头孢菌素酰化酶突变体及其编码基因与应用的制作方法
本发明涉及生物
技术领域:
,具体涉及头孢菌素酰化酶突变体及其编码基因与应用。
背景技术:
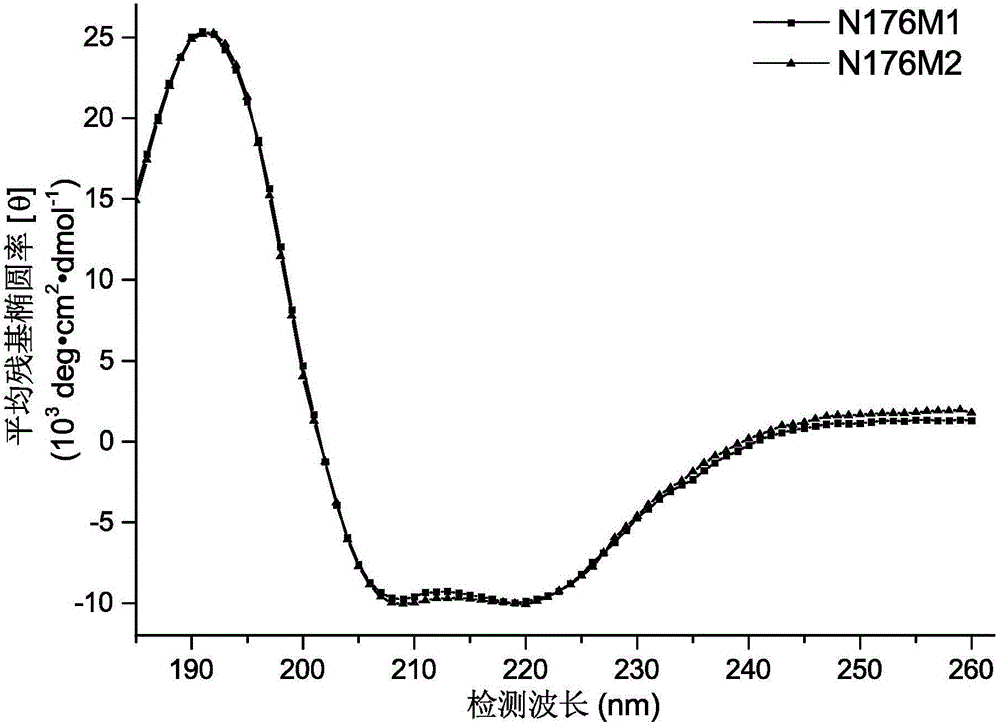
:β-内酰胺类抗生素作为医药行业中最广泛使用的抗生素,每年产量达3万吨,占整个抗生素市场的65%。在抗生素生产过程中,抗生素母核的获得至关重要,如6-氨基青霉烷酸(6-aminopenicillanicacid,6-APA)和7-氨基头孢烷酸(7-aminocephalosporanicacid,7-ACA)是合成β-内酰胺类抗生素的起点。通常,抗生素母核通过由细菌发酵得到的天然抗生素水解得到,如6-APA可由青霉素G(PenicillinG,PG)水解得到,7-ACA可由头孢菌素C(CephalosporinC,CPC)水解得到。以7-ACA作为母核的头孢类抗生素是一类高效、低毒、临床广泛应用的β-内酰胺类抗生素。但是,传统的化学法制备7-ACA工艺流程复杂,通常都需要活化、缩合、保护和去保护的过程,而且反应条件苛刻,能耗高,容易引入杂质。相比而言,生物法采用酶作为催化剂,在水环境中进行反应,条件温和,无需中间产物的提纯过程,且由于酶具有很好的特异性和选择性,并不要求使用纯的反应物,从而大大降低工艺的复杂性和生产成本,因此生物法在β-内酰胺类抗生素工业生产中逐渐成为主导方法。目前工业中广泛采用的CPC水解工艺是由D-氨基酸氧化酶(D-aminoacidoxidase,DAAO)和戊二酰基转移酶(glutaryl-7-ACAacylase,GA)混合的两步酶法。反应步骤为:首先DAAO将CPC氧化成α-酮己二酰-7-氨基头孢烷酸(AKA-7-ACA),并产生过氧化氢和氨,随后过氧化氢迅速与AKA-7-ACA反应生成戊二酰基-7-氨基头孢烷酸(glutaryl-7-aminocephalosporanicacid,GL-7-ACA);最后,GA将GL-7-ACA水解得到7-ACA。由于两步酶法水解CPC的第一步反应中会生成副产物过氧化氢,对酶的稳定性造成不可忽视的影响,造成酶的使用批次降低。因此,探索一步酶法直接催化CPC水解的研究对工业生产意义重大。但是,实现一步酶法水解CPC制备7-ACA并不容易。目前,自然界中尚未发现以CPC为天然底物的酶,而已知能够水解CPC的酰化酶均以GL-7-ACA为天然底物,其催化CPC水解的活性和特异性都非常低,远远达不到工业生产的要求。例如,从假单胞菌N176中提取的戊二酰基转移酶,对CPC的水解活性只有对其天然底物GL-7-ACA的1/250(二者的Vmax/Km分别为0.06和15.1U·mg-1·mM-1)(LiY,ChenJ,JiangW,MaoX,ZhaoG,WangE.Invivopost-translationalprocessingandsubunitreconstitutionofcephalosporinacylasefromPseudomonassp.130.EuropeanJournalofBiochemistry1999;262:713-719.),尽管如此,该酶也已经是天然酶中对CPC活性较高的了。在过去二十多年的研究中,不断有学者测定出新的头孢菌素酰化酶的晶体结构,并对已有基因序列进行突变,以此改善对CPC的水解活性,使其能够应用于β-内酰胺类抗生素的工业生产中来。1991年,Aramori等人成功测定了头孢菌素酰化酶N176的基因序列,其属于第III类头孢菌素酰化酶。1995年,Ishii等人基于已有的头孢菌素酰化酶N176的基因序列,得到了CPC催化活性相对于天然酶提高了1.7倍的单点突变Metβ31Phe,对头孢菌素C的水解活性(Vmax/Km)为0.06U·mg-1·mM-1。