低聚芴二醇、低聚芴二芳基酯及它们的制造方法与流程
本申请是分案申请,其原申请的国际申请号为pct/jp2013/078009,中国国家申请号为201380051990.5,申请日为2013年10月15日,发明名称为“树脂组合物、拉伸膜、圆偏振片和图像显示装置”。
本发明涉及光学特性、耐热性以及熔融加工性优异的树脂组合物、使用其得到的拉伸膜、圆偏振片和图像显示装置。
背景技术:
近年来,有人报告了由在侧链具有芴环的二羟基化合物衍生出的聚碳酸酯树脂、聚酯树脂,提出了发挥源于芴环的光学特性及耐热性之类的特征,将其作为在光学用途中有用的材料(例如,参见专利文献1)。在专利文献2、3中公开了下述内容:通过将聚碳酸酯树脂中的具有芴环的重复单元的含量控制在特定的范围,由该聚碳酸酯树脂形成的拉伸膜显示出波长越短则相位差越小的逆波长分散性,因而作为相位差膜具有优异的性能。显示出波长越短则相位差越小的所谓逆波长分散性的相位差膜在可见区域的各波长下能够得到理想的相位差特性,作为圆偏振片,对图像显示装置的防止外光反射或可视角校正等是有用的。
另外,在专利文献4中公开了下述内容:通过将聚酰亚胺树脂中的具有芴环的重复单元的含量控制在特定的范围,由该聚酰亚胺树脂形成的拉伸膜显示出了从短波长侧直到长波长侧的宽带域几乎没有相位差变化的平坦分散性,作为相位差膜具有优异的性能。显示出平坦分散性的相位差膜对va(垂直取向)模式的液晶的校正是有用的。
作为在侧链具有芴环的二羟基化合物,目前经常使用专利文献2或3中记载的9,9-双(4-(2-羟基乙氧基)苯基)芴、9,9-双(4-羟基-3-甲基苯基)芴之类。另外,在专利文献4中公开了在侧链具有芴环的二胺化合物,进一步记载了使用该二胺化合物的聚酰亚胺树脂的拉伸膜。在专利文献5中公开了在同一分子内具有2个芴环的二酯化合物,进一步记载了使用该二酯化合物的聚酯树脂。在专利文献6中公开了在同一分子内具有2个芴环的二羟基化合物或二酯化合物,进一步记载了使用该二羟基化合物或二酯化合物的聚酯树脂的拉伸膜。
现有技术文献
专利文献
专利文献1:日本特开平10-101786号公报
专利文献2:国际公开第2006/041190号
专利文献3:国际公开第2011/149073号
专利文献4:日本特开2008-112124号公报
专利文献5:美国专利第3324084号说明书
专利文献6:美国专利申请公开第2012/0170118号说明书
技术实现要素:
发明所要解决的课题
fpd领域的发展惊人,要求其兼具各种特性。例如,作为在逆波长分散膜、1/4λ板、平坦分散的相位差膜中使用的材料,要求光弹性系数低、具有所期望的光学特性、同时兼具充分的耐热性与熔融加工性、机械强度等各种物性的材料。
已知由专利文献2、专利文献3中的具有芴环的聚碳酸酯树脂形成的拉伸膜作为显示出逆波长分散性的相位差膜、用于防止图像显示装置的外光反射的圆偏振片是有用的。但是,根据本发明人的研究结果,为了利用使用了专利文献2的9,9-双[4-羟基-3-甲基苯基]芴的树脂或使用了专利文献3的9,9-双[4-(2-羟基乙氧基)苯基]芴的树脂表现出所期望的逆波长分散性,需要增高具有芴环的重复单元的比例,基于共聚的分子设计的自由度低,进而难以兼顾耐热性、熔融加工性、机械强度等诸物性与光学物性。此外,出于校正图像显示装置的漏色(色漏れ)的目的,希望根据装置的特性调整逆波长分散性,因此要求维持各种特性,同时相位差比非常小,可显示出很强的逆波长分散性。但是,上述的专利文献2、专利文献3中记载的在侧链具有芴环的二羟基化合物所表现出的逆波长分散较弱,在由使用它们的树脂制作相位差膜的情况下,难以在维持其它必要物性的同时显示出较强的逆波长分散性。因此,为了进一步改良树脂的光学物性与机械强度等各种物性,要求将光学物性与机械强度等各种物性的平衡优异的新颖化合物用于原料。专利文献5中记载的聚酯并未制成膜使用,光学特性等也不明确。此外,在专利文献6记载了其聚酯显示出了拉伸方向的双折射为负性的负折射率各向异性。作为相位差膜,需要在拉伸方向具有正折射率各向异性,上述聚酯拉伸膜不满足该要件。并且相位差的波长依赖性也不明确。
另一方面,作为平坦分散的相位差膜中所用的材料,人们要求材料具有相位差的波长分散性小、呈平坦分散的优异的光学特性,同时光弹性系数低。已知专利文献4的由具有芴骨架的聚酰亚胺树脂形成的拉伸膜在va模式液晶的校正中是有用的。但是,该膜的对于相位差膜重要的光弹性系数亦不明确。
此外,在液晶显示装置的偏振片保护膜、光学透镜等广泛的用途中,人们要求使用在零双折射下平坦分散的光学材料、即所谓宽带域零双折射材料。
本发明的第1目的在于提供一种树脂组合物和用于这样的树脂组合物的低聚芴二酯单体、以及该低聚芴二酯单体的制造方法,该树脂组合物在成型为膜时显示出优异的光学特性,通过使用即使在树脂中的比例低也可有效表现出所期望的光学特性的结构单元,可提高树脂设计的自由度,兼顾耐热性、熔融加工性、机械强度等各种物性。进一步地,本发明的第2目的在于提供一种二醇单体,该二醇单体用于具有光弹性系数低、相位差的波长分散性小之类的优异的光学特性的树脂组合物。
解决课题的手段
本发明人为了解决上述课题反复进行了深入研究,结果发现,含有具有特定的2价低聚芴作为重复单元的聚合物且具有特定光学特性的树脂组合物容易进行物性的调整,在成型为膜时显示出优异的光学特性、优异的机械特性,能够实现这样的第1目的,从而完成了本发明。
进而,本发明人为了解决上述课题反复进行了深入研究,结果发现,特定的低聚芴二醇在制成树脂组合物时具有光弹性系数低、相位差的波长分散性小之类的优异的光学特性,能够实现这样的第2目的,从而完成了本发明。
即,本发明要点如下。
〔1〕
一种树脂组合物,该树脂组合物包含具有2价低聚芴作为重复单元的聚合物,其特征在于,
上述2价低聚芴包含具有或不具有取代基的2个以上芴单元,该芴单元的9位碳原子彼此直接键合或者藉由亚烷基、亚芳香基或亚芳烷基进行键合,该亚烷基具有或不具有取代基,该亚芳香基具有或不具有取代基,该亚芳烷基具有或不具有取代基,并且
在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比满足下式(2)。
re450/re550≦1.0(2)
〔2〕
一种树脂组合物,该树脂组合物包含具有2价低聚芴作为重复单元的聚合物,其特征在于,
上述2价低聚芴包含具有或不具有取代基的2个以上芴单元,该芴单元的9位碳原子彼此直接键合或者藉由亚烷基、亚芳香基或亚芳烷基进行键合,该亚烷基具有或不具有取代基,该亚芳香基具有或不具有取代基,该亚芳烷基具有或不具有取代基;
上述2价低聚芴相对于上述聚合物的摩尔分数为1%以上;并且
该树脂组合物具有正折射率各向异性。
〔3〕
如上述〔1〕或〔2〕所述的树脂组合物,其特征在于,上述聚合物为聚碳酸酯。
〔4〕
一种树脂组合物,其为包含具有2价低聚芴作为重复单元的聚碳酸酯聚合物的树脂组合物,其特征在于,
上述2价低聚芴包含具有或不具有取代基的2个以上芴单元,上述芴单元的9位碳原子彼此直接键合或者藉由亚烷基、亚芳香基或亚芳烷基进行键合,该亚烷基具有或不具有取代基,该亚芳香基具有或不具有取代基,该亚芳烷基具有或不具有取代基。
〔5〕
如上述〔2〕~〔4〕的任一项所述的树脂组合物,其特征在于,该树脂组合物在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比满足下式(2)。
re450/re550≦1.0(2)
〔6〕
如上述〔1〕~〔5〕的任一项所述的树脂组合物,其特征在于,上述2价低聚芴由下述通式(1)表示。
[化1]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或具有或不具有取代基的碳原子数6~10的亚芳烷基,或者r1和r2各自独立地为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团,
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基、或者氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
n表示1~5的整数值。)
〔7〕
如上述〔1〕~〔6〕的任一项所述的树脂组合物,其特征在于,其进一步具有下述通式(3)所表示的2价有机基团作为重复单元。
[化2]
(式中,r10表示具有或不具有取代基的碳原子数2~20的亚烷基、具有或不具有取代基的碳原子数4~20的亚芳香基、具有或不具有取代基的碳原子数6~20的亚芳烷基、具有或不具有取代基的碳原子数2~100的亚烷基醚基、具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团、或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团。)
〔8〕
如上述〔7〕所述的树脂组合物,其特征在于,上述通式(3)所表示的2价有机基团为下式(4)~(9)中的至少一种。
[化3]
(上述式(5)中,r11表示具有或不具有取代基的碳原子数0~18的直链状亚烷基。)
[化4]
(上述式(6)中,r12表示具有或不具有取代基的碳原子数4~20的环亚烷基。)
[化5]
(上述式(7)中,r13表示具有或不具有取代基的碳原子数2~10的亚烷基,p为1~40的整数。)
[化6]
(上述式(8)中,r14表示具有或不具有取代基的碳原子数2~10的亚烷基,r15表示具有或不具有取代基的碳原子数12~30的亚芳香基。)
[化7]
(上述式(9)中,r16表示具有或不具有取代基的具有碳原子数2~20的乙缩醛环的基团。
〔9〕
如上述〔1〕~〔8〕的任一项所述的树脂组合物,其特征在于,该树脂组合物的玻璃化转变温度为90℃以上、170℃以下。
〔10〕
如上述〔1〕~〔9〕的任一项所述的树脂组合物,其特征在于,该树脂组合物在测定温度240℃、剪切速度91.2sec-1的条件下的熔融粘度为500pa·s以上、5000pa·s以下。
〔11〕
如上述〔1〕~〔10〕的任一项所述的树脂组合物,其特征在于,该树脂组合物的光弹性系数为45×10-12pa-1以下。
〔12〕
