盐酸厄罗替尼关键中间体的制备方法与流程
本发明涉及医药化学领域,具体的说涉及一种盐酸厄罗替尼关键中间体2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法。
背景技术:
盐酸厄罗替尼(erlotinibhydrochloride)商品名为特罗凯(tarceva),是由美国osipharmaceuticals开发,后经罗氏制药公司分别于2004年11月、2005年9月和2006年4月在美国、欧洲和中国批准上市的替尼类抗肿瘤药物。作为一种小分子化合物,盐酸厄罗替尼属于表皮生长因子受体(egfr)家族络氨酸激酶抑制剂,适用于既往接受过至少一个化疗方案失败后的局部晚期或转移的非小细胞肺癌(nsclc)的治疗。盐酸厄洛替尼还是一种靶向药物制剂,能够瞄准特定的病变部位,在目标部位处蓄积或释放有效成分,形成相对较高的浓度,从而在提高药效的同时降低毒副作用,减少对正常组织、细胞的伤害,具有优良的抗肿瘤活性。
在盐酸厄罗替尼的合成工艺中,化合物“2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯”是一个关键中间体,对于它的合成方法有较多的文献报道,主要分为两类。
第一类合成方法以美国辉瑞公司(pfizerinc.)报道的合成方法(us5747498)和cancerreaearchtechnology,ltd.报道的合成方法(wo2009053694)为代表,其合成路线如下:
该方法以乙酸为溶剂,浓硝酸为硝化剂进行硝化反应,反应时间24~48小时。jyothiprasad、nageshwarrao等在专利wo2007060691a2中描述到该硝化方法在放大生产时,反应的收率和纯度都会大幅下降,我们在重复该专利方法 时同样也发现了该问题。此外,我们还发现该硝化方法即使在3,4-二甲氧乙氧基苯甲酸乙酯的投料量较小时,反应速率也非常慢,在10g规模级的反应时,反应进行48小时,依然无法完全转化,并且该方法的转化率不高,反应时间过长,因此,该方法难以用于工业化大生产。
第二合成方法以ubeindustries,ltd.报道的合成方法(ep1650196)为代表,其合成路线如下:
该方法以乙酸作溶剂,浓硝酸(3.0eq)和浓硫酸(0.5eq)组成的混酸做硝化试剂进行硝化反应,反应时间为2小时。重复该硝化方法时发现该硝化条件太强而导致三个较为严重的问题,1.由于浓硫酸的量较大,浓硫酸加入反应液时极易造成原料3,4-二(2-甲氧乙氧基)的碳化;2.滴加硝酸时的反应温度难以控制,温度极易迅速升高至100℃以上,导致副反应急剧增加,使该反应的纯度降低至60%,收率降低至45%;3.由于使用的浓硫酸量很大,导致该方法在处理废液时需消耗大量的碱水,产生大量的废水,增加废液处理成本也不绿色环保。上述三个原因导致该硝化方法不适合用于进行工业化大生产。
技术实现要素:
本发明的目的在于解决现有技术的不足,旨在提供一种制备盐酸厄罗替尼关键中间体“2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯”的方法。本发明提供的技术方案如下。
