):C90H116F10N2O4S7[M]+,理论值:1702.68;实测值:1702.70其结构式如下所示:实施例20氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.13g,0.10mmol),2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-十五氟辛基2-氰基乙酸酯(0.467g,1.00mmol)和50mL干燥三氯甲烷双口瓶中滴入三滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-石油醚为洗脱剂,过柱分离,得0.16g墨绿色固体,产率73%。MALDI-TOFMS(m/z):C100H116F30N2O4S7[M]+,理论值:2203.65;实测值:2203.71其结构式如下所示:实施例21氩气保护下,在盛有双醛基七噻吩7T(CHO)2(0.13g,0.10mmol),(E)-5-(3-乙基-5-羰基噻唑啉2-叶立德)-3-辛基-2,4-二羰基噻唑啉(0,356g,1.00mmol),乙酸铵(0.077g,1mmol)中100mL双口瓶中,加入30mL干燥氯苯和20mL冰乙酸双口瓶,室温搅拌过夜。倒入100mL水中,二氯甲烷(30mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以二氯甲烷-石油醚为洗脱剂,过柱分离,得0.16g蓝黑色固体,产率为80%。MALDI-TOFMS(m/z):C110H156N4O4S13[M]+,理论值:2013.85;实测值:2013.83其结构式如下所示:实施例22氩气保护下,在盛有双醛基双噻吩三噻吩并噻吩DFD3TBT(230mg,0.20mmol)和25mL三氯甲烷的50mL双口瓶中加入0.8mL氰基乙酸正辛酯,氩气保护下,搅拌回流过夜。降到室温,倒入100mL冰水中,二氯甲烷(30mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚和二氯甲烷的混合液(体积比2∶1)为洗脱剂,过柱分离。1HNMR(400MHz,CDCl3):8.21(s,2H),7.60(s,2H),7.31(m,4H),7.14(d,J=3.6Hz,2H),7.07(s,2H),3.64(t,J=6.4Hz,4H),2.84(t,J=7.6Hz,4H),2.79(t,J=7.6Hz,4H),1.68(m,12H),1.28(m,60H),0.89(m,18H).其结构式如下所示:实施例23氩气保护下,在盛有(147mg,0.104mmol)双(醛基三噻吩)噻咯,十倍摩尔当量的腈基乙酸辛酯和50mL干燥三氯甲烷双口瓶中滴入几滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(20mL×3)萃取。有机相依次用水(50mL),饱和食盐水(50mL)和水(50mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚-二氯甲烷=1∶1为洗脱剂,过柱分离,得到褐色固体,产率为90%。MALDI-TOFMS(m/z):C90H124N4O6S7[M]+,理论值:1773.9;实测值:1773.9。其结构式如下所示:实施例24氩气保护下,在盛有(1.0g,0.71mmol)3T(BDT)3T(CHO)2,十倍摩尔当量的腈基乙酸辛酯和70mL干燥三氯甲烷双口瓶中滴入几滴三乙胺,室温40小时。倒入100mL水中,二氯甲烷(50mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚-二氯甲烷=2∶3为洗脱剂,过柱分离,得到黑色固体,产率为70%。MALDI-TOFMS(m/z):C106H148N2O4S8[M]+,理论值:1768.92;实测值:1768.93。其结构式如下所示:实施例25氩气保护下,在盛有(173mg,0.1mmol)双(醛基三噻吩)氧异辛基苯并双噻吩,十倍摩尔当量的3-乙基洛丹宁和乙酸铵(2mg,0.012mmol),搅拌加热回流过夜。降到室温,倒入200mL水中,加入80mL二氯甲烷萃取,有机相加入80mL水洗(三次)。有机相无水硫酸镁干燥,过滤,旋干,以二氯甲烷和石油醚(2∶1)为淋洗剂,柱色谱分离,产率为70%,MALDI-TOFMS(m/z):C94H124N2O4S12[M]+,理论值:1728.62;实测值:1728.61。其结构式如下所示:实施例26氩气保护下,在盛有双(醛基三噻吩)噻咯(170mg,0.1mmol),十倍摩尔当量的3-乙基洛丹宁(20mg,0.12mmol)和乙酸铵(20mg,0.12mmol),搅拌加热回流过夜。