如上述〔1〕~〔11〕的任一项所述的树脂组合物,其特征在于,上述2价低聚芴相对于上述聚合物的摩尔分数为1%以上且小于50%。
〔13〕
如上述〔3〕或〔4〕所述的树脂组合物,其特征在于,上述聚合物是将下述通式(10a)所表示的二羟基化合物与下述通式(11)所表示的碳酸二酯熔融缩聚得到的。
[化8]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、具有或不具有取代基的碳原子数6~10的亚芳烷基,或者r1和r2各自独立地为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团;
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子,或者为选自由具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基以及氰基组成的组中的取代基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
n表示1~5的整数值。)
[化9]
(式中,a1和a2分别为取代或无取代的碳原子数1~18的脂肪族烃基、或者取代或无取代的芳香族烃基,a1与a2可以相同、也可以不同。
〔14〕
如上述〔13〕所述的树脂组合物,其特征在于,na、k、cs以及fe的含有比例的合计为3质量ppm以下。
〔15〕
如上述〔13〕或〔14〕所述的树脂组合物,其特征在于,由上述通式(11)所表示的碳酸二酯生成的单羟基化合物的含有比例为1500质量ppm以下。
〔16〕
如上述〔13〕~〔15〕的任一项所述的树脂组合物,其特征在于,上述通式(10a)中,r1和r2为亚甲基。
〔17〕
一种成型体,其特征在于,其是将上述〔1〕~〔16〕的任一项所述的树脂组合物成型得到的。
〔18〕
一种光学部件,其特征在于,其由上述〔1〕~〔16〕的任一项所述的树脂组合物形成。
〔19〕
一种膜,其特征在于,其由上述〔1〕~〔16〕的任一项所述的树脂组合物形成。
〔20〕
一种拉伸膜,其特征在于,其是将上述〔19〕所述的膜沿至少一个方向拉伸得到的。
〔21〕
一种1/4λ板,其特征在于,其由上述〔20〕所述的拉伸膜形成。
〔22〕
一种圆偏振片,其特征在于,其具有上述〔21〕所述的1/4λ板。
〔23〕
一种图像显示装置,其特征在于,其具备上述〔22〕所述的圆偏振片。
〔24〕
一种低聚芴二醇,其由下述通式(19)表示。
[化10]
(式中,r3为具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基、或者氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
n表示1~5的整数值。)
〔25〕
如上述〔24〕所述的低聚芴二醇,其特征在于,氯含有比例以cl换算质量计为100质量ppm以下。
〔26〕
如上述〔24〕或〔25〕所述的低聚芴二醇,其特征在于,长周期型周期表第1族与第2族金属的含有比例为500质量ppm以下。
〔27〕
如上述〔24〕~〔26〕的任一项所述的低聚芴二醇,其特征在于,其在热重测定中的5%失重温度为250℃以上。
〔28〕
如如上述〔24〕~〔27〕的任一项所述的低聚芴二醇,其特征在于,上述通式(19)中,r3为亚甲基、亚乙基、正亚丙基、正亚丁基、或2,2-二甲基亚丙基,r4~r9为氢原子,n为1或2。
〔29〕
下述通式(vii-1)所表示的低聚芴二酯的制造方法,其特征在于,其具有在碱存在下使下述通式(ii)所表示的低聚芴化合物与下述通式(vi-1)所表示的不饱和羧酸酯发生反应的工序。
[化11]
(式中,r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基、或者氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
riii为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基或者具有或不具有取代基的碳原子数6~10的芳烷基;
r17是碳原子数1~10的有机取代基。
n表示1~5的整数值。)
〔30〕
一种低聚芴二芳基酯,其由下述通式(10d)所表示。
[化12]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,或者r1和r2各自独立地为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团;
r3为具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基或氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
ar1为具有或不具有取代基的碳原子数4~10的芳基。
n表示1~5的整数值。)
〔31〕
如上述〔30〕所述的低聚芴二芳基酯,其特征在于,其在热重测定中的5%失重温度为250℃以上。
〔32〕
如上述〔30〕或〔31〕所述的低聚芴二芳基酯,其特征在于,上述通式(10d)中,r3为亚甲基、亚乙基、正亚丙基、正亚丁基、或2,2-二甲基亚丙基,r4~r9为氢原子,ar1为苯基,n为1~2的整数值。
〔33〕
如上述〔30〕~〔32〕的任一项所述的低聚芴二芳基酯的制造方法,其特征在于,其具有在酯交换反应催化剂存在下使下述通式(10f)所表示的低聚芴二烷基酯与下述通式(11a)所表示的二芳基碳酸酯类发生反应的工序。
[化13]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,或者r1和r2为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团;
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基、或者氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
r18为氢原子或具有或不具有取代基的碳原子数1~10的烷基。
n表示1~5的整数值。)
[化14]
(式中,ar1为具有或不具有取代基的碳原子数4~10的芳基。)
发明的效果
本发明的树脂组合物在成型为膜时显示出优异的光学特性,通过使用即使在树脂中的比例低也可有效表现出所期望的光学特性的重复单元,可提高树脂设计的自由度,兼顾耐热性、熔融加工性、机械强度等各种物性,因而其在光学用途中、特别是作为相位差膜用的材料是有用的。本发明中还能够提供一种低聚芴二酯单体和该单体的制造方法,该低聚芴二酯单体和该制造方法能够适合地用于这样的树脂组合物。此外,本发明的低聚芴二醇在制成树脂组合物时具有光弹性系数低、相位差的波长分散性小之类的优异光学特性,因而在光学用途中、特别是作为相位差膜用的材料是有用的。
附图说明
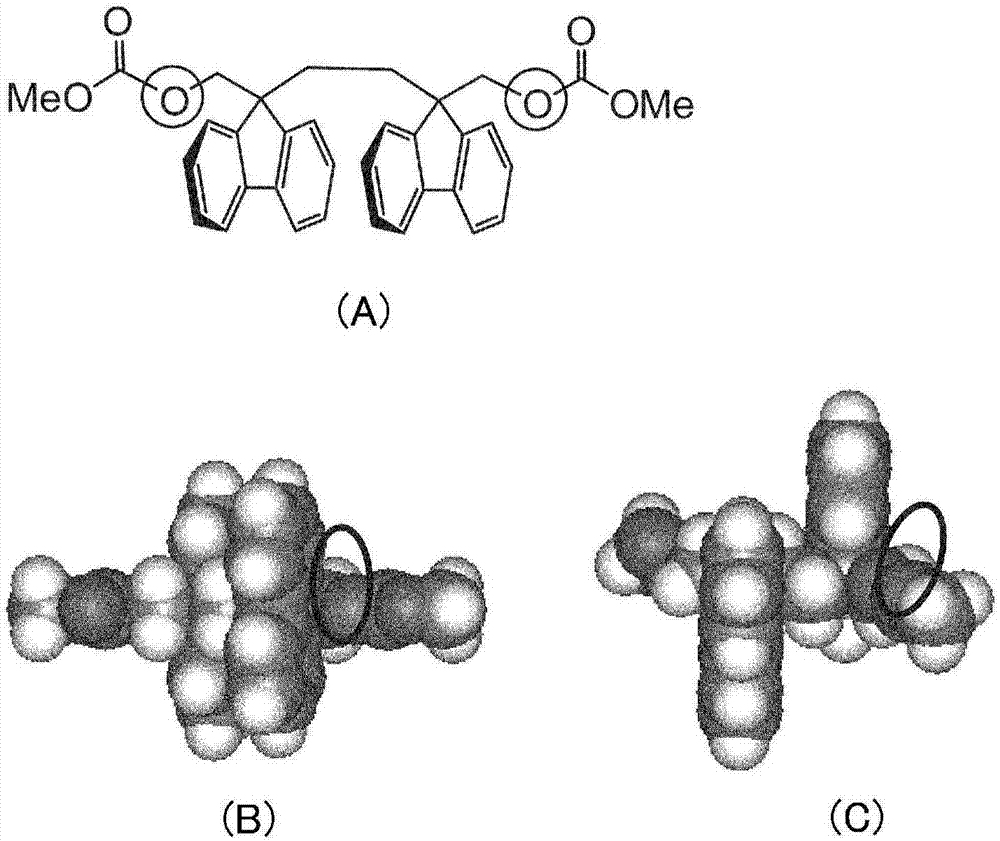
图1中,图1(a)~图1(c)表示利用碳酸甲酯进行了末端基团改性的化合物7b,图1(a)表示该化合物的结构式,图1(b)表示该化合物的反式构象的空间填充模型,图1(c)表示该化合物的歪扭构象的空间填充模型。
图2中,图2(a)~图2(c)表示利用碳酸甲酯进行了末端基团改性的化合物3b,图2(a)表示该化合物的结构式,图2(b)表示该化合物的反式构象的空间填充模型,图2(c)表示该化合物的歪扭构象的空间填充模型。
具体实施方式
下面详细说明本发明的实施方式,但以下记载的构成要件的说明为本发明实施方式的一例(代表例),只要不超出其要点,本发明并不限于以下的内容。在本发明中,“重量”与“质量”的含义相同。此外,在本发明的聚碳酸酯树脂组合物中,不仅含有具有碳酸酯结构的聚合物,还含有其它聚合物、以及含有在该聚合物的制造时生成的各种化合物,还包含在该聚合物中混配的各种添加剂等。
本发明中的重复单元表示的是在聚合物中被夹在任意的连接基团之间的部分结构。在聚合物的末端部分也包括一侧为连接基团、另一侧为聚合反应性基团的部分结构。需要说明的是,本发明中的结构单元与重复单元的含义相同。
此外,本发明中,“具有或不具有取代基”与“可以被取代”含义相同。
本发明的树脂组合物为包含具有2价低聚芴作为重复单元的聚合物的树脂组合物。
如此,本发明的树脂组合物除了含有具有2价低聚芴作为重复单元的聚合物以外,还可以含有后述的其它聚合物,此外还可含有添加剂等。并且,本发明的树脂组合物也可以由具有2价低聚芴作为重复单元的聚合物形成。
<1.低聚芴>
本发明的树脂组合物所含有的聚合物具有2价低聚芴作为重复单元。
2价低聚芴包含具有或不具有取代基的2个以上芴单元,并且该芴单元的9位碳原子彼此直接键合,或者该芴单元的9位碳原子彼此藉由具有或不具有取代基的亚烷基、具有或不具有取代基的亚芳香基或具有或不具有取代基的亚芳烷基进行键合。
上述亚烷基中的碳原子数没有特别限定,从提高后述芴比例的方面出发,通常为1以上,并且通常为10以下、优选为5以下、更优选为3以下。
另外,上述亚芳香基中的碳原子数没有特别限定,从提高后述芴比例的方面出发,通常为4以上,并且通常为10以下、优选为8以下、更优选为6以下。
进而,上述亚芳烷基中的碳原子数没有特别限定,从提高后述芴比例的方面出发,通常为6以上,并且通常为10以下、优选为9以下、更优选为8以下。
另外,作为上述芴单元可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
关于上述的2价低聚芴,在2个以上芴单元中,也可使位于两末端的芴单元的9位碳原子为2价基团(即后述通式(1)中的r1和r2直接键合的情况);另一方面,也可以使位于两末端的芴单元的9位碳原子分别键合取代基α1和α2、该取代基α1和α2为2价基团。在后者的情况下,α1与α2可以相同、也可以不同。
