一种β‑葡萄糖苷酶突变体蛋白的表达载体及表达工程菌和表达方法与流程
本发明涉及生物工程领域,具体地说涉及一种β-葡萄糖苷酶突变体蛋白的表达载体及表达工程菌和表达方法。
背景技术:
β-葡萄糖苷酶(EC 3.1.2.21,β-glucosidase)属于纤维水解酶类,主要水解糖苷或者寡糖中的β-1,4-糖苷键,同时释放出末端非还原性的葡萄糖以及糖苷配体,在现代工业生物技术如食品、医药、饲料加工、能源炼制等领域具有重要应用价值。目前细菌β-葡萄糖苷酶普遍采用异源表达的生产方法,又以大肠杆菌为主要表达宿主菌。然而,常用的大肠杆菌诱导表达方法存在诸多问题,如诱导剂IPTG价格昂贵、存在一定的细胞毒性;大量蛋白多在胞内表达,需要专用仪器对细胞壁和细胞膜进行破碎,增加了后续纯化步骤和纯化难度。因此,开发新的β-葡萄糖苷酶在大肠杆菌中的诱导表达方法,不仅有利于促进β-葡萄糖苷酶的生产和开发,也能为其它蛋白在大肠杆菌中的异源表达提供思路和借鉴。
光照是一种易于控制的诱导方法,利用光照的方法调控目的基因的表达能够减少发酵过程中的操作步骤,减少可能造成的外源污染,降低目的蛋白的生产成本。
技术实现要素:
本发明所要解决的技术问题是提供一种诱导简单、表达量高、无需外源添加物的β-葡萄糖苷酶突变体蛋白的表达载体。
为了解决上述技术问题,本发明采用如下技术方案:一种β-葡萄糖苷酶突变体蛋白的表达载体,按以下方法构建而成:
(1)克隆β-葡萄糖苷酶突变体蛋白的DNA序列,该β-葡萄糖苷酶突变体蛋白的DNA序列如SEQ ID NO.1所示;
(2)克隆光敏调控单元的DNA序列,该光敏调控单元的DNA序列如SEQ ID NO.2所示;
(3)通过双酶切的方法分别切割上述克隆得到的β-葡萄糖苷酶突变体蛋白的DNA和光敏调控单元的DNA,并将切割得到的β-葡萄糖苷酶突变体蛋白的DNA酶切片段和光敏调控单元的DNA酶切片段连接至pET22b(+)载体上,即得所述β-葡萄糖苷酶突变体蛋白的表达载体。
本发明所表达的β-葡萄糖苷酶突变体蛋白相较于野生型β-葡萄糖苷酶,其乙醇耐受浓度IC50提高1.8倍,且葡萄糖耐受浓度提高2倍,pH及温度稳定性也有所提高,该突变体蛋白能够在较短时间内高效水解大豆异黄酮糖苷制备苷元,具有潜在应用价值。
而本发明通过插入光敏调控单元构建得到可光诱导的表达载体,该表达载体可通过光照手段对β-葡萄糖苷酶突变体蛋白进行诱导表达,与传统的诱导表达方法相比,光照诱导方法具有表达量高、无外源添加物的特点,此外光照诱导方法对蛋白的表达强度可通过光照强度进行控制,且不影响后续纯化步骤。
本发明还提供一种表达β-葡萄糖苷酶突变体蛋白的工程菌,是通过将上述β-葡萄糖苷酶突变体蛋白的表达载体转化至大肠杆菌BL21(DE3)感受态细胞中所获得。
大肠杆菌由于其遗传学、生物化学和分子生物学等方面已充分被人们了解而成为表达异源蛋白的首选表达系统。大肠杆菌容易培养且费用低、对多种蛋白具有良好的耐受能力、能高水平的表达异源蛋白,而且光照对大肠杆菌无毒性;同时,大肠杆菌可进行高密度发酵,可进一步降低生产成本。因此,利用大肠杆菌工程菌异源表达β-葡萄糖苷酶具有单位产量高、生产周期短等优势。