2005年,Pollegioni等人通过易错PCR突变、分子建模方法、饱和突变及定向进化等方法,在引入已有突变Metβ31Phe的基础上,进一步筛选出了4倍于起始突变体(单点突变Metβ31Phe)催化活性的两点突变Hisβ57Ser/Hisβ70Ser,对头孢菌素C的水解活性(Vmax/Km)为0.227U·mg-1·mM-1。但是,上述突变体中,酶的催化活性远远达不到工业生产的要求,酶的稳定性也较差。技术实现要素:本发明所要解决的技术问题是如何生产7-氨基头孢烷酸。为解决上述技术问题,本发明首先提供了一种蛋白质。本发明所提供的蛋白质,可为如下a1)或a2)或a3)或a4)或a5)或a6)或a7):a1)氨基酸序列是序列表中序列2所示的蛋白质;a2)在序列表中序列2所示的蛋白质的N端或/和C端连接标签得到的融合蛋白质;a3)氨基酸序列是序列表中序列4所示的蛋白质;a4)氨基酸序列是序列表中序列6所示的蛋白质;a5)在序列表中序列6所示的蛋白质的N端或/和C端连接标签得到的融合蛋白质;a6)氨基酸序列是序列表中序列8所示的蛋白质;a7)将a1)或a2)或a3)或a4)或a5)或a6)所示的蛋白质经过一个或几个氨基酸残基的取代和/或缺失和/或添加得到的具有相同功能的蛋白质。序列表中序列2可由774个氨基酸残基组成。序列表中序列4可由784个氨基酸残基组成。序列表中序列6可由774个氨基酸残基组成。序列表中序列8可由784个氨基酸残基组成。为了使a1)或a4)中的蛋白质便于纯化,可在序列表中序列2或序列表中序列6所示的蛋白质的氨基末端或羧基末端连接上如表1所示的标签。表1.标签的序列标签残基序列Poly-Arg5-6(通常为5个)RRRRRPoly-His2-10(通常为6个)HHHHHHFLAG8DYKDDDDKStrep-tagII8WSHPQFEKc-myc10EQKLISEEDL上述a7)中的蛋白质,所述一个或几个氨基酸残基的取代和/或缺失和/或添加为不超过10个氨基酸残基的取代和/或缺失和/或添加。上述a7)中的蛋白质可人工合成,也可先合成其编码基因,再进行生物表达得到。上述a7)中的蛋白质的编码基因可通过将序列表中序列1自5’末端起第9至2330位、序列表中序列3自5’末端起第3至2354位、序列表中序列5自5’末端起第9至2330位或序列表中序列7自5’末端起第3至2354位所示的DNA序列中缺失一个或几个氨基酸残基的密码子,和/或进行一个或几个碱基对的错义突变,和/或在其5’端和/或3’端连上表1所示的标签的编码序列得到。编码所述蛋白质的核酸分子也属于本发明的保护范围。编码所述蛋白质的核酸分子,具体可为如下b1)或b2)或b3)或b4)或b5)或b6)所示的DNA分子:b1)核苷酸序列是序列表中序列1自5’末端起第9至2330位所示的DNA分子;b2)核苷酸序列是序列表中序列3自5’末端起第3至2354位所示的DNA分子;b3)核苷酸序列是序列表中序列5自5’末端起第9至2330位所示的DNA分子;b4)核苷酸序列是序列表中序列7自5’末端起第3至2354位所示的DNA分子;b5)与b1)或b2)或b3)或b4)限定的核苷酸序列具有75%或75%以上同一性,且编码权利要求1所述蛋白质的DNA分子;b6)在严格条件下与b1)或b2)或b3)或b4)限定的核苷酸序列杂交,且编码权利要求1所述蛋白质的DNA分子。其中,所述核酸分子可以是DNA,如cDNA、基因组DNA或重组DNA;所述核酸分子也可以是RNA,如mRNA或hnRNA等。