降到室温,倒入200mL水中,加入80mL二氯甲烷萃取,有机相加入50mL水洗(三次)。有机相无水硫酸镁干燥,过滤,旋干,以二氯甲烷和石油醚(1∶1)为淋洗剂,柱色谱分离,得褐色固体,产率为82%,MALDI-TOFMS(m/z):C92H124N2O2S13Si[M]+,理论值:1701.61;实测值:1701.60。其结构式如下所示:实施例27方法同实施例2。用丙二睛代替氰基乙酸乙酯,产率为83%。MALDI-TOFMS(m/z):C106H148N2O4S8[M]+,理论值:1400.69;实测值:1400.69。其结构式如下所示:氩气保护下,在盛有中间体HOC3T(BDT)3TCHO(1.0g,0.71mmol),十倍摩尔当量的腈基乙酸辛酯和70mL干燥三氯甲烷双口瓶中滴入几滴三乙胺,室温40小时。倒入100mL水中,二氯甲烷(50mL×3)萃取。有机相依次用水(100mL),饱和食盐水(100mL)和水(100mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚-二氯甲烷=2∶3为洗脱剂,过柱分离,得到黑色固体,产率为70%。MALDI-TOFMS(m/z):C106H148N2O4S8[M]+,理论值:1768.92;实测值:1768.93。其结构式如下所示:实施例28氩气保护下,在盛有(360mg,0.2mmol)3T(OEH-BDT)3T(CHO)2,十倍摩尔当量的腈基乙酸辛酯和100mL干燥三氯甲烷双口瓶中滴入几滴三乙胺,室温搅拌过夜。倒入100mL水中,二氯甲烷(30mL×3)萃取。有机相依次用水(60mL),饱和食盐水(60mL)和水(60mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚-二氯甲烷=1∶2为洗脱剂,过柱分离,得到褐色固体,产率为91%。MALDI-TOFMS(m/z):C106H148N2O4S8[M]+,理论值:1800.91;实测值:1800.90。其结构式如下所示:实施例29氩气保护下,在盛有中间体5T(OEH-BDT)5T(CHO)2(0.18g,0.08mmol),十倍摩尔当量的腈基乙酸辛酯和60mL干燥三氯甲烷双口瓶中滴入几滴三乙胺,室温搅拌48小时。倒入100mL水中,二氯甲烷(50mL×3)萃取。有机相依次用水(80mL),饱和食盐水(80mL)和水(80mL)洗,无水硫酸钠干燥。减压除去溶剂,以石油醚-二氯甲烷=2∶3为洗脱剂,过柱分离,得到黑色固体,产率为76%。MALDI-TOFMS(m/z):C154H220N2O6S12[M]+,理论值:2577.36;实测值:2577.35。其结构式如下所示:实施例30与实施例14的合成方法类似,只是用3-辛基洛丹宁替代3-乙基洛丹宁,反应收率90%,MALDI-TOFMS(m/z):C100H145N2O2S11[M]+,理论值:1758.83;实测值:1758.82。其结构式如下所示:实施例31与实施例14的合成方法类似,只是用3-甲基洛丹宁替代3-乙基洛丹宁,反应收率96%,MALDI-TOFMS(m/z):C86H118N2O2S11[M]+,理论值:1562.61;实测值:1562.62。其结构式如下所示:实施例32与实施例14的合成方法类似,只是用3T(DPP)3T(CHO)2代替7T(CHO)2,反应收率76%,MALDI-TOFMS(m/z):C82H105N3O2S11[M]+,理论值:1547.50;实测值:1547.52。其结构式如下所示:实施例33方法同实施例2,只是用3T(DPP)3T(CHO)2代替7T(CHO)2,反应收率76%,MALDI-TOFMS(m/z):C82H105N3O2S11[M]+,理论值:1547.50;实测值:1547.52。其结构式如下所示:实施例34与实施例14的合成方法类似,只是用5T(CHO)2代替7T(CHO)2,反应收率96%,MALDI-TOFMS(m/z):C65H90N2O2S9[M]+,理论值:1218.44;实测值:1218.45。其结构式如下所示:实施例35与实施例14合成方法类似,3-(1-乙酰乙酯)甲基洛丹宁替代3-乙基洛丹宁,反应收率87%,MALDI-TOFMS(m/z):C92H126N2O6S11[M]+,理论值:1706.65;实测值:1706.64。其结构式如下所示:实施例36方法同实施例2。用1-璜酰丁基-2,3,3-三甲基吲哚内盐代替氰基乙酸乙酯,反应收率87%,MALDI-TOFMS(m/z):C106H146N2O6S9[M]+,理论值:1830.86;实测值:1830.87。