本发明还提供一种利用上述表达β-葡萄糖苷酶突变体蛋白的工程菌进行低密度发酵生产β-葡萄糖苷酶突变体蛋白的方法,包括以下步骤:将表达β-葡萄糖苷酶突变体蛋白的工程菌接种在TB培养基中,避光培养至OD600为0.4-1.0时,再加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达。
通过上述低密度发酵方法获得的β-葡萄糖苷酶酶活与使用T7启动子、IPTG诱导方法获得的酶活基本保持一致,但无需加入IPTG,节省原料成本,在后续的纯化过程中避免了IPTG的污染。
进一步地,所述TB培养基中含有氨苄青霉素,氨苄青霉素的浓度为100μg·mL-1,避光培养的培养温度为28℃,光照照度为1000-6000lx,光的颜色为白色。设计上述条件适于工程菌的生长发酵,而且不需要添加诱导物,诱导操作简单方便,诱导表达量高。
本发明还提供一种利用上述表达β-葡萄糖苷酶突变体蛋白的工程菌高进行密度发酵生产β-葡萄糖苷酶突变体蛋白的方法,包括以下步骤:将表达β-葡萄糖苷酶突变体蛋白的工程菌接种在TB培养基中进行避光培养,当培养基中甘油耗尽、发酵液溶氧DO上升幅度超过30%时,添加补料培养基,当发酵液OD600为15-25时,再加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达。
这种高密度发酵方法能够诱导获得大量目标蛋白,而且操作简单方便,同时也避免了额外添加诱导物对后续纯化蛋白产生的影响。
本发明还提供了另外一种具有裂解功能的β-葡萄糖苷酶突变体蛋白的表达载体,按以下方法构建而成:
(1)克隆λ噬菌体裂解蛋白SRRz的DNA序列;
(2)克隆权利要求1的β-葡萄糖苷酶突变体蛋白的表达载体中的β-葡萄糖苷酶突变体蛋白以及光敏调控单元的DNA序列;
(3)通过双酶切的方法分别切割上述克隆得到的λ噬菌体裂解蛋白SRRz的DNA和β-葡萄糖苷酶突变体蛋白以及光敏调控单元的DNA,并将切割得到的λ噬菌体裂解蛋白SRRz的DNA酶切片段和β-葡萄糖苷酶突变体蛋白以及光敏调控单元的DNA酶切片段连接至pET22b(+)载体上,即得所述具有裂解功能的β-葡萄糖苷酶突变体蛋白的表达载体。
这种表达载体是在第一种表达载体的基础上增加了裂解蛋白SRRz的DNA,使用可表达裂解蛋白SRRz的载体,在添加IPTG或添加乳糖进入培养基进行诱导后,其诱导表达出的裂解蛋白能够使宿主菌发生裂解,释放出胞内的目标蛋白到外部环境。该裂解方法简单方便,目标蛋白回收效率高。
相应的,本发明还提供一种可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌,是通过将上述具有裂解功能的β-葡萄糖苷酶突变体蛋白的表达载体转化至大肠杆菌BL21(DE3)pLysS感受态细胞中所获得。
相应的,本发明还提供一种利用上述可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌进行低密度发酵生产β-葡萄糖苷酶突变体蛋白的方法,包括以下步骤:将可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌接种在TB培养基中,避光培养至OD600为0.4-0.6时,加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达,当发酵液OD600达到8.0时,添加IPTG或乳糖。
