一种埃洛石表面引发硼亲和印迹聚合物吸附剂的制备方法与流程
本发明涉及一种埃洛石表面引发硼亲和印迹聚合物吸附剂的制备方法,属环境功能材料制备技术领域。
背景技术:
分子印迹技术由德国的Wulff在1972年提出的,分子印迹聚合物是具有预定识别功能结合位点三维交联高分子,制备的分子印迹聚合物(MIPs)能对模板分子产生特异性吸附。由于分子印迹技术具有构效预定性、特异识别性和广泛实用性。ATRP(原子自由基聚合)作为分子印迹技术的一种形式,是以简单的有机卤化物为引发剂、过渡金属配合物为卤原子载体,通过氧化还原反应,从而实现对反应的进行控制。埃洛石纳米管(HNTs)它是通过中间的铝、硅胶体凝聚而成的一种粘土质硅酸盐矿物。由于其较大的比表面积、优良的化学稳定性能,埃洛石纳米管可以广泛用于表面引发印迹的基质材料。
木犀草素(LTL)作为黄酮类的一种重要的天然有机化合物。木犀草素具有很大的药用价值,如强抗氧化性、抗菌、抗病毒、抗肿瘤、抗炎等。目前,花生壳中木犀草素常用的分离纯化的方法主要有柱层析法、薄层层析法和大孔树脂吸附分离法等。这些方法虽然各有独特优点,但也各有其局限性,其中共性的缺陷是方法选择性差、纯化成本太高、纯化率低下。因此,构建选择性识别与分离纯化花生壳提取液中木犀草素的新方法、增加产品得率的同时获得较高纯度吸引了很大的关注。
目前,吸附木犀草素的材料很少被研究,最近有带有双重识别功能的石墨烯分子印迹材料用于吸附木犀草素(Shucheng Liu, Jianming Pan, Hengjia Zhu, Guoqing Pan, Fengxian Qiu, Minjia Meng, Juntong Yao, Dong Yuan, Graphene oxide based molecularly imprinted polymers with double recognition abilities: The combination of covalent boronic acid and traditional non-covalent monomers, Chemical Engineering Journal 290 (2016) 220–231.),但是它们无法快速识别和分离木犀草素并且具有差的再生性能。
因此,本工作利用硼亲和占主导的ATRP分子印迹聚合物的特异性识别诱导作用制备出埃洛石表面引发印迹聚合物,最后特异性吸附木犀草素。
技术实现要素:
本发明的目的在于克服现有技术中的缺陷而提供一种埃洛石表面引发硼亲和印迹聚合物吸附剂的制备方法。
本发明首先将埃洛石纳米管包裹了多巴胺,然后引进2-溴异丁酰溴从而接上溴引发剂,再通过ATRP技术接上乙烯基咪唑,然后吸附锌离子,再吸附木犀草素,最后通过ATRP分子印迹技术把氟苯硼酸接到埃洛石表面,最后在甲醇和醋酸(v:v,11:1)的混合溶液中洗脱木犀草素;洗脱后的埃洛石纳米管就可以形成木犀草素留下来的特异性空穴,该印迹聚合物可以用来特异性回收木犀草素。
本发明采用的技术方案是:
(1)制备乙烯基氟苯硼酸 (F-BA):
在冰水浴中,将氟苯硼酸加入到100mol/L的碳酸钠溶液中,然后再将50mmol/L的丙烯酰氯加入到上述冰水浴溶液中,用0.1mol/L的氢氧化钠溶液把pH调至中性(pH=7)反应12小时,真空60℃烘干。
其中步骤(1)中所述的氟苯硼酸和碳酸钠的用量为:2-4g:100-200mL;所述的丙烯酰氯和氟苯硼酸的比例为:3-5 mL:2-4g。
(2)制备羧基化的埃洛石纳米管(HNTs):
块状埃洛石纳米管研磨粉碎过100目筛,在100℃高温下煅烧24h,煅烧后的HNTs被放置在硫酸和盐酸的混合溶液(v:v, 1:3)中并在80℃下搅拌6h;然后用大量的蒸馏水冲洗直至中性,最后在真空60℃下烘干。
其中步骤(2)中所述的埃洛石,硫酸和盐酸的混合溶液的比例为:0.1-0.3 g: 50-70mL。
(3)制备多巴胺修饰后的埃洛石纳米管(HNTs@PDA):
将活化后的埃洛石纳米管放到乙醇和水的混合溶液中(v:v,1:1),再将盐酸多巴胺加到上述溶液中并搅拌5min,然后将预先配好的100mmol/L的三(羟甲基)氨基甲烷溶液(pH为8.5,用0.1mol L-1氢氧化钠调节)逐滴滴加到上述溶液中在25℃下搅拌24小时。
其中步骤(3)中所述的活化后的埃洛石纳米管,盐酸多巴胺,乙醇和水的混合溶液的比例为:100-200 mg:50-150 mg:40-60mL。
其中步骤(3)中所述的三(羟甲基)氨基甲烷溶液和盐酸多巴胺的比例为:40-60 mL:50-150mg。