序列表中序列1由2336个核苷酸组成,序列表中序列1自5’末端起第9至2330位的核苷酸编码序列表中序列2所示的氨基酸序列。序列表中序列3由2357个核苷酸组成,序列表中序列3自5’末端起第3至2354位的核苷酸编码序列表中序列4所示的氨基酸序列。序列表中序列5由2336个核苷酸组成,序列表中序列5自5’末端起第9至2330位的核苷酸编码序列表中序列6所示的氨基酸序列。序列表中序列7由2357个核苷酸组成,序列表中序列7自5’末端起第3至2354位的核苷酸编码序列表中序列8所示的氨基酸序列。本领域普通技术人员可以很容易地采用已知的方法,例如定向进化和点突变的方法,对本发明的编码所述蛋白质的核苷酸序列进行突变。那些经过人工修饰的,具有与本发明分离得到的所述蛋白质的核苷酸序列75%或者更高同一性的核苷酸,只要编码所述蛋白质,均是衍生于本发明的核苷酸序列并且等同于本发明的序列。这里使用的术语“同一性”指与天然核酸序列的序列相似性。“同一性”包括与本发明的编码序列表的序列2、序列表的序列4、序列表的序列6或序列表的序列8所示的氨基酸序列组成的蛋白质的核苷酸序列具有75%或更高,或80%或更高,或85%或更高,或90%或更高,或95%或更高同一性的核苷酸序列。同一性可以用肉眼或计算机软件进行评价。使用计算机软件,两个或多个序列之间的同一性可以用百分比(%)表示,其可以用来评价相关序列之间的同一性。含有所述编码所述蛋白质的核酸分子的表达盒、重组载体、重组微生物或转基因细胞系也属于本发明的保护范围。所述重组载体可为将所述蛋白质的编码基因插入出发质粒,得到的重组质粒。所述出发质粒可为克隆载体。所述克隆载体可为载体pET28a(+)。所述重组载体可为将所述蛋白质的编码基因通过多克隆位点插入出发质粒,得到的重组质粒。所述重组载体具体可为将所述蛋白质的编码基因(序列表的序列1自5’末端起第9至2330位所示的DNA分子)插入载体pET28a(+)的NcoI和XhoI识别位点之间得到的重组质粒pET28a-N176M1。所述重组载体具体可为将所述蛋白质的编码基因(序列表的序列5自5’末端起第9至2330位所示的DNA分子)插入载体pET28a(+)的NcoI和XhoI识别位点之间得到的重组质粒pET28a-N176M2。所述重组微生物可为将所述重组载体导入出发微生物得到重组菌。所述重组微生物具体可为将所述重组质粒pET28a-N176M1导入出发微生物得到重组菌。所述重组微生物具体可为将所述重组质粒pET28a-N176M2导入出发微生物得到重组菌。所述出发微生物可为酵母、细菌、藻类或真菌。所述细菌可为大肠杆菌。所述大肠杆菌具体可为大肠杆菌BL21(DE3)。所述转基因植物细胞系均不包括繁殖材料。所述蛋白质的应用也属于本发明的保护范围。所述蛋白质的应用可为如下d1)或d2):d1)作为头孢菌素酰化酶的应用;d2)在制备具有头孢菌素酰化酶功能的产品中的应用。所述蛋白质的应用可为如下e1)或e2):e1)在水解头孢菌素C中的应用;e2)在制备用于水解头孢菌素C的产品中的应用。所述蛋白质的应用可为如下f1)或f2):f1)在生产7-氨基头孢烷酸中的应用;f2)在制备用于生产7-氨基头孢烷酸的产品中的应用。所述f2)中,所述“生产7-氨基头孢烷酸”是以头孢菌素C作为底物的。实验证明,本发明所提供的蛋白质(氨基酸序列是序列表中序列4所示)对头孢菌素C的水解活性(Vmax/Km)为0.228U·mg-1·mM-1,比纯化的对照蛋白1(氨基酸序列是序列表中序列10所示)对头孢菌素C的水解活性提高了60.6%;蛋白质(氨基酸序列是序列表中序列4所示)水解后形成7-氨基头孢烷酸的产量68.67μg/mL,而纯化的对照蛋白1水解后形成7-氨基头孢烷酸的产量为42.65μg/mL。