其结构式如下所示:实施例37与实施例14的合成方法类似,用二醛基苯并噻唑二噻吩代替7T(CHO)2,反应收率79%,MALDI-TOFMS(m/z):C42H50N4O2S7[M]+,理论值:866.19;实测值:866.19。其结构式如下所示:实施例38实施例2到6中的含受体端基的给受体型寡聚噻吩的热稳定性测试含受体端基的给受体型寡聚噻吩的热稳定性用热重分析(TG)在TAinstrumentSDT-TGQ600热重分析仪上进行,差示扫描量热(DSC)在TAinstrumentDSC-2910分析仪上进行分析。氮气流下加热扫描速率为10℃/min。实施例39实施例4到6中化合物的循环伏安法测试通过循环伏安测试可以了解分子的能级结构,以估算最高占有轨道(HOMO)和最低空轨道(LUMO)的值的大小。我们采用LK98BII电化学工作站进行电化学性质的测试,电解池为三电极体系(玻碳电极为工作电极,铂丝电极为辅助电极,甘汞电极为参比电极),以二茂铁做内标,干燥过的二氯甲烷为溶剂,0.1M的四丁基六氟磷酸胺(n-Bu4NPF6)为支持电解质,扫描速度为100mVs-1。在氩气保护下,扫描得到的循环伏安曲线如附图3所示。按参考文献(Li,Y.F.;Cao,Y.;Gao,J.;Wang,D.L.;Yu,G.;Heeger,A.J.Synth.Met.1999,99,243.)换算得到分子的HOMO和LUMO能级:实施例4:E(HOMO)=-5.09eV,E(LUMO)=-3.33eV。实施例5:E(HOMO)=-5.13eV,E(LUMO)=-3.29eV。实施例6:E(HOMO)=-5.10eV,E(LUMO)=-3.26eV。实施例40以实施例4到6中的化合物为电子给体的太阳能电池器件的制备器件结构为ITO/PEDOT:PSS/donor:PC61BM/Ca/Al,其中donor为实施例4到6中的化合物中的任何一种。具体制备过程为:首先将ITO(氧化铟锡,阳极)玻璃进行预处理,具体步骤如下:首先用清洗剂擦洗ITO玻璃,去离子水冲洗干净,然后将ITO玻璃依次用丙酮、异丙醇溶剂超声清洗各20分钟,取出后放入烘箱中烘干。然后再预处理过的ITO玻璃上旋涂一层PEDOT:PSS(BaytronPVPAl4083)作为阳极修饰层(40nm),待PEDOT:PSS在120℃加热20分钟完全干燥后,将donor:PC61BM混合物的氯仿溶液(donor:PC61BM质量比为1∶0.5,donor浓度为8mg/mL)旋涂在PEDOT:PSS表面作为活性层(140nm),然后再蒸镀Ca(20nm)及金属电极Al(80nm)。在蒸镀过程中保持真空度低于4×10-4Pa。在标准太阳光(AM1.5G)辐照条件下,使用计算机控制的Keithley2400数字源表对器件性能进行测试。器件的电流密度-电压曲线如附图4所示,性能参数列于表1。表1:实施例4、5、6材料制备的化合物太阳能电池性能比较(光强为100mW/cm2AM1.5G照射条件下测量)由表1可知,利用本申请的化合物制备的溶液处理的本体异质结太阳能电池器件的紫外可见吸收可以达到800nm,太阳能器件开路电压达到0.85V以上,短路电流达到9mA/cm2以上,最大光电转换效率可达到5%以上。实施例41实施例14中的含受体端基的给受体寡聚噻吩的紫外可见光谱测试将实施例14的化合物分别配成10-5和10-2mol/L的氯仿溶液,前者溶液测量溶液紫外吸收,后者溶液于1200rpm在石英片上甩膜,测量膜的紫外吸收,扫描范围均为300-1000nm,测量仪器为JascoV-570UV/VIS/NIRSpectrophotometer。紫外可见吸收光谱如图5所示。实施例42实施例14中的化合物的溶液和膜的循环伏安法测试通过循环伏安测试可以了解分子的能级结构,以估算最高占有轨道(HOMO)和最低空轨道(LUMO)的值的大小。我们采用LK98BII电化学工作站进行电化学性质的测试,电解池为三电极体系(玻碳电极为工作电极,铂丝电极为辅助电极,甘汞电极为参比电极),溶液法以二茂铁做内标,干燥过的二氯甲烷为溶剂,0.1M的四丁基六氟磷酸胺(n-Bu4NPF6)为支持电解质,扫描速度为100mV.s-1;膜的电化学以干燥的乙腈为溶剂,0.1M的四丁基六氟磷酸胺(n-Bu4NPF6)为支持电解质,将实施例14的化合物的溶液滴到玻碳电极上成膜测量。均在氩气保护下,扫描得到的循环伏安曲线如图6所示。按参考文献(Li,Y.F.;Cao,Y.;Gao,J.;Wang,D.L.;Yu,G.;Heeger,A.J.Synth.Met.1999,99,243.)