(4)制备负载溴引发剂的埃洛石纳米管(HNTs@PDA@Br):
将HNTs@PDA 加入到甲醇中,随后加入三乙胺,并搅拌10min,再加入预先配好的2-溴异丁酰溴和N,N-二甲基甲酰胺(v:v, 1:4)的混合溶液,并在氮气保护下25℃搅拌24h。
其中步骤(4)中所述的HNTs@PDA,甲醇,三乙胺,2-溴异丁酰溴和N,N-二甲基甲酰胺混合溶液的比例为:50-150 mg:10-30 mL:1.5-2.0 mL:5-15 mL。
(5)咪唑印迹聚合物的制备(HNTs@PDA@Br@VLD):
将HNTs@PDA@Br加入到甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1),然后通入氮气10min,再加入乙烯基咪唑,氯化亚铜,五甲基二乙烯三胺,然后通入氮气10min,加入预先准备的抗败坏血酸,最后加入甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液(v:v:v, 1:1:1),并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。
其中步骤(5)中所述的HNTs@PDA@Br和甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液的比例为:25-75 mg:2-4mL。
其中步骤(5)中所述的HNTs@PDA@Br,乙烯基咪唑,氯化亚铜,五甲基二乙烯三胺,抗败坏血酸,甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液的比例为:25-75 mg:50-100 mg:0.015-0.02mmol:0.015-0.02mmol:25-75 mg:1-5 mL。
(6)锌离子络合物 (HNTs@PDA@Br@VLD@Zn(Ⅱ)) 的制备:
将HNTs@PDA@Br@VLD加入到乙醇和水的混合溶液中(v:v, 1:1),然后加入六水合硝酸锌,在25℃下搅拌12h后在真空条件下40℃12小时烘干。
其中步骤(6)中所述的HNTs@PDA@Br@VLD, 六水合硝酸锌, 乙醇和水的混合溶液的比例为50-100 mg:0.4-0.6 g:20-30 mL。
(7)预聚合溶液 (HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL) 的制备:
将HNTs@PDA@Br@VLD@Zn(Ⅱ) 加入到乙醇和水的混合溶液中(v:v, 1:1),然后加入木犀草素,在25℃下搅拌12h并在真空40℃12小时烘干。
其中步骤(7)中所述的HNTs@PDA@Br@VLD@Zn(Ⅱ),木犀草素, 乙醇和水的混合溶液的比例为50-100 mg:20-30 mg:20-30 mL。
(8)分子印迹聚合物(MIPs)的制备:
将HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL、氟苯硼酸加入到甲醇,N,N-二甲基甲酰胺,苯甲醚的混合溶液(v:v:v, 1:1:1)中,然后通入氮气10min,再加入氯化亚铜,五甲基二乙烯三胺,然后通入氮气10min,加入预先准备的抗败坏血酸,最后加入甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液(v:v:v, 1:1:1)中并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。得到的产品浸泡在甲醇和醋酸(v:v,11:1)的混合溶液中12小时,然后甲醇洗3次,离心并在60℃下6小时烘干。
其中步骤(8)中所述的HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL, 氟苯硼酸甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液(v:v:v, 1:1:1)的比例为50-100 mg:20-30 mg:4-8 mL。
其中步骤(8)中所述的抗败坏血酸,氯化亚铜,五甲基二乙烯三胺,甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液,甲醇和醋酸的混合溶液的比例为25-75 mg:0.015-0.02 mmol:0.015-0.02 mmol:4-8 mL:20-30 mL。