通过上述低密度发酵方法能够高效大量释放β-葡萄糖苷酶酶到发酵液中,极大地简化了后续纯化步骤,不需要额外的细胞破碎仪器,节省原成本,同时当以乳糖为裂解诱导剂时,在后续的纯化过程中避免了IPTG的污染。
相应的,本发明还提供一种利用上述可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌进行高密度发酵生产β-葡萄糖苷酶突变体蛋白的方法,包括以下步骤:将可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌接种在TB培养基中进行避光培养,当培养基中甘油耗尽、发酵液溶氧DO上升幅度超过30%时,添加补料培养基,当发酵液OD600为10时,再加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达,当发酵液OD600达到30时,添加IPTG或乳糖。
这种高密度发酵方法能够大量诱导目标蛋白在大肠杆菌胞内表达,而诱导出来的裂解蛋白SRRz能够裂解大肠杆菌,将目标蛋白释放至外部环境,简化后续对目标蛋白的纯化步骤,酶活回收率高。
进一步地,所述TB培养基的组成为:甘油15-20g·L-1,酵母粉20-26g·L-1,胰蛋白胨10-12g·L-1,KH2PO4 17mM,K2HPO4 72mM,氨苄青霉素100μg·mL-1,氯霉素12.5μg·mL-1;所述补料培养基的组成为:甘油450-500g·L-1,酵母粉40-50g·L-1,蛋白胨40-50g·L-1,氨苄青霉素100μg·mL-1,氯霉素12.5μg·mL-1。本发明上述初始TB培养基和补料培养基能够满足大肠杆菌高密度发酵所需营养物质,同时其具有一定pH缓冲能力,适用于高密度发酵。
附图说明
图1是实施例2的发酵结果的SDS-PAGE电泳图谱检测,M泳道为蛋白分子量Marker,1泳道为无bgl1A(A24S/F297Y)片段插入的空载体诱导情况,2泳道为含有bgl1A(A24S/F297Y)片段插入后光照诱导情况。
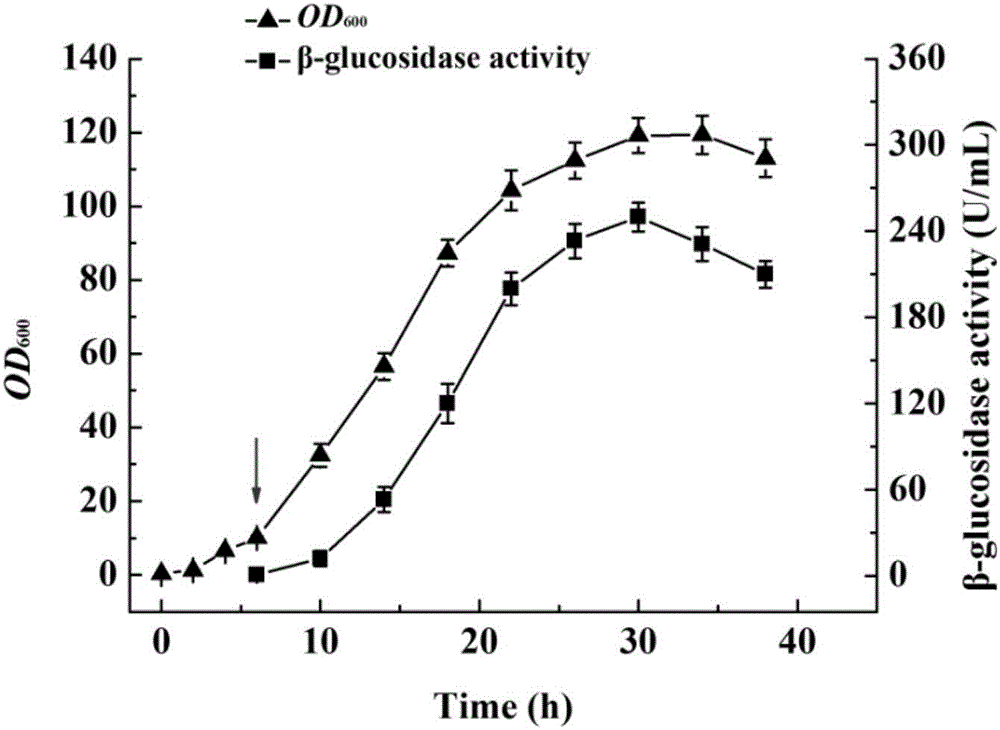
图2是实施例3发酵过程中生物量(OD600)及β-葡萄糖苷酶突变体蛋白酶活(U/mL)变化图。
具体实施方式
下面结合实施例对本发明作进一步描述:
以下实施例中所涉及到的培养基的配方如下:
1、LB液体培养基:酵母粉5g·L-1,胰蛋白胨10g·L-1,NaCl 10g·L-1,氨苄青霉素100μg·mL-1(氨苄青霉素使用时添加);
2、TB培养基:甘油15g·L-1,酵母粉20-26g·L-1,胰蛋白胨10-12g·L-1,KH2PO417mM,K2HPO4 72mM,氨苄青霉素100μg·mL-1(氨苄青霉素使用时添加);
3、补料培养基:甘油450-500g·L-1,酵母粉40-50g·L-1,胰蛋白胨40-50g·L-1,氨苄青霉素100μg·mL-1(氨苄青霉素使用时添加);
上述LB液体培养基、TB培养基和补料培养基用于可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌时,也即用在实施例5和6中时,均再添加氯霉素12.5μg·mL-1,且氯霉素也是在使用时加入。
实施例1
表达β-葡萄糖苷酶突变体蛋白的工程菌的构建