换算得到实施例14的化合物的溶液和膜的HOMO和LUMO能级:实施例14的化合物(溶液中)E(HOMO)=-5.00eVE(LUMO)=-3.28eV实施例14的化合物(膜中)E(HOMO)=-5.21eVE(LUMO)=-3.74eV实施例43以实施例14中的化合物为电子给体的太阳能电池器件的制备器件结构为ITO/PEDOT:PSS/donor:PC61BM/LiF/Al,其中donor为实施例14的化合物。具体制备过程为:首先将ITO(氧化铟锡,阳极)玻璃进行预处理,具体步骤如下:首先用清洗剂擦洗ITO玻璃,去离子水冲洗干净,然后将ITO玻璃依次用丙酮、异丙醇溶剂超声清洗各20分钟,取出后放入烘箱中烘干。然后再预处理过的ITO玻璃上旋涂一层PEDOT:PSS(BaytronPVPAl4083)作为阳极修饰层(40nm),待PEDOT:PSS在140℃加热20分钟完全干燥后,冷却后将实施例14的化合物:PC61BM混合物的氯仿溶液(实施例14的化合物:PC61BM质量比分别为1∶0.8,1∶0.5,1;0.3,实施例14的化合物浓度为8mg/mL)旋涂在PEDOT:PSS表面作为活性层(80nm),然后蒸镀LiF(0.8nm)及金属电极Al(60nm)。在蒸镀过程中保持真空度低于3×10-4Pa。在标准太阳光(AM1.5G)辐照条件下,使用计算机控制的Keithley2400数字源表对器件性能进行测试。器件的电流密度-电压曲线如图7所示,性能参数列于表2。表2:实施例14中化合物以不同给受体比制备的太阳能电池性能比较(光强为100mW/cm2AM1.5G照射条件下测量)由表2可知,利用本发明的化合物制备的溶液处理的本体异质结太阳能电池器件的紫外可见吸收可以达到780nm,太阳能器件开路电压达到0.90V以上,短路电流达到14mA/cm2以上,最大光电转换效率可达到6%以上。实施例44以实施例25中的化合物为电子给体的太阳能电池器件的制备与测试器件结构为ITO/PEDOT:PSS/91:PC71BM/LiF/Al。ITO玻璃的清洗处理以及PSS-PEDOT旋涂与实施例43相同。待PEDOT:PSS在140℃加热20分钟完全干燥后,冷却后将91∶PC71BM混合物的氯仿溶液(91∶PC71BM质量比分别为1∶0.6,1∶0.8,1∶0.1)旋涂在PEDOT:PSS表面作为活性层(80nm),然后蒸镀LiF(0.8nm)及金属电极Al(60nm)。在蒸镀过程中保持真空度低于3×10-4Pa。在标准太阳光(AM1.5G)辐照条件下,使用计算机控制的Keithley2400数字源表对器件性能进行测试。器件的电流密度-电压曲线如图8所示,性能参数列于表3。表3:实施例25中化合物以不同给受体比制备的太阳能电池性能比较(光强为100mW/cm2AM1.5G照射条件下测量)另外,上述器件活性层混合过程中加入少量的聚二甲基硅氧烷(polydimethylsiloxane,PDMS)可以使得能量转换效率进一步提高到7.46%,电流密度-电压曲线曲线见图9,具体数值见表4。表4:以实施例25中化合物-C71PCBM(1∶0.8)为活性层时添加不同量PDMS的太阳能电池性能比较(光强为100mW/cm2AM1.5G照射条件下测量)实施例45实施例13、17、21、23、24、27、28、29、30、34和35中化合物作为电子给体的有机太阳能电池器件的制备ITO玻璃的清洗处理以及PSS-PEDOT旋涂与实施例43相同。待PEDOT:PSS在140℃加热20分钟完全干燥后,冷却后将donor:PC61BM混合物的氯仿溶液旋涂在PEDOT:PSS表面作为活性层,然后蒸镀LiF(0.8nm)及金属电极Al(60nm)。在蒸镀过程中保持真空度低于3×10-4Pa。在标准太阳光(AM1.5G)辐照条件下,使用计算机控制的Keithley2400数字源表对器件性能进行测试。器件的电流密度-电压曲线如图10-21所示,性能参数列于表5。表5:实施例13、17、21、23、24、27、28、29、30、34和35中化合物作为给体制备的有机太阳能电池性能参数(光强为100mW/cm2AM1.5G照射条件下测量)由上可知,利用本发明的化合物制备的溶液处理的本体异质结太阳能电池器件最大光电转换效率可达到7%以上。并且本发明的化合物具有精确的分子量、结构可控、易纯化,适用于制备具有高开路电压、稳定性好、柔性、大面积的高性能有机太阳能电池。从前述中可以理解,尽管为了示例性说明的目的描述了本发明的具体实施方案,但是在不偏离本发明的精神和范围的条件下,本领域所述技术人员可以作出各种变形或改进。这些变形或修改都应落入本申请所附权利要求的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1