特别是在位于两末端的芴单元的9位碳原子为2价基团的情况下,或者是与位于两末端的芴单元的9位碳原子分别键合的取代基α1和α2为2价基团且α1与α2中的至少1者的碳原子数为2以上的情况下,由于芴环(芴单元)相对于主链大致垂直地取向,因而即使树脂组合物中的2价低聚芴的比例为少量,也具有容易表现出逆波长分散性的倾向。在后者的情况下,从同样的方面出发,优选α1和α2这两者的碳原子数为2以上。另一方面,在与两末端的芴单元的9位碳原子分别键合的α1和α2为2价基团且α1和α2这两者的碳原子数为1(即为具有或不具有取代基的亚甲基)的情况下,芴环(芴单元)相对于主链并未大致垂直地取向、而有较大倾斜地进行取向,因而即使树脂组合物中的2价低聚芴的比例在较宽范围内进行变化,也具有如下倾向:容易呈在宽带域内相位差的差异较小的平坦分散性。
此外,作为碳原子数为1的α1和α2,可以举出无取代的亚甲基或具有取代基的亚甲基,从提高2价低聚芴的重复单元中的芴比例的方面出发,优选无取代的亚甲基。作为可以具有的取代基,可以举出甲基、乙基、氟原子、氯原子、甲氧基等;从提高芴比例的方面出发,优选甲基。此外,具有取代基的亚甲基中,亚甲基所具有的2个氢原子中,可以两者被取代基取代,也可以任意之一被取代基取代,从提高芴比例的方面出发,优选任意一个被甲基取代基所取代。
如此,本发明的树脂组合物通过含有具有2以上的芴单元的9位碳原子彼此经特定的碳-碳键连接而成的重复单元的聚合物,能够更为有效地得到源于芴环的光学特性。
作为上述2价低聚芴,具体地说,可优选使用下述通式(1)所表示的物质。
[化15]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或具有或不具有取代基的碳原子数6~10的亚芳烷基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团;
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基、或者氰基[其中,r4~r9中,相邻的至少2个基团可以相互键合形成环]。
n表示1~5的整数值。)
需要说明的是,r1和r2与下述含义相同。
r1和r2各自独立地为选自如下示出的(a)~(e)组成的组中的任意一者。
(a)直接键合
(b)具有或不具有取代基的碳原子数1~10的亚烷基
(c)具有或不具有取代基的碳原子数4~10的亚芳香基
(d)具有或不具有取代基的碳原子数6~10的亚芳烷基
(e)选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团
<1-1.取代基的具体例>
下面举出r1~r3中的“具有或不具有取代基的碳原子数1~10的亚烷基”的具体结构,但并不限于这些,可以举出:亚甲基、亚乙基、正亚丙基、正亚丁基、正亚戊基、正亚己基等直链状的亚烷基;甲基亚甲基、二甲基亚甲基、乙基亚甲基、丙基亚甲基、丁基亚甲基、(1-甲基乙基)亚甲基、1-甲基亚乙基、2-甲基亚乙基、1-乙基亚乙基、2-乙基亚乙基、1-甲基亚丙基、2-甲基亚丙基、1,1-二甲基亚乙基、2,2-二甲基亚丙基、3-甲基亚丙基等包含支链的亚烷基(r1和r2中的取代位置的数值自芴环侧的碳起计数);在下述[c]组中示出的脂环结构的任意2处具有直链状或支链状的亚烷基结合键的脂环式亚烷基
[化16]
[c]
(上述[c]组中示出的各环结构中,2个结合键的取代位置为任意的,也可以在同一个碳上取代2个结合键。);在下述[d]组中示出的杂环结构的任意2处具有直链状或支链状的亚烷基结合键的杂环式亚烷基
[化17]
[d]
(上述[d]组中示出的各环结构中,2个结合键的取代位置为任意的,也可以在同一个碳上取代2个结合键。)。
上述[c]组中示出的脂环结构、或上述[d]组中示出的杂环结构中,在任意2处具有的直链状或支链状的亚烷基结合键的具体结构可以举出如下结构,但并不限于这些:亚甲基、亚乙基、正亚丙基、正亚丁基、正亚戊基、正亚己基等直链状的亚烷基;1-甲基亚乙基、2-甲基亚乙基、1-乙基亚乙基、2-乙基亚乙基、1-甲基亚丙基、2-甲基亚丙基、1,1-二甲基亚乙基、2,2-二甲基亚丙基、3-甲基亚丙基等包含支链的亚烷基(此处取代位置的数值自与上述环结构键合的碳起计数)。
此外,具有或不具有取代基的碳原子数1~10的亚烷基中的碳原子数优选为6以下、更优选为3以下。
特别是从表现出逆波长分散性的方面出发,在r1和/或r2为具有或不具有取代基的亚烷基的情况下,该碳原子数优选为2以上。另外,从表现出平坦分散性的方面出发,在r1和r2为具有或不具有取代基的亚烷基的情况下,该碳原子数优选为1。
进一步地,在表现出逆波长分散性时的r1和/或r2为具有或不具有取代基的亚烷基的情况下,从容易使芴环的取向相对于主链固定、有效地得到逆波长分散特性的方面出发,碳原子数优选为5以下、更优选为4以下、进一步优选为3以下、特别优选为2以下。另外,从对树脂组合物赋予柔软性的方面出发,该碳原子数优选为2以上、更优选为3以上、进一步优选为4以上。
此外,从提高芴比例的方面出发,在r3为具有或不具有取代基的亚烷基的情况下,该碳原子数优选为4以下、更优选为2以下。
作为该亚烷基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的亚烷基的具体例,可以举出:环丁基亚甲基、环戊基亚甲基、环己基亚甲基、1-环己基亚丙基等烷基取代亚烷基;苯基亚甲基、1-苯基亚乙基、1-苯基亚丙基等芳基取代亚烷基;1,1,2,2-四氟亚乙基、三氯甲基亚甲基、三氟甲基亚甲基等卤原子取代亚烷基;2-甲氧基甲基-2-甲基亚丙基等烷氧基取代亚烷基等。(r1和r2中的取代位置的数值自芴环侧的碳起计数)
下面举出r1~r3中的“具有或不具有取代基的碳原子数4~10的亚芳香基”的具体结构,但并不限于这些,可以举出:1,2-亚苯基、1,3-亚苯基、1,4-亚苯基等亚苯基;1,5-亚萘基、2,6-亚萘基等亚萘基;2,5-亚吡啶基、2,4-亚噻吩基、2,4-亚呋喃基等杂亚芳香基。
具有或不具有取代基的碳原子数4~10的亚芳香基中的碳原子数优选为8以下、更优选为6以下。
此外,从提高芴比例的方面出发,在r3为具有或不具有取代基的亚芳香基的情况下,其碳原子数优选为8以下、更优选为6以下。
作为该亚芳香基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的亚芳香基的具体例,可以举出2-甲基-1,4-亚苯基、3-甲基-1,4-亚苯基、3,5-二甲基-1,4-亚苯基、3-甲氧基-1,4-亚苯基、3-三氟甲基-1,4-亚苯基、2,5-二甲氧基-1,4-亚苯基、2,3,5,6-四氟-1,4-亚苯基、2,3,5,6-四氯-1,4-亚苯基、3-硝基-1,4-亚苯基、3-氰基-1,4-亚苯基等。
r1~r3中,“具有或不具有取代基的碳原子数6~10的亚芳烷基”的具体结构如下,可以举出下述[e]组中所示的亚芳烷基,
[化18]
[e]
但并不限于这些。
具有或不具有取代基的碳原子数6~10的亚芳烷基中的碳原子数优选为8以下。
此外,从提高芴比例的方面出发,在r3为具有或不具有取代基的亚芳烷基的情况下,其碳原子数优选为8以下。
作为该亚芳烷基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的亚芳烷基的具体例,可以举出2-甲基-1,4-亚二甲苯基、2,5-二甲基-1,4-亚二甲苯基、2-甲氧基-1,4-亚二甲苯基、2,5-二甲氧基-1,4-亚二甲苯基、2,3,5,6-四氟-1,4-亚二甲苯基、α,α-二甲基-1,4-亚二甲苯基、α,α,α’,α’-四甲基-1,4-亚二甲苯基等。
r1和r2中,“选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团”的具体结构如下,可以举出下述[f]组所示的2价基团,
[化19]
[fl
但并不限于这些。它们之中,优选在能够保持树脂组合物的透明性和稳定性的同时赋予柔软性的、选自亚烷基、亚芳香基或亚芳烷基中的2个以上基团经氧原子连接而成的基团,更优选可在赋予柔软性的同时提高树脂组合物的玻璃化转变温度的下述[g]组中示出的亚烷基经氧原子连接而成的基团。
[化20]
[g]
此外,从提高芴比例的方面出发,在r3为这些经连接而成的基团的情况下,其碳原子数优选为2以上,并且优选为6以下、更优选为4以下。
关于r1和r2,它们之中,优选为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团,更优选为直接键合、直链状的亚烷基、包含支链的亚烷基、在上述[c]组中示出的脂环结构的任意2处具有直链状或者支链状的亚烷基结合键的脂环式亚烷基、亚苯基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子连接而成的基团,进一步优选的是,由于不具有芳香环而具有能够实现光学膜所要求的低光弹性系数的倾向的、直接键合、亚甲基、亚乙基、正亚丙基、正亚丁基、甲基亚甲基、1-甲基亚乙基、2-甲基亚乙基、2,2-二甲基亚丙基、2-甲氧基甲基-2-甲基亚丙基或下述[h]组中示出的脂环式亚烷基
[化21]
[h]
(上述[h]组中示出的各环结构中的2个结合键的取代位置为任意的,可以在同一个碳上取代有2个结合键。),更进一步优选直接键合、亚甲基、亚乙基、正亚丙基、正亚丁基、甲基亚甲基、1-甲基亚乙基、2-甲基亚乙基、或2,2-二甲基亚丙基,特别优选亚甲基、亚乙基、或者正亚丙基。由于链长较长时具有玻璃化转变温度降低的倾向,因而优选较短的链状基团,例如优选碳原子数为2以下的基团。进一步地,由于分子结构变小,因而具有能够提高重复单元中的芴环的浓度(芴比例)的倾向,从而能够效率良好地表现出所期望的光学物性。最优选为亚甲基,该基团即使相对于树脂组合物整体的质量以任意质量含有,也具有可得到相位差的波长分散性小的平坦分散的倾向,进而还具有能够以较短的过程且工业上低成本地进行导入的优越性。
另一方面,出于改善所得到的膜的机械强度、高温下的可靠性的目的,优选能够提高树脂组合物的玻璃化转变温度的、碳原子数4~10的亚芳香基、或者选自由具有或不具有取代基的碳原子数1~10的亚烷基和具有或不具有取代基的碳原子数4~10的亚芳香基组成的组中的2个以上基团经氧原子连接而成的基团,更优选1,4-亚苯基、1,5-亚萘基、2,6-亚萘基、或者下述[f2]组中示出的2价基团。
[化22]
[f2]