本发明所提供的蛋白质(氨基酸序列是序列表中序列8所示)对头孢菌素C的水解活性(Vmax/Km)为0.363U·mg-1·mM-1,比纯化的对照蛋白2(氨基酸序列是序列表中序列12所示)对头孢菌素C的水解活性提高了59.5%;蛋白质(氨基酸序列是序列表中序列8所示)水解后形成7-氨基头孢烷酸的产量85.32μg/mL,而纯化的对照蛋白2水解后形成7-氨基头孢烷酸的产量为53.66μg/mL。可见,本发明所提供的蛋白质催化头孢菌素C的活性增强、形成7-氨基头孢烷酸的产量更高。本发明所提供的蛋白质在水解头孢菌素C和/或生产7-氨基头孢烷酸中具有重要的应用价值。附图说明图1为N176M1粗酶、N176M2粗酶、N176M1纯酶和N176M2纯酶的SDS-PAGE。图2为重组头孢菌素酰化酶甲和重组头孢菌素酰化酶乙的吸光度曲线。图3为重组头孢菌素酰化酶甲的熔化温度曲线。图4为头孢菌素C酰化酶催化头孢菌素C水解生成7-氨基头孢烷酸的反应式。图5为头孢菌素C酰化酶乙的高效液相色谱检测图谱。具体实施方式下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。以下实施例中的定量试验,均设置三次重复实验,结果取平均值。结合缓冲液A:含500mMNaCl和20mM咪唑的pH8.0、100mMTris-HCl缓冲液。结合缓冲液B:含500mMNaCl和50mM咪唑的pH8.0、100mMTris-HCl缓冲液。洗脱缓冲液:含500mMNaCl和250mM咪唑的pH8.0、100mMTris-HCl缓冲液。载体pET28a(+)为Novagen公司的产品。大肠杆菌BL21(DE3)为宝生物工程(大连)有限公司的产品。层析柱为北京韦氏博慧色谱科技有限公司公司产品,产品目录号为CS-A01S-0B。10kDaUltra-0.5超滤离心管为Millipore公司产品。脱盐柱为北京韦氏博慧色谱科技有限公司产品,产品目录号为CC-G01-50。C18反相柱为日本岛津公司产品,规格为5μm、150×4.6mm。高效液相色谱仪为日本岛津公司产品,型号为LC-20AT。头孢菌素C为石药集团公司产品。7-氨基头孢烷酸为Sigma-Aldrich公司产品,产品目录号为191140。头孢菌素C溶液:用去离子水溶解头孢菌素C至其浓度为20mg/mL。7-氨基头孢烷酸溶液:用去离子水溶解7-氨基头孢烷酸至其浓度为0.1mg/mL。实施例1、重组头孢菌素酰化酶甲和重组头孢菌素酰化酶乙的制备一、重组质粒的构建1、重组质粒pET28a-N176M1的构建(1)人工合成序列表中序列1所示的双链DNA分子1。(2)用限制性内切酶NcoI和XhoI双酶切双链DNA分子1,回收酶切产物1。(3)用限制性内切酶NcoI和XhoI双酶切载体pET28a(+),回收约5300bp的载体骨架1。(4)将酶切产物1与载体骨架1连接,得到重组质粒pET28a-N176M1。根据测序结果,对重组质粒pET28a-N176M1进行结构描述如下:将载体pET28a(+)的NcoI识别序列和XhoI识别序列间的DNA小片段替换为核苷酸序列是序列表的序列1自5’末端起第9至2330位所示的双链DNA分子。重组质粒pET28a-N176M1中,序列表的序列1所示的DNA分子与载体骨架上的His-tag标签(由6个组氨酸残基组成)的编码序列融合,形成序列表的序列3所示的融合基因,表达序列表的序列4所示的具有His-tag标签的融合蛋白。2、重组质粒pET28a-N176M2的构建(1)人工合成序列表中序列5所示的双链DNA分子2。(2)用限制性内切酶NcoI和XhoI双酶切双链DNA分子2,回收酶切产物2。