本发明提供了一种制备盐酸厄罗替尼关键中间体2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的方法,所述方法包括催化量浓硫酸存在条件下以乙酸和乙酸酐为溶剂将3,4-二(甲氧乙氧基)苯甲酸乙酯与硝酸反应。
优选的,本发明所述的2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法,浓硫酸的用量与3,4-二(甲氧乙氧基)苯甲酸乙酯摩尔比为0.01~0.2:1,优选0.01~0.1:1,更优选0.01~0.05:1。
优选的,发明所述的2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法,以乙酸和乙酸酐混合物为溶剂,乙酸与乙酸酐的体积比为100:1~1:100,优选乙 酸:乙酸酐比例为10:1~1:10,更优选乙酸:乙酸酐比例为10:1~1:1。
优选的,本发明所述的2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法,以乙酸和乙酸酐混合物为溶剂,与3,4-二(甲氧乙氧基)苯甲酸乙酯体积质量为1~10:1,优选比例为3~4:1。
除非有另外申明,本发明所使用的体积质量比的单位为l/kg。
优选的,本发明所述的2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法,以硝酸为硝化剂,优选硝酸的浓度66%。
优选的,本发明所述的2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的制备方法,以硝酸为硝化剂,其用量与3,4-二(甲氧乙氧基)苯甲酸乙酯摩尔比为1.5~10:1,优选为2~5:1。
优选的,本发明所述的“2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯”的制备方法,在10~100℃的反应温度条件进行硝化反应,优选的温度范围为35~55℃。
本发明的优势在于,通过创造性的选择冰醋酸和乙酸酐为溶剂,解决了冰醋酸在低温时易结冰的问题,生产可操作性更强;并通过对二者体积比的优化,大幅度的减少了浓硫酸的用量,反应条件温和;反应速率快,缩短了反应时间,在2小时内可使反应进行完全。由于浓硫酸的用量大幅减少,防止原料发生碳化,产物收率和纯度达到98%以上;另一方面,反应结束处理废液时,可以减少碱液的用量,进而减少废液的产生,绿色环保,节约生产成本,更适合工业化大生产。本发明所述的方法,放大至20kg级别规模时,仍然可以使反应时间在2小时内使反应转化率达到98%以上,收率达到96%以上,易于进行工业化生产。
附图说明
图1为2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的1hnmr图谱;
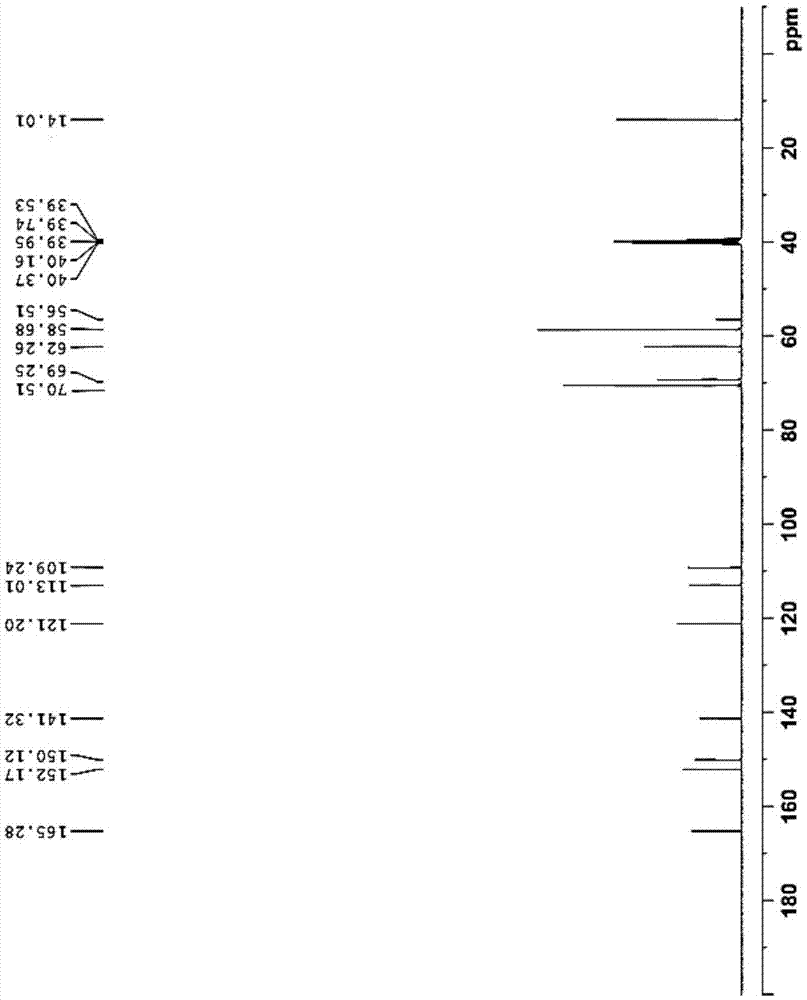
图2为2-硝基-4,5-二(2-甲氧乙氧基)苯甲酸乙酯的13cnmr图谱。
具体实施方式
为体现本发明的技术方案及其所取得的效果,下面将结合具体实施例对本发明做进一步说明,但本发明的保护范围并非局限于具体实施例。
实例一