此外,在应用于具有逆波长分散性的相位差膜的情况下,对于r1和r2的适当选择是重要的。例如,由于以亚甲基为代表的碳原子数为1的基团具有预料不到的逆波长分散性低的倾向,因而r1和r2优选为直接键合、或其至少任意一个是碳原子数为2以上的基团。
更优选为直接键合、具有或不具有取代基的碳原子数2~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团。
进一步优选为直接键合、直链状的亚烷基、包含支链的亚烷基、在上述[c]组中示出的脂环结构的任意2处具有直链状或者支链状的亚烷基结合键的脂环式亚烷基、亚苯基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子连接而成的基团。
更进一步优选的是,由于不具有芳香环而能够实现光学膜所要求的低光弹性系数的、直接键合、亚乙基、正亚丙基、正亚丁基、甲基亚甲基、1-甲基亚乙基、2-甲基亚乙基、2,2-二甲基亚丙基、2-甲氧基甲基-2-甲基亚丙基或上述[h]组中示出的脂环式亚烷基;或者能够增高树脂组合物的玻璃化转变温度的、1,4-亚苯基、选自由具有或不具有取代基的碳原子数1~10的亚烷基和具有或不具有取代基的碳原子数4~10的亚芳香基组成的组中的2个以上基团经氧原子连接而成的基团。
特别优选为直接键合、亚乙基、正亚丙基、正亚丁基、甲基亚甲基、1-甲基亚乙基、2-甲基亚乙基、或2,2-二甲基亚丙基。
最优选为亚乙基、或者正亚丙基。由于链长较长时具有玻璃化转变温度降低的倾向,因而优选较短的链状基团,例如优选碳原子数为3以下的基团。进一步地,由于分子结构减小,因而具有能够提高重复单元中的芴环的浓度(芴比例),从而能够效率良好地表现出所期望的光学物性。
此外,出于制造容易的原因,优选r1和r2相同。
关于r3,其中优选具有或不具有取代基的碳原子数1~10的亚烷基、或者具有或不具有取代基的碳原子数4~10的亚芳香基,更优选直链状的亚烷基、包含支链的亚烷基、在上述[c]组中示出的脂环结构的任意2处具有直链状或者支链状的亚烷基结合键的脂环式亚烷基、或者亚苯基,进一步优选的是,由于不具有芳香环而具有能够实现光学膜所要求的低光弹性系数的倾向的亚甲基、亚乙基、正亚丙基、正亚丁基、甲基亚甲基、二甲基亚甲基、乙基亚甲基、丙基亚甲基、丁基亚甲基、(1-甲基乙基)亚甲基、2,2-二甲基亚丙基、苯基亚甲基、三氯甲基亚甲基、三氟甲基亚甲基、上述[h]组中示出的脂环式亚烷基、或杂环式亚烷基,或者能够增高树脂组合物的玻璃化转变温度的1,4-亚苯基,特别优选亚甲基、甲基亚甲基、亚乙基、正亚丙基、或2,2-二甲基亚丙基。长链状基团具有降低树脂组合物的玻璃化转变温度的倾向。
下面举出r4~r9中的“具有或不具有取代基的碳原子数1~10的烷基”的具体结构,但并不限于这些,可以举出:甲基、乙基、正丙基、正丁基、正戊基、正己基、正癸基等直链状的烷基;异丙基、2-甲基丙基、2,2-二甲基丙基、2-乙基己基等包含支链的烷基;环丙基、环戊基、环己基、环辛基等环状的烷基。
具有或不具有取代基的碳原子数1~10的烷基中的碳原子数优选为4以下、更优选为2以下。该碳原子数为该范围内时,芴环彼此之间不易产生空间位阻,具有可得到源于芴环的所期望的光学特性的倾向。
作为该烷基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的烷基的具体例,可以举出三氟甲基、苄基、4-甲氧基苄基、甲氧基甲基等。
下面举出r4~r9中的“具有或不具有取代基的碳原子数4~10的芳基”的具体结构,但并不限于这些,可以举出:苯基、1-萘基、2-萘基等芳基;2-吡啶基、2-噻吩基、2-呋喃基等杂芳基。
具有或不具有取代基的碳原子数4~10的芳基中的碳原子数优选为8以下、更优选为7以下。该碳原子数为该范围内时,芴环彼此之间不易产生空间位阻,具有可得到源于芴环的所期望的光学特性的倾向。
作为该芳基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的芳基的具体例,可以举出2-甲基苯基、4-甲基苯基、3,5-二甲基苯基、4-苯甲酰基苯基、4-甲氧基苯基、4-硝基苯基、4-氰基苯基、3-三氟甲基苯基、3,4-二甲氧基苯基、3,4-亚甲基二羟苯基、2,3,4,5,6-五氟苯基、4-甲基呋喃基等。
下面举出r4~r9中的“具有或不具有取代基的碳原子数1~10的酰基”的具体结构,但并不限于这些,可以举出:甲酰基、乙酰基、丙酰基、2-甲基丙酰基、2,2-二甲基丙酰基、2-乙基己酰基等脂肪族酰基;苯甲酰基、1-萘基羰基、2-萘基羰基、2-呋喃基羰基等芳香族酰基。
具有或不具有取代基的碳原子数1~10的酰基中的碳原子数优选为4以下、更优选为2以下。该碳原子数为该范围内时,芴环彼此之间不易产生空间位阻,具有可得到源于芴环的所期望的光学特性的倾向。
作为该酰基可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有或不具有取代基的酰基的具体例,可以举出氯乙酰基、三氟乙酰基、甲氧基乙酰基、苯氧基乙酰基、4-甲氧基苯甲酰基、4-硝基苯甲酰基、4-氰基苯甲酰基、4-三氟甲基苯甲酰基等。
下面举出r4~r9中的“具有或不具有取代基的碳原子数1~10的烷氧基或芳氧基”的具体结构,但并不限于这些,可以举出:甲氧基、乙氧基、异丙氧基、叔丁氧基、三氟甲氧基、苯氧基等烷氧基;乙酰氧基、苯甲酰基氧基等酰氧基。
具有或不具有取代基的碳原子数1~10的烷氧基或芳氧基中的碳原子数优选为4以下、更优选为2以下。碳原子数为该范围内时,芴环彼此之间不易产生空间位阻,具有可得到源于芴环的所期望的光学特性的倾向。
下面举出r4~r9中的“具有或不具有取代基的氨基”的具体结构,但并不限于这些,可以举出:氨基;n-甲基氨基、n,n-二甲氨基、n-乙基氨基、n,n-二乙基氨基、n,n-甲基乙基氨基、n-丙基氨基、n,n-二丙基氨基、n-异丙基氨基、n,n-二异丙基氨基等脂肪族氨基;n-苯基氨基、n,n-二苯基氨基等芳香族氨基;甲酰胺基、乙酰胺基、癸酰胺基、苯甲酰胺基、氯乙酰胺基等酰基氨基;苄氧基羰基氨基、叔丁氧基羰基氨基等烷氧基羰基氨基等。
它们之中,出于不具有酸性度高的质子、分子量小、具有能够提高芴比例的倾向的原因,优选n,n-二甲氨基、n-乙基氨基、或者n,n-二乙基氨基,更优选n,n-二甲氨基。
下面举出r4~r9中的“具有取代基的硫原子”的具体结构,但并不限于这些,可以举出:磺基;甲基磺酰基、乙基磺酰基、丙基磺酰基、异丙基磺酰基等烷基磺酰基;苯磺酰基、对甲苯基磺酰基等芳基磺酰基;甲基亚磺酰基、乙基亚磺酰基、丙基亚磺酰基、异丙基亚磺酰基等烷基亚磺酰基;苯基亚磺酰基、对甲苯基亚磺酰基等芳基亚磺酰基;甲硫基、乙硫基等烷硫基;苯硫基、对甲苯基硫基等芳硫基;甲氧基磺酰基、乙氧基磺酰基等烷氧基磺酰基;苯氧基磺酰基等芳氧基磺酰基;氨基磺酰基;n-甲基氨基磺酰基、n-乙基氨基磺酰基、n-叔丁基氨基磺酰基、n,n-二甲氨基磺酰基、n,n-二乙基氨基磺酰基等烷基磺酰基;n-苯基氨基磺酰基、n,n-二苯基氨基磺酰基等芳基氨基磺酰基等。需要说明的是,磺基可以与锂、钠、钾、镁、铵等形成盐。
它们之中,出于不具有酸性度高的质子、分子量小、具有能够提高芴比例的倾向的原因,优选甲基亚磺酰基、乙基亚磺酰基或者苯基亚磺酰基,更优选甲基亚磺酰基。
作为r4~r9中的“卤原子”,可以举出氟原子、氯原子、溴原子、碘原子。
它们之中,出于比较容易导入、由于为吸电子性的取代基因而具有提高芴9位的反应性的倾向的原因,优选氟原子、氯原子或者溴原子,更优选氯原子或溴原子。
相邻的r4~r9可以相互键合形成环。作为其具体例,可以举出下述[i]组中示出的取代芴结构。
[化23]
[]
如此,通过使r4~r9为上述那样的特定的原子或取代基,在主链与芴环之间、芴环彼此之间的空间位阻少,具有能够得到源于芴环的所期望的光学特性的倾向。
这些r4~r9中,优选全部为氢原子,或者r4和/或r9为选自由卤原子、酰基、硝基、氰基以及磺基组成的组中的任意一种、且r5~r8为氢原子。在全部为氢原子的情况下,在工业上也能够由低成本的芴衍生出。另外,在r4和/或r9为选自由卤原子、酰基、硝基、氰基以及磺基组成的组中的任一种、且r5~r8为氢原子的情况下,由于芴9位的反应性提高,因而具有能够适应各种诱导反应的倾向。更优选全部为氢原子,或者r4和/或r9为选自由氟原子、氯原子、溴原子以及硝基组成的组中的任意一种、且r5~r8为氢原子,特别优选全部为氢原子的情况。此外,通过为上述基团,能够提高芴比例,并且芴环彼此间不易产生空间位阻,还具有可得到源于芴环的所期望的光学特性的倾向。
此外,上述通式(1)中的n表示1~5的整数值,从合成上的容易性的方面出发,优选为4以下、更优选为3以下。
需要说明的是,如上所述,上述2价低聚芴包含2个以上芴单元,该芴的9位碳原子彼此直接键合或者藉由亚烷基、亚芳香基或亚芳烷基进行键合,该亚烷基具有或不具有取代基,该亚芳香基具有或不具有取代基,该亚芳烷基具有或不具有取代基;更具体地说,2个以上芴单元的9位碳原子彼此可以藉由通式(1)中的作为r3的上述基团进行键合。此外,作为亚烷基可以具有的取代基、亚芳香基可以具有的取代基、亚芳烷基可以具有的取代基,可使用通式(1)中的作为r3中的取代基而示例出的基团。另外,作为上述取代基α1和/或α2,可使用通式(1)中的r1和/或r2。
<1-2.具体的结构>
作为上述通式(1)所表示的2价低聚芴的具体结构,可以举出下述[j]组中示出的结构。
[化24]
[j]
[化25]
它们之中优选的2价低聚芴可以举出下述[k]组中示出的结构。
[化26]
[k]
它们之中,下图所示的2价低聚芴包含2个无取代的芴单元,芴的9位碳原子彼此直接键合,且芴单元的9位碳原子为2价基团。
[化27]
并且,它们之中,下图所示的2价低聚芴包含3个无取代的芴单元,3个芴单元的9位碳原子彼此藉由亚甲基呈直线状键合,并且分别键合在位于两末端的芴单元的9位碳原子上的亚甲基为2价基团。
[化28]
<2.聚合物>
本发明的树脂组合物中所含有的聚合物具有2价低聚芴作为重复单元。例如可以举出2价低聚芴彼此通过任意的连接基团进行连接而成的聚合物。此外,该聚合物也可以为具有2价低聚芴以外的任意重复单元的共聚物。