(3)用限制性内切酶NcoI和XhoI双酶切载体pET28a(+),回收约5300bp的载体骨架1。(4)将酶切产物2与载体骨架1连接,得到重组质粒pET28a-N176M2。根据测序结果,对重组质粒pET28a-N176M2进行结构描述如下:将载体pET28a(+)的NcoI识别序列和XhoI识别序列间的DNA小片段替换为核苷酸序列是序列表的序列5自5’末端起第9至2330位所示的双链DNA分子。重组质粒pET28a-N176M1中,序列表的序列5所示的DNA分子与载体骨架上的His-tag标签(由6个组氨酸残基组成)的编码序列融合,形成序列表的序列7所示的融合基因,表达序列表的序列8所示的具有His-tag标签的融合蛋白。3、重组质粒pET28a-d1的构建(1)人工合成序列表中序列9所示的双链DNA分子3。(2)用限制性内切酶NcoI和XhoI双酶切双链DNA分子3,回收酶切产物3。(3)用限制性内切酶NcoI和XhoI双酶切载体pET28a(+),回收约5300bp的载体骨架1。(4)将酶切产物3与载体骨架1连接,得到重组质粒pET28a-d1。根据测序结果,对重组质粒pET28a-d1进行结构描述如下:将载体pET28a(+)的NcoI识别序列和XhoI识别序列间的DNA小片段替换为核苷酸序列是序列表的序列9自5’末端起第9至2330位所示的双链DNA分子。重组质粒pET28a-d1中,序列表的序列9所示的DNA分子与载体骨架上的His-tag标签(由6个组氨酸残基组成)的编码序列融合形成融合基因,表达序列表的序列10所示的具有His-tag标签的融合蛋白。4、重组质粒pET28a-d2的构建(1)人工合成序列表中序列11所示的双链DNA分子4。双链DNA分子4的核苷酸序列记载于如下文献中:PollegioniL,LorenziS,RosiniE,MarconeGL,MollaG,VergaR,CabriW,PiloneMS.EvolutionofanacylaseactiveoncephalosporinC.ProteinSci.2005;14:3064-3076.(2)用限制性内切酶NcoI和XhoI双酶切双链DNA分子4,回收酶切产物4。(3)用限制性内切酶NcoI和XhoI双酶切载体pET28a(+),回收约5300bp的载体骨架1。(4)将酶切产物4与载体骨架1连接,得到重组质粒pET28a-d2。根据测序结果,对重组质粒pET28a-d2进行结构描述如下:将载体pET28a(+)的NcoI识别序列和XhoI识别序列间的DNA小片段替换为核苷酸序列是序列表的序列11自5’末端起第9至2330位所示的双链DNA分子。重组质粒pET28a-d2中,序列表的序列11所示的DNA分子与载体骨架上的His-tag标签(由6个组氨酸残基组成)的编码序列融合形成融合基因,表达序列表的序列12所示的具有His-tag标签的融合蛋白。二、蛋白的表达1、重组头孢菌素酰化酶甲的表达(1)将重组质粒pET28a-N176M1导入大肠杆菌BL21(DE3),得到重组菌,将该重组菌命名为BL21(DE3)-N176M1。(2)取BL21(DE3)-N176M1单克隆,接种至100mLLB液体培养基(含50μg/mL卡那霉素),37℃、200rpm振荡培养7h,得到培养菌液。(3)取培养菌液,按体积比为1:100接种至1LLB液体培养基(含50μg/mL卡那霉素),37℃、200rpm振荡培养至OD600nm值0.6~0.8,然后加入IPTG并使其浓度为0.5mM,28℃、120rpm振荡培养20h,4℃、10000rpm离心10min,收集菌体沉淀。