与现有技术相比较,本发明的有益效果如下:
本发明通过制备的埃洛石表面印迹聚合物来识别回收木犀草素,传统的技术,一般都是单一的识别位点,并且不具有选择性,从而无法高效快速吸附木犀草素。本发明的方法使用了双重引发技术(ATRP)的概念,通过层层修饰引入双重识别位点。制备的材料中埃洛石表面印迹聚合物具有硼酸特异性识别基团和锌离子特别识别位点,具有快速的吸附动力学,同时对木犀草素具有良好的特异性吸附能力。此外,材料化学稳定性良好,具有pH响应功能可以简化吸附脱附操作,大大提高吸附效率。
附图说明
图1 为实施例1中HNTs(a1)、HNTs@PDA(a2)、HNTs@PDA@Br(a3)、HNTs@PDA@Br@VLD(a4)、MIPs(a5)的透射电镜图;图中标尺大小均为50nm。
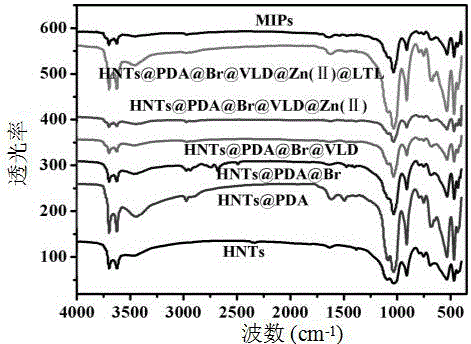
图2为实施例1中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、HNTs@PDA@Br@VLD@Zn(Ⅱ)、HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL、MIPs的红外光谱图。
图3为实施例1中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、MIPs的X射线衍射谱图。
图4为实施例1中F-BA的核磁图。
图5为实施例1中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、MIPs的s的拉曼光谱图。
图6为实施例1中所得材料的吸附等温线模型。
图7为实施例1中所得材料的吸附动力学模型。
图8为实施例1中所得材料对对硝基苯酚、间苯二酚、槲皮素和木犀草素的竞争物吸附结果图。
具体实施方式
本发明具体实施方式中识别性能评价按照下述方法进行:利用静态吸附实验完成。将10ml一定浓度的LTL溶液加入到离心管中,加入一定量的表面印迹纳米颗粒吸附剂MIPs,放在25oC恒温水域中静置若干小时,吸附后LTL含量用紫外可见分光光度计测定,并根据结果计算出吸附容量;饱和吸附后,表面印迹纳米颗粒吸附剂MIPs用高速离心收集,选择几种结构和性质类似的羟基类化合物,作为竞争吸附物,参与研究MIPs聚合物的识别性能。
下面结合具体实施实例对本发明做进一步说明。
实施例1:
(1) 乙烯基氟苯硼酸的制备(F-BA):
在冰水浴中,将2g氟苯硼酸(4-(aminoethylcarbamoyl)-3-fluorophenylboronic acid)加入到100mL碳酸钠溶液中,然后将3mL丙烯酰氯加入到上述溶液中,然后用0.1mol/L的氢氧化钠溶液把pH调节到中性反应12小时,最后真空60℃烘干。
(2) 埃洛石纳米管(HNTs)的活化:
将0.1g块状埃洛石纳米管研磨粉碎过100筛,在100℃高温下煅烧24h,
将煅烧后的0.1g 的HNTs放置在50mL的硫酸和盐酸的混合溶液(v:v, 1:3)中,并在80℃下搅拌6h;然后用大量的蒸馏水冲洗直至中性,在真空60℃下烘干。
(3) 多巴胺负载在埃洛石纳米管(HNTs@PDA)的制备:
将100mg的活化后的埃洛石纳米管被放到40mL的乙醇和水的溶液中(v:v,1:1),其次将50mg的盐酸多巴胺加入上述溶液中并搅拌5min,再将预先配好的40mL三(羟甲基)氨基甲烷溶液(酸碱值为8.5用0.1mol L-1调节)逐滴滴加到上述溶液中,在25℃下搅拌24小时。
(4) 埃洛石的负载溴引发剂(HNTs@PDA@Br)的制备:
将50mg HNTs@PDA 加入到10mL的N,N-二甲基甲酰胺溶液中,随后加入1.5mL的三乙胺,并搅拌10min,再加入预先配好的5mL的2-溴异丁酰溴和N,N-二甲基甲酰胺的混合溶液(v:v, 1:4)中并在氮气保护下搅拌24h在25℃下。
(5) 埃洛石的溴引发剂表面上修饰乙烯基咪唑(HNTs@PDA@Br@VLD)的制备:
将25mg HNTs@PDA@Br加入到2mL甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1),然后通入氮气10min,再加入50mg的乙烯基咪唑,0.015mmol的氯化亚铜,0.015mmol的五甲基二乙烯三胺,然后通入氮气10min,再加入预先准备好的25mg的抗败坏血酸,最后加入 1mL的甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1),并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。