<2-1.连接基团>
下面举出上述聚合物中所用的连接基团的具体结构,但并不限于这些,可以举出:下述[a]组中示出的连接基团
[化29]
[a]
(上述[a]组中示出的各连接基团中,z表示重复单元所连接的部位,y表示与其它连接基团的键合部位、或者表示使连接基团彼此键合的任意结构单元所连接的部位。),可以将这些连接基团中的两种以上合用。此外,在连接基团为非对称的情况下,连接基团可以按照任意方向与重复单元连接。它们之中,优选构成耐热性与熔融加工性、机械强度的平衡优异的聚酯、聚碳酸酯、聚酯碳酸酯的、下述[b]组中示出的连接基团。
[化30]
[b]
(上述[b]组中示出的各连接基团中,z表示重复单元所连接的部位。)
连接基团可以单独使用一种基团,也可将两种以上的连接基团合用。
作为重复单元经连接基团连接而成的聚合物,具体地说,可以举出包含聚烯烃、聚酯、聚碳酸酯、聚酰胺、聚酰亚胺、聚氨酯、环氧树脂、聚丙烯酸酯、聚甲基丙烯酸酯或聚砜的聚合物以及将它们合用得到的聚合物,优选包含通常为高透明性的聚烯烃、聚酯、聚碳酸酯、环氧树脂或聚丙烯酸酯的聚合物,特别优选包含耐热性与熔融加工性、机械强度的平衡优异的聚酯或聚碳酸酯的聚合物,特别优选包含通常耐热性、耐化学药品性优异的聚碳酸酯的聚合物。
在制成将两种以上连接基团合用的聚合物的情况下,关于连接基团的组合没有特别限定,例如可以举出合用碳酸酯结构与酯结构作为连接基团的聚合物、合用碳酸酯结构与氨基甲酸酯结构作为连接基团的聚合物、合用酯结构与酰胺作为连接基团的聚合物等,优选可以举出合用碳酸酯结构与酯结构作为连接基团的聚合物。此处,作为合用两种以上的连接基团的聚合物的具体例,可以举出聚酯碳酸酯、具有碳酸酯键的聚氨酯、聚酯酰胺、聚酯酰亚胺等,它们之中,优选耐热性与熔融加工性、机械强度的平衡优异的聚酯碳酸酯。本说明书中,将具有碳酸酯键的聚合物称为聚碳酸酯,除了仅具有碳酸酯键作为连接基团的聚合物外,还包含聚酯碳酸酯(具有酯键和碳酸酯键的聚合物)、具有碳酸酯键的聚氨酯等。此处,包含聚碳酸酯的聚合物中的碳酸酯键的比例可以为任意值,但为了对树脂组合物赋予由碳酸酯键带来的耐热性、耐化学药品性等优异的特性,优选其为一定比例以上,碳酸酯键在全部连接基团中的摩尔分数优选为30%以上、更优选为50%以上、进一步优选为60%以上、特别优选为70%以上,通常为100%以下。
<2-2.共聚物>
具有2价低聚芴作为重复单元的聚合物可以为进一步含有任意的2价有机基团作为重复单元的共聚物。这种情况下,优选重复单元彼此通过上述的连接基团进行连接。
共聚物中,作为可以与2价低聚芴合用的任意的2价有机基团,从控制在树脂组合物中所需要的光学特性和物性的范围的方面出发,可优选使用下述通式(3)所表示的2价有机基团。这种情况下,作为任意的2价有机基团,可以进一步合用通式(3)所表示的2价有机基团以外的2价有机基团。
[化31]
(式中,r10表示具有或不具有取代基的碳原子数2~20的亚烷基、具有或不具有取代基的碳原子数4~20的亚芳香基、具有或不具有取代基的碳原子数2~100的亚烷基醚基、具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团。)
聚合物含有上述通式(3)所表示的2价有机基团的情况下,除了能够承担对树脂组合物赋予正折射率各向异性的功能以外,还可将相位差的波长分散性或光弹性系数之类的光学物性以及机械强度、耐热性、熔融加工性等各种树脂物性控制在优选的范围内等,具有能够任意地控制树脂组合物的物性的倾向。
需要说明的是,不具有相对于主链垂直取向的芳香环、或者即使具有这样的芳香环其在整体中的比例也较少的树脂组合物通常显示出正折射率各向异性,这是众所周知的。上述通式(3)所表示的2价有机基团的重复单元中,除了在侧链具有芳香环的结构以外,全部为显示出正折射率各向异性的结构,因而可认为,含有50摩尔%以上的上述通式(3)所表示的2价有机基团的树脂组合物显示出正折射率各向异性。
<2-3.有机基团的具体例>
如上所述,通式(3)中的r10表示具有或不具有取代基的碳原子数2~20的亚烷基、具有或不具有取代基的碳原子数4~20的亚芳香基、具有或不具有取代基的碳原子数2~100的亚烷基醚基、具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团、或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团。
下面举出“具有或不具有取代基的碳原子数2~20的亚烷基”的具体结构,但并不限于这些,可以举出:亚乙基、正亚丙基、正亚丁基、正亚戊基、正亚己基等直链状的亚烷基;1-甲基亚乙基、2-甲基亚乙基、1-乙基亚乙基、2-乙基亚乙基、1-甲基亚丙基、2-甲基亚丙基、2,2-二甲基亚丙基、3-甲基亚丙基等包含支链的亚烷基。其碳原子数优选为2以上、并且为12以下,更优选为6以下。它们之中,优选具有适度的疏水性与柔软性并具有赋予低光弹性系数的倾向的、下述通式(5)所表示的直链状的亚烷基。
[化32]
(式中,r11表示具有或不具有取代基的碳原子数0~18的直链状亚烷基。)
下面举出“具有或不具有取代基的碳原子数0~18的直链状亚烷基”的具体结构,但并不限于这些,可以举出:亚乙基、正亚丙基、正亚丁基、正亚戊基、正亚己基等。其碳原子数优选为2以上,并且优选为10以下、更优选为4以下。
下面举出“具有或不具有取代基的碳原子数4~20的亚芳香基”的具体结构,但并不限于这些,可以举出:1,2-亚苯基、1,3-亚苯基、1,4-亚苯基等亚苯基;、1,5-亚萘基、2,6-亚萘基等亚萘基;2,5-亚吡啶基、2,4-亚噻吩基、2,4-亚呋喃基等杂亚芳香基。其碳原子数优选为4以上、并且为8以下,更优选为6以下。它们之中,从能够在工业上低成本地获得的方面出发,优选1,2-亚苯基、1,3-亚苯基或1,4-亚苯基。
作为“具有或不具有取代基的碳原子数2~20的亚烷基”、“具有或不具有取代基的碳原子数2~20的直链状亚烷基”和“具有或不具有取代基的碳原子数4~20的亚芳香基”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同,也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有取代基的亚烷基的具体例,可以举出苯基亚乙基、1-苯基亚丙基、1-环己基亚丙基、1,1,2,2-四氟亚乙基等。
作为具有取代基的亚芳香基的具体例,可以举出2-甲基-1,4-亚苯基、5-甲基-1,3-亚苯基、2,5-二甲基-1,4-亚苯基、2-甲氧基-1,4-亚苯基、2-三氟甲基-1,4-亚苯基、2,5-二甲氧基-1,4-亚苯基、2,3,5,6-四氟-1,4-亚苯基等取代亚芳香基。
下面举出“具有或不具有取代基的碳原子数6~20的亚芳烷基”的具体结构,但并不限于这些,可以举出:下述[m]组中示出的亚芳烷基。
[化33]
[m]
其碳原子数优选为6以上、并且为10以下,更优选为8以下。它们之中,从能够在工业上低成本地获得的方面出发、优选邻亚二甲苯基、间亚二甲苯基或对亚二甲苯基。
作为“具有或不具有取代基的碳原子数6~20的亚芳烷基”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
“具有或不具有取代基的碳原子数2~100的亚烷基醚基”是指具有1个以上的亚烷基与醚性氧原子的2价基团。其碳原子数优选为4以上、更优选为6以上,并且优选为60以下、更优选为50以下、进一步优选为40以下、特别优选为30以下。更具体地说,可以举出:下述通式(7)
[化34]
(式中,r13表示具有或不具有取代基的碳原子数2~10的亚烷基,p为1~40的整数。)所表示的基团、或者下述通式(8)
[化35]
(式中,r14表示具有或不具有取代基的碳原子数2~10的亚烷基,r15表示具有或不具有取代基的碳原子数12~30的亚芳香基。)所表示的基团。
式(7)和式(8)中,r13和r14表示具有或不具有取代基的碳原子数2~10的亚烷基。下面举出其具体结构,但并不限于这些,可以举出:亚乙基、正亚丙基、正亚丁基、正亚戊基、正亚己基等直链状的亚烷基;1-甲基亚乙基、2-甲基亚乙基、1-乙基亚乙基、2-乙基亚乙基、1-甲基亚丙基、2-甲基亚丙基、2,2-二甲基亚丙基、3-甲基亚丙基等包含支链的亚烷基(此处取代位置的数值自末端侧的碳起计数)等。
其碳原子数优选为2以上、并且为8以下,更优选为4以下。
作为“具有或不具有取代基的碳原子数2~100的亚烷基醚基”和“具有或不具有取代基的碳原子数2~10的亚烷基”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
作为具有取代基的亚烷基的具体例,可以举出苯基亚乙基、1-苯基亚丙基、1-环己基亚丙基、1,1,2,2-四氟亚乙基等。
这些r13和r14中,优选直链状的亚烷基,直链状的亚烷基由于不具有不对称点因而容易进行单体的品质管理,更优选能够在工业上低成本地导入、可赋予柔软性与吸水性的亚乙基。
式(7)中,p为1~40的整数,优选为1以上、更优选为2以上,并且优选为30以下、更优选为20以下。
式(8)中,r15表示具有或不具有取代基的碳原子数12~30的亚芳香基。下面举出其具体结构,但并不限于这些,从能够提高树脂组合物的玻璃化转变温度的方面出发,可以举出下述[n]组中示出的亚芳香基。
[化36]
[n]
作为“具有或不具有取代基的碳原子数6~20的亚芳香基”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
下面举出式(7)的具体结构,但并不限于这些,可以举出:下述[o]组中示出的亚烷基醚基
[化37]
[o]
(上述[o]组中,关于可能为非对映体的结构,可以为任意一种非对映体,也可以为非对映体混合物。)。
下面举出式(8)的具体结构,但并不限于这些,可以举出:下述[p]组中示出的亚烷基醚基
[化38]
[p]
(上述[p]组中,关于可能为非对映体的结构,可以为任意一种非对映体,也可以为非对映体混合物。)。