(4)完成步骤(3)后,取菌体沉淀,加入100mLpH8.0、100mM的Tris-HCl缓冲液,重悬后超声破碎(超声波功率600W,循环程序为:破碎4s,停6s,共20min),然后4℃、10000rpm离心10min,收集上清液。(5)完成步骤(4)后,取上清液,4℃、12000rpm离心10min(目的是进一步去除超声破碎带来的细胞杂质),得到重组头孢菌素酰化酶甲的粗酶液,以下简称N176M1粗酶。2、重组头孢菌素酰化酶乙的表达按照上述步骤1的步骤,将重组质粒pET28a-N176M1替换为重组质粒pET28a-N176M2,其它步骤均不变,得到重组头孢菌素酰化酶乙的粗酶液,以下简称N176M2粗酶。3、对照蛋白1的表达按照上述步骤1的步骤,将重组质粒pET28a-N176M1替换为重组质粒pET28a-d1,其它步骤均不变,得到对照蛋白1的粗酶液。4、对照蛋白2的表达按照上述步骤1的步骤,将重组质粒pET28a-N176M1替换为重组质粒pET28a-d2,其它步骤均不变,得到对照蛋白2的粗酶液。三、蛋白的纯化1、重组头孢菌素酰化酶甲的粗酶液的纯化采用镍琼脂糖凝胶为层析柱填料,采用层析柱(规格为20mL)进行纯化,具体步骤如下:(1)先用2~4个柱体积的结合缓冲液A平衡层析柱(流速为2mL/min)。(2)完成步骤(1)后,将N176M1粗酶上样至层析柱(流速为1.5mL/min)。(3)完成步骤(2)后,用结合缓冲液B冲洗层析柱(流速为2mL/min),直至洗脱液在280nm处的吸光值与结合缓冲液B在280nm处的吸光值基本相同。(4)完成步骤(3)后,用洗脱缓冲液冲洗层析柱(流速为2mL/min),收集保留体积为18-40mL的过柱后溶液,再用10kDaUltra-0.5超滤离心管(Millipore公司产品)浓缩,得到浓缩蛋白。(5)完成步骤(4)后,将浓缩蛋白用脱盐柱透析除盐两次(以除去浓缩蛋白中的咪唑和NaCl组分),再置换入pH8.0、100mM的Tris-HCl缓冲液中,得到N176M1纯酶。N176M1纯酶的浓度通过Bradford法检测(使用牛血清蛋白作为标准试剂,检测时使用的吸光度为595nm),然后置于4℃冷藏保存。2、重组头孢菌素酰化酶乙的粗酶液的纯化按照上述步骤1,将N176M1粗酶替换为N176M2粗酶,其它步骤均不变,得到N176M2纯酶。3、对照蛋白1的粗酶液的纯化按照上述步骤1,将N176M1粗酶替换为对照蛋白1的粗酶液,其它步骤均不变,得到纯化的对照蛋白1。4、对照蛋白2的粗酶液的纯化按照上述步骤1,将N176M1粗酶替换为对照蛋白2的粗酶液,其它步骤均不变,得到纯化的对照蛋白2。将上述得到的各酶进行SDS-PAGE,部分实验结果见图1(M为高分子量标准蛋白质)。结果表明,N176M1粗酶、N176M2粗酶、N176M1纯酶、N176M2纯酶、对照蛋白1的粗酶液、对照蛋白2的粗酶液、纯化的对照蛋白1和纯化的对照蛋白2均含有两条分子量条带,且对应分子量为22kDa(α链)和58kDa(β链)。实施例2、蛋白的稳定性检测1、取N176M1纯酶、N176M2纯酶、纯化的对照蛋白1或纯化的对照蛋白2,用脱盐柱透析除盐两次(目的为去除减少对圆二色光谱吸光度信号的干扰),并用10kDaUltra-0.5超滤离心管浓缩,使浓缩后的蛋白样品中蛋白的浓度为0.4-0.5mg/mL。2、取浓缩后的蛋白样品,置于厚度为0.1cm的石英比色皿中,然后在远紫外区(波长为180-260nm)测定圆二色光谱的吸光度信号,以确定其特征吸收峰,得到吸光度曲线。吸光度信号数据用平均残基椭圆率表示,由圆二色谱仪(英国应用光物理公司产品)测定并收集。