将冰乙酸(70l)、乙酸酐(10l)及3,4-二(2-甲氧乙氧基)苯甲酸乙酯(23kg,77.1mol)加入200l反应釜内,搅拌下缓慢滴加硝酸(12l,269.85mol)。另取浓硫酸(200ml,3.85mol)稀释于冰醋酸(2l)中。待硝酸滴加完毕,将稀释好的浓硫酸的冰乙酸液缓慢滴加入反应釜中,滴毕,升温至40℃反应2小时。反应毕,将反应液降温至20℃,搅拌下缓慢倾入冰水(80l)和二氯甲烷(80l)的混合液中,分出下层的二氯甲烷层,上层的水层加入二氯甲烷(80l)萃取1次,放出下层的有机层,合并有机相。有机相依次用纯化水(80l)、碳酸氢钠水液(100l)、饱和食盐水(100l)洗涤,分出下层有机层。有机层用无水硫酸钠(20kg)干燥3小时,甩虑,滤液50℃减压浓缩得浅棕黄色油状物25.9kg,收率:98%,hplc:99.39%。其1hnmr图谱基本与图1相同,13c-nmr图谱基本与图2相同。
1hnmr(500mhz,dmso-d6,ppm)δ:1.267-1.302(t,3h,j=7.2),3.309(s,6h),3.699-3.721(t,4h,j=4.4),4.273-4.313(m,6h,j=4.8,7.3),7.344(s,1h),7.674(s,1h)。
实例二
将冰乙酸(80l)、乙酸酐(15l)及3,4-二(2-甲氧乙氧基)苯甲酸乙酯(27kg,90.5mol)加入200l反应釜内,搅拌下缓慢滴加硝酸(14l,316.75mol)。另取浓硫酸(10ml,0.18mol)稀释于冰醋酸(1l)中。待硝酸滴加完毕,将稀释好的浓硫酸的冰乙酸液缓慢滴加入反应釜中,滴毕,升温至40℃反应2小时。反应毕,将反应液降温至20℃,搅拌下缓慢倾入冰水(80l)和二氯甲烷(80l)的混合液中,分出下层的二氯甲烷层,上层的水层加入二氯甲烷(80l)萃取1次,放出下层的有机层,合并有机相。有机相依次用纯化水(80l)、碳酸氢钠水液(100l)、饱和食盐水(100l)洗涤,分出下层有机层。有机层用无水硫酸钠(20kg)干燥3小时,甩虑,滤液50℃减压浓缩得浅棕黄色油状物26.0kg,收率:98.3%,hplc:99.64%。其1hnmr图谱基本与图1相同,13c-nmr图谱基本与图2相同。
实例三
将冰乙酸(80l)、乙酸酐(15l)及3,4-二(2-甲氧乙氧基)苯甲酸乙酯(25kg,83.8mol)加入200l反应釜内,搅拌下缓慢滴加硝酸(13l,293.3mol)。另取浓硫 酸(460ml,8.4mol)稀释于冰醋酸(1l)中。待硝酸滴加完毕,将稀释好的浓硫酸的冰乙酸液缓慢滴加入反应釜中,滴毕,升温至40℃反应2小时。反应毕,将反应液降温至20℃,搅拌下缓慢倾入冰水(80l)和二氯甲烷(80l)的混合液中,分出下层的二氯甲烷层,上层的水层加入二氯甲烷(80l)萃取1次,放出下层的有机层,合并有机相。有机相依次用纯化水(80l)、碳酸氢钠水液(100l)、饱和食盐水(100l)洗涤,分出下层有机层。有机层用无水硫酸钠(20kg)干燥3小时,甩虑,滤液50℃减压浓缩得浅棕黄色油状物27.9kg,收率:96.3%,hplc:99.0%。其1hnmr图谱基本与图1相同,13c-nmr图谱基本与图2相同。
- 还没有人留言评论。精彩留言会获得点赞!