下面举出“具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团”的具体结构,但并不限于这些,出于具有能够提高玻璃化转变温度、能够降低光弹性系数的倾向的原因,可以优选地举出在下述[q]组中示出的脂环结构或杂环结构的任意2处具有直链状或支链状的亚烷基结合键的有机基团。
[化39]
[q]
(上述[q]组中示出的各环结构中的2个结合键的取代位置为任意的,也可以在同一个碳上取代有2个结合键。)此处的结合键为直接键合、或者是碳原子数1~5的直链状或者支链状的亚烷基,2个结合键的长度可以不同。优选的结合键为玻璃化转变温度的降低少的直接键合、或者亚甲基。
作为“具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
下面举出“具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团”的优选具体结构,但并不限于这些,可以举出:具有可赋予高透明性与玻璃化转变温度、吸水性、双折射、低光弹性系数的倾向的、下述通式(4)
[化40]
所表示的基团、下述通式(6)
[化41]
(式中,r12表示具有或不具有取代基的碳原子数4~20的环亚烷基。)所表示的基团、或者下述通式(9)
[化42]
(式中,r16表示具有或不具有取代基的具有碳原子数2~20的乙缩醛环的基团。)所表示的基团。
式(6)中,r12表示具有或不具有取代基的碳原子数4~20的环亚烷基。下面举出其具体结构,但并不限于这些,出于具有能够提高玻璃化转变温度、能够降低光弹性系数的倾向的原因,优选可以举出下述[r]组中示出的环亚烷基
[化43]
[r]
(上述[r]组中,关于可能为非对映体的结构,可以为任意一种非对映体,也可以为非对映体混合物。)。
作为“具有或不具有取代基的碳原子数4~20的环亚烷基”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
下面举出式(6)的具体结构,但并不限于这些,可以举出下述[s]组中示出的具有脂环结构的有机基团
[化44]
[s]
(上述[s]组中,关于可能为非对映体的结构,可以为任意一种非对映体,也可以为非对映体混合物。)。它们之中,从能够在工业上低成本地获得的方面出发,优选下述[s-2]组中示出的具有脂环结构的有机基团。
[化45]
[s-2]
式(9)中,r16表示具有或不具有取代基的碳原子数2~20的乙缩醛环。下面举出其具体结构,但并不限于这些,出于具有能够提高玻璃化转变温度与双折射、能够降低光弹性系数的倾向的原因,优选可以举出下述[t]组中示出的具有乙缩醛环的基团
[化46]
[t]
(上述[t]组中,关于可能为非对映体的结构,可以为任意一种非对映体。)。
作为“具有或不具有取代基的碳原子数2~20的乙缩醛环”可以具有的取代基,可以举出:卤原子(例如氟原子、氯原子、溴原子、碘原子);碳原子数1~10的烷基(例如甲基、乙基、异丙基等);碳原子数1~10的烷氧基(例如甲氧基、乙氧基等);碳原子数1~10的酰基(例如乙酰基、苯甲酰基等);碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等);硝基;氰基;可以具有选自卤原子(例如氟原子、氯原子、溴原子、碘原子)、碳原子数1~10的烷基(例如甲基、乙基、异丙基等)、碳原子数1~10的烷氧基(例如甲氧基、乙氧基等)、碳原子数1~10的酰基(例如乙酰基、苯甲酰基等)、碳原子数1~10的酰氨基(例如乙酰胺基、苯甲酰基酰胺基等)、硝基、氰基等中的1~3个取代基的碳原子数6~10的芳基(例如苯基、萘基等)等。该取代基的个数没有特别限定,优选为1~3个。取代基为2个以上的情况下,取代基的种类可以相同、也可以不同。此外,从能够低成本地进行工业制造的方面出发,优选无取代。
通式(3)所表示的2价有机基团中的优选基团为具有或不具有取代基的亚烷基、具有或不具有取代基的亚烷基醚基、具有或不具有取代基的具有脂环结构的有机基团或者具有或不具有取代基的具有杂环结构的有机基团,这些优选基团在主链中不具有芳香环或者在主链中大量含有芳香环以外的部分结构,因而具有能够实现光学膜中所要求的低光弹性系数的倾向。更优选为选自下述基团中的至少一种:
具有可赋予高透明性与玻璃化转变温度、吸水性、双折射、低光弹性系数的倾向的下述通式(4)、
[化47]
或具有适度的疏水性与柔软性、具有可赋予低光弹性系数的倾向的下述通式(5)
[化48]
(式中,r11表示具有或不具有取代基的碳原子数0~18的直链状亚烷基。)、
或者具有可赋予较高的透明性与玻璃化转变温度、适度的柔软性的倾向的下述通式(6)
[化49]
(式中,r12表示具有或不具有取代基的碳原子数4~20的环亚烷基。)、或者具有可赋予柔软性与吸水性、低光弹性系数的倾向的下述通式(7)
[化50]
(式中,r13表示具有或不具有取代基的碳原子数2~10的亚烷基,p为1~40的整数。)、或者可赋予较高的透明性与玻璃化转变温度的倾向的下述通式(8)
[化51]
(式中,r14表示具有或不具有取代基的碳原子数2~10的亚烷基,r15表示具有或不具有取代基的碳原子数12~30的亚芳香基。)、或者具有可赋予较高的透明性与玻璃化转变温度、双折射的倾向的下述通式(9)
[化52]
(式中,r16表示具有或不具有取代基的具有碳原子数2~20的乙缩醛环的基团。)。进一步优选上述通式(4)所表示的基团,上述通式(4)所表示的基团通过赋予较高的透明性与玻璃化转变温度、吸水性、低光弹性系数而具有可赋予作为相位差膜优异的物性的倾向。
通式(3)所表示的2价有机基团可以可以单独使用一种,也可将两种以上合用。从减少批次间光学物性、机械物性的波动等之类的品质管理的方面出发,优选单独使用一种。另一方面,从兼顾光学特性、机械物性的方面出发,优选将两种以上合用来使用,并且通常为4种以下、优选为3种以下。
通式(3)所表示的2价有机基团合用两种以上的情况下,关于其组合没有特别限定。例如,出于赋予较高的透明性与玻璃化转变温度、双折射的目的,优选上述通式(4)所表示的有机基团或上述通式(9)所表示的有机基团,出于赋予柔软性的目的,优选上述通式(5)所表示的有机基团或者上述通式(7)所表示的有机基团,另一方面,出于赋予较高的透明性与玻璃化转变温度、适度的柔软性的目的,优选上述通式(6)所表示的有机基团,它们之中,可以根据所要求的目的的组合选择与其对应的有机基团的组合即可。具体地说,优选为相当于上述通式(4)所表示的有机基团的isb来源的重复单元与相当于上述通式(6)所表示的有机基团的chdm来源的重复单元的组合、或者相当于上述通式(9)所表示的有机基团的spg来源的重复单元与相当于上述通式(6)所表示的有机基团的chdm来源的重复单元的组合。
<2-4.共聚组成>
如此,在使用含有2价低聚芴以及通式(3)所表示的2价有机基团的至少两种以上作为重复单元的共聚物的情况下,只要在可表现出后述的光学物性的范围内,即可在上述共聚物中以任意质量含有2价低聚芴与通式(3)所表示的2价有机基团。
关于2价低聚芴的优选含有比例,为了表现出逆波长分散性且确保熔融加工性、机械强度,相对于上述共聚物整体的质量,该含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下。从同样的方面出发,2价低聚芴的优选摩尔分数相对于上述共聚物所含有的全部重复单元优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、更进一步优选为35%以下、特别优选为30%以下、最优选为20%以下。并且,从同样的方面出发,通式(3)所表示的2价有机基团的优选含有比例相对于上述共聚物整体的质量优选为10质量%以上、更优选为20质量%以上、进一步优选为30质量%以上、特别优选为40质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为88质量%以下、特别优选为85质量%以下、最优选为80质量%以下。从同样的方面出发,通式(3)所表示的2价有机基团的优选摩尔分数相对于上述共聚物所含有的全部重复单元优选为10%以上、更优选为20%以上、进一步优选为30%以上、特别优选为40%以上,并且优选为98%以下、更优选为95%以下、进一步优选为92%以下、更进一步优选为90%以下、特别优选为85%以下、最优选为80%以下。
另一方面,在使用r1和r2为具有或不具有取代基的亚甲基的2价低聚芴的情况下,即使相对于共聚物整体的质量以任意质量含有2价低聚芴,也具有能够表现出相位差的波长分散性小的平坦分散性的倾向。另一方面,通过提高含有比例,具有能够在维持平坦分散性的同时降低相位差、双折射的倾向,因而其也能够用作在全波长区域不易表现出相位差、双折射的宽带域零双折射材料。其优选含有比例相对于上述共聚物整体的质量优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,2价低聚芴的优选摩尔分数相对于上述共聚物整体所含有的全部重复单元优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。另外,从同样的方面出发,通式(3)所表示的2价有机基团的优选含有比例相对于上述共聚物整体的质量优选为5质量%以上、更优选为10质量%以上、进一步优选为15质量%以上、特别优选为20质量%以上,并且优选为70质量%以下、更优选为60质量%以下、进一步优选为50质量%以下、特别优选为40质量%以下。从同样的方面出发,通式(3)所表示的2价有机基团的优选摩尔分数相对于上述共聚物整体所含有的全部重复单元优选为20%以上、更优选为25%以上、进一步优选为30%以上、特别优选为35%以上,并且优选为90%以下、更优选为85%以下、进一步优选为80%以下、特别优选为75%以下。
<3.树脂组合物>
本发明的树脂组合物包含具有2价低聚芴作为重复单元的聚合物。此外,本发明的树脂组合物除了该聚合物外,还可以进一步含有其它成分。
<3-1.聚合物共混>
本发明的树脂组合物中,期待表现出由共混所带来的其它效果,也可以含有任意的聚合物作为其它成分。即,除了具有2价低聚芴作为重复单元的聚合物以外,还可以共存有除此以外的任意聚合物。