结果表明(N176M1纯酶和N176M2纯酶的吸光度曲线见图2,其中N176M1为N176M1纯酶,N176M2为N176M2纯酶),N176M1纯酶、N176M2纯酶、纯化的对照蛋白1和纯化的对照蛋白2的特征吸收波长均固定于220nm,温度区间为4-85℃(精度为1℃)。测量过程中的升温速率为1℃/min,并在随后的1min内使目的蛋白样品达到热力学平衡。3、完成步骤2后,使用Pro-DataViewer(英国应用光物理公司产品)分析吸光度曲线,得到浓缩后的蛋白样品的熔化温度(Tm)曲线。结果表明(N176M1纯酶和纯化的对照蛋白1的熔化温度(Tm)曲线见图3),纯化的对照蛋白1的熔化温度为43.8℃,N176M1纯酶的熔化温度为55.5℃,纯化后的对照蛋白2的熔化温度为51.7℃,N176M2纯酶的熔化温度为51.2℃。上述结果表明,与纯化的对照蛋白1相比,N176M1纯酶的热稳定性均有一定程度的提高;N176M2纯酶与纯化的对照蛋白2相比,热稳定性基本保持不变。实施例3、蛋白的催化活性检测头孢菌素酰化酶催化头孢菌素C的生成7-氨基头孢烷酸的反应式如图4所示。为检测实施例1纯化的蛋白对头孢菌素C是否具有催化活性,进行如下实验。一、N176M1纯酶的催化活性检测1、N176M1纯酶的催化活性检测实验重复三次,每次需进行三个平行实验。(1)制备反应体系1a和反应体系1b:反应体系1a(1mL):由pH8.0、100mMTris-HCl缓冲液、头孢菌素C溶液和N176M1纯酶组成;反应体系1a中,头孢菌素C的浓度为0.8mg/mL、1.0mg/mL、1.2mg/mL、2.0mg/mL、3.0mg/mL、5.0mg/mL、7.0mg/mL或10.0mg/mL,N176M1纯酶的浓度为0.1mg/mL。反应体系1b(1mL):由pH8.0、100mMTris-HCl缓冲液和N176M1纯酶组成;反应体系1b中,N176M1纯酶的浓度为0.1mg/mL。作为参照组。(2)将步骤(1)制备的反应体系1a或反应体系1b置于37℃水浴条件下,反应8min。(3)完成步骤(2)后,加入3mL终止液(含15%(体积百分比)乙酸和15mMNaOH的水溶液)终止反应,得到待测溶液。(4)取步骤(3)得到的待测溶液、头孢菌素C溶液和7-氨基头孢烷酸溶液,分别进行HPLC检测。使用配有C18反相柱的高效液相色谱仪进行HPLC检测。流动相由15%(v/v)甲醇(A)、7.5%(v/v)乙腈(B)、1%(v/v)乙酸和76.5%(v/v)的水组成,流速为0.8mL/min。检测波长为280nm。柱温箱为30℃。通过Lineweaver-Burk法将实验数据拟合为经典的米氏方程(Michaelis-Mentenequation)得到酶催化动力学参数Vmax/Km。2、空白对照(1)制备反应体系2反应体系2(1mL):由pH8.0、100mMTris-HCl缓冲液和N176M1纯酶组成;反应体系2中,N176M1纯酶的浓度为0.1mg/mL。(2)将步骤(1)制备的反应体系2置于37℃水浴条件下,反应8min。(3)完成步骤(2)后,加入3mL终止液(含15%(体积百分比)乙酸和15mMNaOH的水溶液)终止反应。(4)完成步骤(3)后,加入500μL的头孢菌素C溶液(含头孢菌素C0.8mg、1.0mg、1.2mg、2.0mg、3.0mg、5.0mg、7.0mg或10.0mg),得到待测溶液a。(5)完成步骤(3)后,加入500μL的pH8.0、100mMTris-HCl缓冲液,得到待测溶液b。作为参照组。(6)取步骤(5)得到的待测溶液a、待测溶液b、头孢菌素C溶液和7-氨基头孢烷酸溶液,分别进行HPLC检测。使用配有C18反相柱的高效液相色谱仪进行HPLC检测。