(6) 埃洛石的溴引发剂表面上修饰乙烯基咪唑吸附金属离子(HNTs@PDA@Br@VLD@Zn(Ⅱ)) 的制备:
将50mg HNTs@PDA@Br@VLD加入到20mL乙醇和水的混合溶液中(v:v, 1:1),然后加入0.4g六水合硝酸锌,然后在25℃下搅拌12h并在真空40℃12小时烘干。
(7) 洛石的溴引发剂表面上修饰乙烯基咪唑与金属离子络合物吸附木犀草素 (HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL) 的制备:
将50mg HNTs@PDA@Br@VLD@Zn(Ⅱ) 加入到20mL乙醇和水的混合溶液中(v:v, 1:1),然后加入20mg的木犀草素,然后在25℃搅拌12h并在真空40℃12小时烘干。
(8) 分子印迹聚合物(MIPs)的制备:
将50mg HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL和20mg 4-乙烯基苯硼酸加入到4mL甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1),然后通入氮气10min,再加入0.015mmol氯化亚铜,0.015mmol五甲基二乙烯三胺,然后通入氮气10min,再加入25mg的抗败坏血酸,4mL甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1)并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。得到的产品浸泡在20mL的甲醇和醋酸(v:v,11:1)的混合溶液中12小时,然后甲醇洗3次,然后离心并在60℃下6小时烘干。
图1 为该实施例中HNTs(a1)、HNTs@PDA(a2)、HNTs@PDA@Br(a3)、HNTs@PDA@Br@VLD(a4)、MIPs(a5)的透射电镜图。从图中可以得出,材料尺寸越来越厚,分别为(0nm, 8nm, 12nm, 18nm,22nm),表明每一步都聚合成功。
图2为该实施例中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、HNTs@PDA@Br@VLD@Zn(Ⅱ)、HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL、 MIPs的红外光谱图,从图中可以看出MIPs在1327cm-1出现一个峰,说明出现硼酸基团,说明MIPs合成成功。
图3为该实施例中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、MIPs的X射线衍射谱图。从图可以得出1340 cm-1有一个明显的特征峰,表明MIPs中存在烯基氟苯硼酸功能单体。
图4为该实施例中F-BA的核磁图,从图中可以得出,上面的氢谱都已经出现,表明F-BA合成成功。
图5为该实施例中HNTs、HNTs@PDA、HNTs@PDA@Br、HNTs@PDA@Br@VLD、MIPs的s的拉曼光谱图,MIPs的拉曼光谱图在189cm-1有一个特征峰,表明MIPs中存在氟苯硼酸功能单体;
实施例2:
(1) 乙烯基氟苯硼酸的制备(F-BA):
在冰水浴中,将3g氟苯硼酸(4-(aminoethylcarbamoyl)-3-fluorophenylboronic acid)加入到150mL碳酸钠溶液中,然后4mL丙烯酰氯加入到上述溶液中,然后用0.1mol/L的氢氧化钠溶液把pH调节到中性反应12小时,最后真空60℃烘干。
(2) 埃洛石纳米管(HNTs)的活化:
将0.2g块状埃洛石纳米管研磨粉碎过100筛,在100℃高温下煅烧24h,煅烧后的0.2g 的HNTs被放置在60mL的硫酸和盐酸的混合溶液中, (v:v, 1:3) 在80℃下搅拌6h。然后用大量的蒸馏水冲洗直至中性,最后在真空60℃下烘干。
(3) 多巴胺负载在埃洛石纳米管(HNTs@PDA)的制备:
将150mg的活化后的埃洛石纳米管被放到50mL乙醇和水的溶液中(v:v,1:1), 其次60mg的盐酸多巴胺加入到上述溶液中并搅拌5min,最后将预先配好的50mL的三(羟甲基)氨基甲烷溶液(酸碱值为8.5用0.1mol L-1调节)逐滴滴加到上述溶液中,在25℃下搅拌24小时。