此处所说的共存是指在树脂组合物中存在2种以上的聚合物,该共存的方法不限,可以举出将2种以上的聚合物以溶液的状态或熔融的状态进行混合的方法、在含有1种以上的聚合物的溶液中或熔融液中进行聚合的方法等。
例如,可以共混具有通式(3)所表示的2价有机基团作为重复单元的聚合物,也可以共混具有任意的重复单元的聚合物。需要说明的是,具有通式(3)所表示的2价有机基团作为重复单元的聚合物可以进一步具有通式(3)以外的2价有机基团作为重复单元,也可以具有两种以上的通式(3)所表示的2价有机基团作为重复单元。此处,作为通式(3)所表示的2价有机基团,可以使用在共聚物中优选示例出的基团。
特别是从适宜作为相位差膜使用的方面出发,优选共存显示出正折射率各向异性的聚合物或低聚物的共混物或者共聚物;出于光学性能良好、具有能够进行熔融制膜或溶液浇注制膜的倾向的原因,更优选共存热塑性树脂。作为共存物,具体地说,可以举出缩聚系聚合物、烯烃系聚合物或者加成聚合系聚合物,优选缩聚系聚合物。作为缩聚系聚合物,可以举出聚酯、聚酰胺、聚酯碳酸酯、聚酰胺、聚酰亚胺等,其中优选聚酯或聚碳酸酯。
更具体地说,可以举出:聚乙烯、聚丙烯等烯烃系聚合物;具有来源于双酚a或双酚z、异山梨醇等的结构单元的聚碳酸酯;聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚萘二甲酸酯、聚环己烷二亚甲基环己烷二羧酸酯、聚对苯二甲酸-1,4-环己烷二甲醇酯等聚酯等,可以合用它们之中的2种以上的聚合物。
关于可以在本发明的树脂组合物中含有的任意的聚合物,只要在可表现出所需要的光学特性和物性的范围内,就可以在本发明的树脂组合物中以任意的比例含有,优选相对于树脂组合物的总质量为5质量%以上、更优选为10质量%以上、进一步优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为60质量%以下。含有比例过少时,具有难以得到由共混带来的优选物性的倾向;另一方面,含有比例过多时,2价低聚芴的比例相对降低,具有难以得到所需要的物性的倾向。
特别是在使具有2价低聚芴作为重复单元的聚合物与具有通式(3)所表示的2价有机基团作为重复单元的聚合物共存的情况下,只要在可表现出后述光学物性的范围内,其含有比例就没有特别限定。
为了表现出逆波长分散性并且确保熔融加工性、机械强度,相对于树脂组合物整体的质量,2价低聚芴的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上、最优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下、最优选为55质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,2价低聚芴的优选摩尔分数优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、更进一步优选为35%以下、特别优选为30%以下、最优选为20%以下。并且,从同样的方面出发,相对于树脂组合物整体的质量,通式(3)所表示的2价有机基团的优选含有比例优选为10质量%以上、更优选为20质量%以上、进一步优选为30质量%以上、特别优选为40质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为88质量%以下、特别优选为85质量%以下、最优选为80质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,通式(3)所表示的2价有机基团的优选摩尔分数优选为10%以上、更优选为20%以上、进一步优选为30%以上、特别优选为40%以上,并且优选为98%以下、更优选为95%以下、进一步优选为92%以下、更进一步优选为90%以下、特别优选为85%以下、最优选为80%以下。
并且,从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物的质量,2价低聚芴的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上、最优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下、最优选为55质量%以下。从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物,2价低聚芴的优选摩尔分数优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、更进一步优选为35%以下、特别优选为30%以下、最优选为20%以下。并且,从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物的质量,通式(3)所表示的2价有机基团的优选含有比例优选为10质量%以上、更优选为20质量%以上、进一步优选为30质量%以上、特别优选为40质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为88质量%以下、特别优选为85质量%以下、最优选为80质量%以下。从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物,通式(3)所表示的2价有机基团的优选摩尔分数优选为10%以上、更优选为20%以上、进一步优选为30%以上、特别优选为40%以上,并且优选为98%以下、更优选为95%以下、进一步优选为92%以下、更进一步优选为90%以下、特别优选为85%以下、最优选为80%以下。
另一方面,在使用r1和r2为具有或不具有取代基的亚甲基的2价低聚芴的情况下,即使相对于树脂组合物整体的质量以任意的含有比例含有该2价低聚芴,也具有相位差的波长分散性小、能够表现出平坦分散性的倾向。特别是通过提高含有比例,具有能够在维持平坦分散性的同时降低相位差、双折射的倾向,因而该组合物也能够用作在全波长区域不易表现出相位差、双折射的宽带域零双折射材料。其优选含有比例相对于树脂组合物整体的质量优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,2价低聚芴的优选摩尔分数优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。另外,从同样的方面出发,相对于树脂组合物整体的质量,通式(3)所表示的2价有机基团的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为15质量%以上、特别优选为20质量%以上,并且优选为70质量%以下、更优选为60质量%以下、进一步优选为50质量%以下、特别优选为40质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,通式(3)所表示的2价有机基团的优选摩尔分数优选为20%以上、更优选为25%以上、进一步优选为30%以上、特别优选为35%以上,并且优选为90%以下、更优选为85%以下、进一步优选为80%以下、特别优选为75%以下。
另外,从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物的质量,2价低聚芴的优选含有比例优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物,2价低聚芴的优选摩尔分数优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。另外,从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物的质量,通式(3)所表示的2价有机基团的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为15质量%以上、特别优选为20质量%以上,并且优选为70质量%以下、更优选为60质量%以下、进一步优选为50质量%以下、特别优选为40质量%以下。从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物,通式(3)所表示的2价有机基团的优选摩尔分数优选为20%以上、更优选为25%以上、进一步优选为30%以上、特别优选为35%以上,并且优选为90%以下、更优选为85%以下、进一步优选为80%以下、特别优选为75%以下。
对本发明的树脂组合物进行膜成型的情况下,由于优选膜为光学透明的,因而优选进行共混的聚合物与具有2价低聚芴作为重复单元的聚合物的折射率相近、或选择具有相容性的组合。
<3-2.树脂组合物的组成>
从表现出逆波长分散性并且确保熔融加工性、机械强度的方面出发,树脂组合物中的2价低聚芴的优选含有比例相对于树脂组合物整体的质量优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上、最优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的全部重复单元,2价低聚芴的优选摩尔分数优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、更进一步优选为35%以下、特别优选为30%以下、最优选为20%以下。另外,从同样的方面出发,相对于树脂组合物整体的质量,通式(3)所表示的2价有机基团的优选含有比例优选为10质量%以上、更优选为20质量%以上、进一步优选为30质量%以上、特别优选为40质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为88质量%以下、特别优选为85质量%以下、最优选为80质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的全部重复单元,通式(3)所表示的2价有机基团的优选摩尔分数优选为10%以上、更优选为20%以上、进一步优选为30%以上、特别优选为40%以上,并且优选为98%以下、更优选为95%以下、进一步优选为92%以下、更进一步优选为90%以下、特别优选为85%以下、最优选为80%以下。