以含有bgl1A(A24S/F297Y)基因编码序列(如SEQ ID NO.1所示)的克隆载体Trans1T1-bgl1A(A24S/F297Y)和含有光敏调控单元pD编码序列(如SEQ ID NO.2所示)的克隆载体Trans1T1-pD为模板,设计如下表1所示的引物:
表1
将上述的引物进行配对,分别为pD-F与pD-R、bgl-F与bgl-R作为PCR引物对,使用的模板分别为克隆载体Trans1T1-bgl1A(A24S/F297Y)和含有光敏调控单元pD编码序列的克隆载体Trans1T1-pD,按照如下表2所示的体系进行PCR扩增,扩增产物依次命名为pD和bgl1A(A24S/F297Y),回收备用;
表2
使用限制性内切酶Xba I和PshA I对扩增产物pD和载体pET22b(+)分别进行双酶切,酶切体系如下表3所示:
表3
对酶切后的片段进行回收和纯化,按照如下表4所示的连接体系进行连接,16℃连接反应8h:
表4
连接产物转化到宿主菌BL21(DE3)中,得到的转化子测序验证是否突变,挑选序列正确的转化组得到含有光敏调控单元pD编码序列的载体,命名为pET22b-pD;
参照上述酶切等反应体系,对获得的载体pET22b-pD和PCR扩增片段bgl1A(A24S/F297Y)使用限制性内切酶Nde I和Xho I分别进行双酶切,回收酶切片段进行连接反应,连接产物即为表达载体pET22b-pD-bgl1A(A24S/F297Y);将表达载体转化至宿主菌BL21(DE3)中,获得表达β-葡萄糖苷酶突变体蛋白的工程菌并测序确认,命名为BL21(DE3)/pET22b-pD-bgl1A(A24S/F297Y)。
实施例2
表达β-葡萄糖苷酶突变体蛋白的工程菌在摇瓶低密度发酵生产β-葡萄糖苷酶突变体蛋白中的应用
将测序确认无误的工程菌BL21(DE3)/pET-22b-pD-bgl1A(A24S/F297Y)接种入5ml LB液体培养基中,接种量1%,28℃下220rpm避光培养8-12h;
按1%的接种量将经过上述培养的工程菌接入400ml TB培养基中,28℃,200rpm避光培养,当发酵液OD600达到0.6时,使用白色光源照射,光照照度为3000lx,诱导表达12-14h;
诱导表达结束后,4℃、8000g离心收集菌体,加入0.1倍菌液体积的破碎缓冲液(Na2PO4-柠檬酸),350W冰浴条件下超声40min破碎细胞,30000g离心收集上清,以pNPG为底物,测定得到粗酶液酶活为33.22U/mL,,电泳结果参见图1。
通过本实施例的方法获得的β-葡萄糖苷酶酶活与不含pD的表达β-葡萄糖苷酶突变体蛋白的工程菌按传统方法(使用LB培养基,T7启动子和IPTG诱导)发酵相比较,酶活产量能够提高2.8倍,且无需加入IPTG,节省了原料成本,在后续的纯化过程中避免了IPTG的污染。
上面所述的不含pD的表达β-葡萄糖苷酶突变体蛋白的工程菌是指将bgl1A(A24S/F297Y和T7启动子连接至载体pET22b(+)上形成表达载体,再将该表达载体转化至宿主菌BL21(DE3)中所得到的工程菌BL21(DE3)/pET22b-T7-bgl1A(A24S/F297Y)。
实施例3
表达β-葡萄糖苷酶突变体蛋白的工程菌在发酵罐高密度发酵生产β-葡萄糖苷酶突变体蛋白中的应用