流动相由15%(v/v)甲醇(A)、7.5%(v/v)乙腈(B)、1%(v/v)乙酸和76.5%(v/v)的水组成,流速为0.8mL/min。检测波长为280nm。柱温箱为30℃。通过Lineweaver-Burk法将实验数据拟合为经典的米氏方程(Michaelis-Mentenequation)得到酶催化动力学参数Vmax/Km。3、纯化的对照蛋白1的催化活性检测按照步骤1的方法,将N176M1纯酶替换为纯化的对照蛋白1,其它步骤均不变,得到纯化的对照蛋白1的催化活性检测结果。部分实验结果见图5(横坐标为保留时间,纵坐标为峰面积,1)为步骤1中加入头孢菌素C10.0mg的催化活性检测结果,2)为步骤1中参照组的催化活性检测结果)。在该色谱条件下,头孢菌素C的保留时间为3.2min,7-氨基头孢烷酸的保留时间为2.7min。N176M1纯酶对头孢菌素C的水解活性(Vmax/Km)为0.228U·mg-1·mM-1(三次实验结果平均值),比纯化的对照蛋白1对头孢菌素C的水解活性提高了60.6%,而空白对照中仅能通过水解形成微量的7-氨基头孢烷酸。N176M1纯酶水解后形成7-氨基头孢烷酸的产量68.67μg/mL,而纯化的对照蛋白1水解后形成7-氨基头孢烷酸的产量为42.65μg/mL。结果表明,N176M1纯酶可催化头孢菌素C形成7-氨基头孢烷酸;与纯化的对照蛋白1相比,N176M1纯酶催化头孢菌素C的活性增强,形成7-氨基头孢烷酸的产量更高。二、N176M2纯酶的催化活性检测1、N176M2纯酶的催化活性检测按照步骤一中1的方法,将反应体系1a替换为反应体系1A,反应体系1b替换为反应体系1B,其它步骤均不变,得到N176M2纯酶的催化活性检测结果。反应体系1A(1mL):由pH8.0、100mMTris-HCl缓冲液、头孢菌素C溶液和N176M2纯酶组成;反应体系1a中,头孢菌素C的浓度为0.8mg/mL、1.0mg/mL、1.2mg/mL、2.0mg/mL、3.0mg/mL、5.0mg/mL、7.0mg/mL或10.0mg/mL,N176M2纯酶的浓度为0.1mg/mL。反应体系1B(1mL):由pH8.0、100mMTris-HCl缓冲液和N176M2纯酶组成;反应体系1B中,N176M2纯酶的浓度为0.1mg/mL。作为参照组。2、空白对照按照步骤一中2的方法,将反应体系2替换为反应体系2A,其它步骤均不变,得到空白对照的催化活性检测结果。反应体系2A(1mL):由pH8.0、100mMTris-HCl缓冲液和N176M2纯酶组成;反应体系2A中,N176M2纯酶的浓度为0.1mg/mL。3、按照步骤1的方法,将N176M2纯酶替换为纯化的对照蛋白2,其它步骤均不变,得到纯化的对照蛋白2的催化活性检测结果。实验结果表明,在该色谱条件下,头孢菌素C的保留时间为3.2min,7-氨基头孢烷酸的保留时间为2.7min。N176M2纯酶对头孢菌素C的水解活性(Vmax/Km)为0.363U·mg-1·mM-1(三次实验结果平均值),比纯化的对照蛋白2对头孢菌素C的水解活性提高了59.5%。N176M2纯酶水解后形成7-氨基头孢烷酸的产量85.32μg/mL,而纯化的对照蛋白2水解后形成7-氨基头孢烷酸的产量为53.66μg/mL。结果表明,N176M2纯酶可催化头孢菌素C形成7-氨基头孢烷酸,而空白对照中仅能通过水解形成微量的7-氨基头孢烷酸。与纯化的对照蛋白2相比,N176M2纯酶催化头孢菌素C的活性增强,形成7-氨基头孢烷酸的产量更高。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1