(4) 埃洛石的负载溴引发剂(HNTs@PDA@Br)的制备:
将100mg HNTs@PDA 加入到20mL的N,N-二甲基甲酰胺溶液中,随后加入1.7mL的三乙胺,并搅拌10min,再加入预先配好的10mL的2-溴异丁酰溴和N,N-二甲基甲酰胺的混合溶液(v:v, 1:4)中并在氮气保护下搅拌24h在25℃下。
(5) 埃洛石的溴引发剂表面上修饰乙烯基咪唑(HNTs@PDA@Br@VLD)的制备:
将50mg HNTs@PDA@Br加入到3mL甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1), 然后通入氮气10min,再加入75mg的乙烯基咪唑,0.018mmol氯化亚铜,0.018mmol五甲基二乙烯三胺,然后通入氮气10min,再加入预先准备好的50mg的抗败坏血酸,最后加入3mL甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1)并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。
(6)埃洛石的溴引发剂表面上修饰乙烯基咪唑吸附金属离子(HNTs@PDA@Br@VLD@Zn(Ⅱ)) 的制备:
将75mg HNTs@PDA@Br@VLD加入到25mL乙醇和水的混合溶液中(v:v, 1:1),然后加入0.5g六水合硝酸锌,然后在25℃下搅拌12h并在40℃真空烘干12小时。
(7)洛石的溴引发剂表面上修饰乙烯基咪唑与金属离子络合物吸附木犀草素 (HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL) 的制备:
将75mg HNTs@PDA@Br@VLD@Zn(Ⅱ) 加入到25mL乙醇和水的混合溶液中(v:v, 1:1),然后加入25mg的木犀草素,然后搅拌12h在25℃并在40℃真空烘干12小时。
(8) 分子印迹聚合物(MIPs)的制备:
将75mg HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL和25mg 氟苯硼酸加入到6mL的甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1),然后通入氮气10min,再加入0.018mmol氯化亚铜,0.018mmol五甲基二乙烯三胺,然后通入氮气10min,再加入预先准备好的含50mg的抗败坏血酸, 最后加入6mL甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1)并在25℃下反应6小时, 然后用水洗多次,最后在60℃下烘干。得到的产品浸泡在25mL的甲醇和醋酸(v:v,11:1)的混合溶液中12小时,然后甲醇洗3次,然后离心并在60℃下6小时烘干。
实施例3:
(1) 乙烯基氟苯硼酸的制备(F-BA):
在冰水浴中,将4g氟苯硼酸(4-(aminoethylcarbamoyl)-3-fluorophenylboronic acid)加入到200mL碳酸钠溶液中,然后将5mL丙烯酰氯加入到上述溶液中,然后用0.1mol/L的氢氧化钠溶液把pH调节到中性反应12小时,最后真空60℃烘干。
(2) 埃洛石纳米管(HNTs)的活化:
将0.3g块状埃洛石纳米管研磨粉碎过100筛,在100℃高温下煅烧24h,煅烧后的0.3g 的HNTs被放置在70mL的硫酸和盐酸的混合溶液(v:v, 1:3)中,在80℃下搅拌6h。然后用大量的蒸馏水冲洗直至中性,最后在真空60℃下烘干。
(3) 多巴胺负载在埃洛石纳米管(HNTs@PDA)的制备:
将200mg的活化后的埃洛石纳米管被放到60mL的乙醇和水的溶液中(v:v,1:1), 其次150mg的盐酸多巴胺加入到上述溶液中并搅拌5min,最后将预先配好的60mL的三(羟甲基)氨基甲烷溶液(酸碱值为8.5用0.1mol L-1调节)逐滴滴加到上述溶液中在25℃下搅拌24小时。
(4) 埃洛石的负载溴引发剂(HNTs@PDA@Br)的制备:
将150mg HNTs@PDA 加入到30mL的N,N-二甲基甲酰胺溶液中,随后加入2.0 mL的三乙胺,并搅拌10min,再加入预先配好的15 mL的2-溴异丁酰溴和N,N-二甲基甲酰胺的混合溶液(v:v, 1:4)中,并在氮气保护下搅拌24h在25℃下。
(5) 埃洛石的溴引发剂表面上修饰乙烯基咪唑(HNTs@PDA@Br@VLD)的制备:
将75mg HNTs@PDA@Br加入到4mL甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1), 然后通入氮气10min,再加入100mg的乙烯基咪唑,0.02mmol氯化亚铜,0.02mmol五甲基二乙烯三胺,然后通入氮气10min,再加入预先准备好的75mg的抗败坏血酸,最后加入 4mL甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1)并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。
(6)埃洛石的溴引发剂表面上修饰乙烯基咪唑吸附金属离子(HNTs@PDA@Br@VLD@Zn(Ⅱ)) 的制备:
将100mg HNTs@PDA@Br@VLD加入到30mL乙醇和水的混合溶液中(v:v, 1:1),然后加入0.6g六水合硝酸锌,然后搅拌12h在25℃下并在40℃真空烘干12小时。
(7)洛石的溴引发剂表面上修饰乙烯基咪唑与金属离子络合物吸附木犀草素 (HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL) 的制备:
将100mg HNTs@PDA@Br@VLD@Zn(Ⅱ) 加入到30mL乙醇和水的混合溶液中(v:v, 1:1),然后加入30mg的木犀草素,然后在25℃搅拌12h并在40℃真空烘干12小时。
(8) 分子印迹聚合物(MIPs)的制备:
将100mg HNTs@PDA@Br@VLD@Zn(Ⅱ)@LTL和50mg 氟苯硼酸加入到8mL的甲醇、N,N-二甲基甲酰胺、苯甲醚的混合溶液中(v:v:v, 1:1:1),然后通入氮气10min,再加入0.02mmol氯化亚铜,0.02mmol五甲基二乙烯三胺,然后通入氮气10min,再加入预先准备好的含75mg的抗败坏血酸,最后加入8mL甲醇、N,N-二甲基甲酰胺、苯甲醚组成的混合溶液中(v:v:v, 1:1:1)并在25℃下反应6小时,然后用水洗多次,最后在60℃下烘干。得到的产品浸泡在30mL的甲醇和醋酸(v:v,11:1)的混合溶液中12小时,然后甲醇洗3次,然后离心并在60℃下6小时烘干。
试验例1:取10mL初始浓度分别为10 mg/L、15 mg/L、20 mg/L、25 mg/L、30 mg/L的LTL溶液分别加入到不同的离心管中,再分别加入10mg实施例1中的分子印迹聚合物(MIPs),把测试液放在25℃的水浴中静置12h后在298K、308K、318K温度下,上层清液用高速离心机分离收集,未吸附的LTL分子浓度用紫外可见分光光度计测定,并根据结果计算出吸附容量,从图6中可以得出结果,当初始浓度为30mg/L时,分子印迹聚合物(MIPs)的吸附趋于平衡。
试验例2:取10mL初始浓度为20mg/L的木犀草素(LTL)溶液分别加入到离心管中,加入10mg实施例1中的分子印迹聚合物(MIPs),把测试液放在25℃的水浴振荡器中,分别在5min,15min,30min,60min,120min,180 min,360min和720min的时候取出;通过离心将分子印迹聚合物(MIPs)吸附剂和溶液分离开,再使用孔径为0.45mm的微孔硝酸纤维素膜对溶液进行过滤去除悬浮的粒子。滤液中的木犀草素浓度由紫外分光光度计在349nm的波长下计算测定,并根据结果计算出吸附容量;从图7中可以得出结果, MIPs的吸附过程可以分为快速阶段(前120min)和缓慢阶段,而MIPs在快速阶段的吸附容量达到平衡容量的98.98%,之后缓慢增加直到平衡,证明了硼酸印迹分子结合位点对吸附的影响,印迹聚合物拥有快的吸附动力学。
试验例3:选择对硝基苯酚、间苯二酚、槲皮素为竞争吸附的羟基类化合物,分别配置以上四种羟基类化合物的水溶液,每种竞争吸附剂的浓度都为20mg/L,取10mL配置好的溶液加入到离心管中,分别加入10mg实施例1中的MIPs吸附剂,把测试液放在25℃的水浴中分别静置12.0h,静置时间完成后,上层清液用高速离心分离收集,未吸附的各种竞争吸附羟基类化合物浓度用高效液相(HPLC)测定,从图8中可以得出结果, MIPs对硝基苯酚、间苯二酚、槲皮素和木犀草素的吸附容量分别为6.85mg/g,1.56mg/g,11.18mg/g,23.58mg/g。 表明MIPs对LTL有显著的专一识别性,吸附容量高于其它羟类化合物。
- 还没有人留言评论。精彩留言会获得点赞!