初始发酵培养基为TB培养基,接种量为5%,用25%(v/v)氨水控制pH在6.9-7.1,通过调节通气量和搅拌速度将DO控制在25-30%,温度控制在28-30℃,当甘油耗尽、发酵液溶氧DO上升幅度超过30%时,按照速率10mL·L-1·h-1流添加补料培养基;
当发酵液OD600为10时,加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达,光照强度为30,000lx,颜色为白色,诱导10-18h后结束;
诱导表达结束后,4℃、8000g离心收集菌体,加入0.1倍菌液体积的破碎缓冲液(Na2PO4-柠檬酸),350W冰浴条件下超声40min破碎细胞,30,000g离心收集上清,以pNPG为底物,测定得到的粗酶液酶活。
图2结果显示,菌体生物量在接种后25h至35h达到稳定期,目标蛋白表达量在诱导24h后达到最高值,测定得到粗酶液酶活为249.92U/mL。
实施例4
可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌的构建
以含有pD-bgl1A(A24S/F297Y)基因编码序列的载体pET22b-pD-bgl1A(A24S/F297Y)和含裂解蛋白SRRz编码序列的克隆载体Trans1T1-SRRz为模板,设计如下表5所示的引物:
表5
将上述的引物进行配对,分别为pD-bgl-F与pD-bgl-R、SRRz-F与SRRz-R作为PCR引物对,使用的模板分别为载体pET22b-pD-bgl1A(A24S/F297Y)和含有含裂解蛋白SRRz编码序列的克隆载体Trans1T1-SRRz,参照实施例1的体系进行PCR扩增,扩增产物依次命名为pD-bgl1A(A24S/F297Y)和SRRz,回收备用;
使用限制性内切酶Nde I和Xho I对回收得到的片段SRRz和载体pET22b(+)分别进行双酶切,酶切体系参照实施例1进行配置;
对酶切后的片段进行回收和纯化,参照实施例1配置连接体系进行连接,16℃连接反应8h,连接产物转化到宿主菌Trans109感受态细胞(Transgen Biotech),得到的转化子测序验证是否突变,挑选序列正确的转化子并提取质粒,将该质粒命名为pET22b-T7-SRRz,
将质粒pET22b-T7-SRRz与pD-bgl1A(A24S/F297Y)分别使用限制性内切酶PshA I进行酶切并回收酶切片段,参照实施例1配置连接体系,将回收到的片段进行连接,连接产物即为表达载体pET22b-pD-bgl1A(A24S/F297Y)-T7-SRRz,将连接产物转化至宿主菌BL21(DE3)pLysS中,获得可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌并测序确认,命名为BL21(DE3)pLysS/pET22b-pD-bgl1A(A24S/F297Y)-T7-SRRz。
实施例5
可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌在摇瓶低密度发酵生产β-葡萄糖苷酶突变体蛋白中的应用
将测序确认无误的工程菌BL21(DE3)pLysS/pET22b-pD-bgl1A(A24S/F297Y)-T7-SRRz接种入5ml LB液体培养基中,接种量1%,28℃下220rpm避光培养8-12h。
按1%的接种量将经过上述培养的工程菌接入400ml TB培养基中,28℃,200rpm避光培养,当发酵液OD600达到0.6时,使用白色光源照射,光照照度为3,000lx,当发酵液液OD600达到8.0时,添加终浓度为1.5mM的IPTG或者添加终浓度为5g·L-1的乳糖,诱导表达12-14h;
诱导表达结束后,将发酵液放4℃静置4h,4℃、8,000g离心收集发酵液上清和发酵菌体。对于发酵菌体,加入0.1倍菌液体积的破碎缓冲液(Na2PO4-柠檬酸),350W冰浴条件下超声40min破碎细胞,30,000g离心收集上清。以pNPG为底物,分别测定诱导裂解后的发酵液酶活为和未裂解菌体的破碎液上清酶活。
当以IPTG为诱导剂时,裂解液酶活为22.5U/mL,未裂解细胞内酶活为1.29U/mL,裂解后酶活回收率为94.57%;当以乳糖为诱导剂时,裂解液酶活为17.1U/mL,未裂解细胞内酶活为3.13U/mL,裂解后酶活回收率为84.52%。
通过本实施例的方法能够高效大量释放β-葡萄糖苷酶酶到发酵液中,极大地简化了后续纯化步骤,不需要额外的细胞破碎仪器,节省原成本,同时当以乳糖为裂解诱导剂时,在后续的纯化过程中避免了IPTG的污染。
实施例6
可裂解的表达β-葡萄糖苷酶突变体蛋白的工程菌在发酵罐高密度发酵生产β-葡萄糖苷酶突变体蛋白中的应用
初始发酵培养基为TB培养基,接种量为5%,用25%(v/v)氨水控制pH在6.9-7.1,通过调节通气量和搅拌速度将DO控制在25-30%,温度控制在28-30℃。当甘油耗尽发酵液溶氧DO上升幅度超过30%时,按照速率10mL·L-1·h-1流添加补料培养基;
当发酵液OD600为10时,加入光照,诱导β-葡萄糖苷酶突变体蛋白的表达,光照强度为30,000lx,颜色为白色,当发酵液OD600为30时,添加终浓度为1.5mM的IPTG或者恒速流加0.9g·L-1·h-1的乳糖,诱导表达12-18h;
诱导表达结束后,将发酵液放4℃静置4-12h,4℃、8000g离心收集发酵液上清和发酵菌体。对于发酵菌体,加入0.1倍菌液体积的破碎缓冲液(Na2PO4-柠檬酸),350W冰浴条件下超声40min破碎细胞,30,000g离心收集上清。以pNPG为底物,分别测定诱导裂解后的发酵液酶活为和未裂解菌体的破碎液上清酶活。
当以IPTG为诱导剂时,裂解液酶活为80.5U/mL,未裂解细胞内酶活为6.94U/mL,裂解后酶活回收率为92.06%;当以乳糖为诱导剂时,裂解液酶活为61.5U/mL,未裂解细胞内酶活为19.0U/mL,裂解后酶活回收率为76.39%。
通过本发明工程菌发酵得到的纯化的β-葡萄糖苷酶突变蛋白的最适pH为6.5,酶在pH 5.5-7.5范围内能够显示80%以上的酶活力。酶在pH 6.0-9.0范围内有较高的稳定性,30℃条件下保温1h仍然能够维持原酶活力的60%以上。突变蛋白在15℃-55℃范围内均能显示催化活性,其最适温度为40℃。
应当理解本文所述的例子和实施方式仅为了说明,本领域技术人员可根据它做出各种修改或变化,在不脱离本发明精神实质的情况下,都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!