另一方面,在使用r1和r2为具有或不具有取代基的亚甲基的2价低聚芴的情况下,即使相对于树脂组合物整体的质量以任意的含有比例含有2价低聚芴,也具有相位差的波长分散性小、能够表现出平坦分散性的倾向。特别是通过提高含有比例,具有能够在维持平坦分散性的同时降低相位差、双折射的倾向,因而该组合物也能够用作在全波长区域不易表现出相位差、双折射的宽带域零双折射材料。其优选含有比例相对于树脂组合物整体的质量优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的全部重复单元,2价低聚芴的优选摩尔分数优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。另外,从同样的方面出发,相对于树脂组合物整体的质量,通式(3)所表示的2价有机基团的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为15质量%以上、特别优选为20质量%以上,并且优选为70质量%以下、更优选为60质量%以下、进一步优选为50质量%以下、特别优选为40质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的全部重复单元,通式(3)所表示的2价有机基团的优选摩尔分数优选为20%以上、更优选为25%以上、进一步优选为30%以上、特别优选为35%以上,并且优选为90%以下、更优选为85%以下、进一步优选为80%以下、特别优选为75%以下。
此外,本发明的树脂组合物可以含有两种以上的上述通式(3)所表示的2价有机基团,例如,在组合使用isb来源的重复单元和chdm来源的重复单元的情况下,关于其含有比例没有特别限定,从较高的玻璃化转变温度与双折射、吸水率的方面出发,isb来源的重复单元相对于树脂组合物以摩尔分数计优选为30摩尔%以上、更优选为40摩尔%以上、进一步优选为50摩尔%以上,并且优选为95摩尔%以下、更优选为90摩尔%以下、进一步优选为85摩尔%以下。
此外,在组合使用isb来源的重复单元与chdm来源的重复单元的情况下,关于其含有比例没有特别限定,从柔软性的方面出发,chdm来源的重复单元相对于树脂组合物以摩尔分数计优选为5摩尔%以上、更优选为10摩尔%以上、进一步优选为15摩尔%以上,并且优选为50摩尔%以下、更优选为40摩尔%以下、进一步优选为30摩尔%以下。
在组合使用spg来源的重复单元与chdm来源的重复单元的情况下,关于其含有比例没有特别限定,从较高的玻璃化转变温度与双折射、吸水率的方面出发,spg来源的重复单元相对于树脂组合物以摩尔分数计优选为30摩尔%以上、更优选为40摩尔%以上、进一步优选为50摩尔%以上,并且优选为95摩尔%以下、更优选为90摩尔%以下、进一步优选为85摩尔%以下。
此外,在组合使用spg来源的重复单元与chdm来源的重复单元的情况下,关于其含有比例没有特别限定,从柔软性的方面出发,chdm来源的重复单元相对于树脂组合物以摩尔分数计优选为5摩尔%以上、更优选为10摩尔%以上、进一步优选为15摩尔%以上,并且优选为50摩尔%以下、更优选为40摩尔%以下、进一步优选为30摩尔%以下。
<3-3.折射率各向异性>
本发明的树脂组合物的折射率各向异性不论是正、负的哪一种,通过满足后述的<3-4.相位差比>中记载的在用于逆波长分散膜的情况下的条件,均显示出逆波长分散性。此处,为了得到具有负折射率各向异性的逆波长分散膜,需要将下述结构单元组合使用:具有正折射率各向异性、波长越短波长分散越大的波长分散性大的结构单元;以及具有较大的负折射率各向异性、波长分散性小的结构单元,但后者的材料通常不为人所知,通常难以得到具有负折射率各向异性的逆波长分散膜。因此,本发明的树脂组合物在作为显示出逆波长分散性、平坦分散性等所期望的光学特性的光学材料使用的情况下,优选具有正折射率各向异性。
本发明中的“具有正折射率各向异性的树脂组合物”是指在成型为拉伸膜时,在下述测定条件下显示出正折射率各向异性的树脂组合物。并且,对于“负折射率各向异性”也同样地定义。
本发明中,树脂组合物的折射率各向异性按下述方法测定。首先,利用热压机对树脂组合物进行压制,制作膜。将该膜切成规定的尺寸,进行自由端单向拉伸,制作拉伸膜。使用相位差测定装置(王子计测机器社制造kobra-wpr),测定拉伸膜的相位差。在相对于拉伸方向表现出正相位差的情况下,该树脂组合物显示出正折射率各向异性;在表现出负相位差的情况下,该树脂组合物显示出负折射率各向异性。详细测定条件在下文叙述。
一般来说,如果聚合物主要含有具有经9位碳原子与主链连接的芴环的重复单元,则该聚合物具有显示出负折射率各向异性的倾向,这是众所周知的。本发明中,作为实现“具有正折射率各向异性的树脂组合物”的方法没有任何限定,例如可使用下述方法a、方法b和方法c中的任一种方法,或者可组合使用任意方法。
方法a:作为聚合物使用含有2价低聚芴与通式(3)所表示的2价有机基团作为重复单元的共聚物的方法;
方法b:除了具有2价低聚芴作为重复单元的聚合物之外,还共存具有上述通式(3)所表示的2价有机基团作为重复单元的聚合物的方法;
方法c:对r1~r3进行适当选择,以使具有2价低聚芴作为重复单元的聚合物具有正折射率各向异性的方法。
它们之中,从所得到的树脂组合物的透明性、均匀性的方面出发,优选使用方法a、或者组合使用方法a与其它方法。此外,在方法a中,优选上述通式(3)所表示的2价有机基团具有显示出正折射率各向异性的结构。
这些方法中,通式(3)所表示的2价有机基团可以使用单一种类的基团,也可以将通式(3)范围内的两种以上的有机基团组合使用。并且,2价低聚芴可以使用单一种类的物质,也可将两种以上组合使用。
<3-3-1.方法a>
用于实现“具有正折射率各向异性的树脂组合物”的方法a为使用含有2价低聚芴与通式(3)所表示的2价有机基团的至少两种以上作为重复单元的共聚物作为聚合物的方法。
方法a中,作为2价低聚芴,可优选采用上述<1.低聚芴>中示例出的低聚芴。进一步地,作为通式(3)所表示的有机基团,可优选采用上述<2-3.有机基团的具体例>中示例出的有机基团。
方法a中,关于本发明的树脂组合物,作为聚合物,只要包含含有2价低聚芴与上述通式(3)所表示的2价有机基团的至少两种以上作为重复单元的共聚物即可,也可以进一步含有该共聚物以外的任意的聚合物。进而,上述共聚物也可以含有2价低聚芴和通式(3)以外的重复单元(其中不包括上述的连接基团)。
<3-3-2.方法b>
用于实现“具有正折射率各向异性的树脂组合物”的方法b为除了具有2价低聚芴作为重复单元的聚合物以外还共存具有通式(3)所表示的2价有机基团作为重复单元的聚合物的方法。
方法b中,作为通式(1)所表示的2价低聚芴,可优选采用上述<1.低聚芴>中示例出的低聚芴。进一步地,作为通式(3)所表示的有机基团,可优选采用上述<2-3.有机基团的具体例>中示例出的有机基团。
此外,具有2价低聚芴作为重复单元的聚合物可以具有2价低聚芴以外的2价有机基团(其中不包括上述连接基团)作为重复单元;另一方面,具有通式(3)所表示的2价有机基团作为重复单元的聚合物可以具有通式(3)以外的2价有机基团(其中不包括上述连接基团)作为重复单元。
在上述方法b的情况下,本发明的树脂组合物例如是通过将具有2价低聚芴作为重复单元的聚合物与具有通式(3)所表示的2价有机基团作为重复单元的聚合物进行共混而得到的。进一步地,也可共混具有2价低聚芴作为重复单元的聚合物和具有通式(3)所表示的2价有机基团作为重复单元的聚合物以外的任意聚合物和/或化合物。
<3-3-3.方法c>
用于实现“具有正折射率各向异性的树脂组合物”的方法c为对r1~r3进行适当选择以使具有2价低聚芴作为重复单元的聚合物具有正折射率各向异性的方法。
关于用于使具有2价低聚芴作为重复单元的聚合物具有正折射率各向异性的r1~r3,只要按照使聚合物具有正折射率各向异性的方式进行选择就没有任何限定,具体地说,r1和r2可以各自独立地举出具有或不具有取代基的碳原子数4~10的亚芳香基、具有或不具有取代基的碳原子数6~10的亚芳烷基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团,r3可以举出具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基。据推测,通过在主链导入芳香环,可抵消与主链正交的芴环所具有的负折射率各向异性,具有能够赋予正折射率各向异性的倾向。因此,r1~r3所具有的芳香环的个数以r1~r3所含有的总和计优选为2个以上、更优选为3个以上、进一步优选为4个以上。
方法c中,在2价低聚芴为通式(1)所表示的物质的情况下,作为r4~r9,可优选采用上述<1.低聚芴>中示例出的低聚芴。此外,具有2价低聚芴作为重复单元的聚合物也可以含有2价低聚芴以外的2价有机基团(其中不包括上述连接基团)。
另一方面,在采用上述方法c的情况下,在本发明的树脂组合物中,由于2价低聚芴自身具有正折射率各向异性,因而只要包含这样的聚合物即可:该聚合物含有具有特定r1~r3的2价低聚芴作为重复单元,也可以含有具有2价低聚芴作为重复单元的聚合物以外的任意聚合物和/或化合物。
方法c中,关于2价低聚芴,只要在可表现出后述的光学物性的范围内,即可在树脂组合物中以任意质量含有。关于上述通式(1)所表示的2价低聚芴的优选含有比例,为了表现出逆波长分散性并且确保熔融加工性、机械强度,相对于树脂组合物整体的质量,其含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上、最优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,2价低聚芴的优选摩尔分数优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、特别优选为30%以下、最优选为20%以下。
另外,从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物的质量,2价低聚芴的优选含有比例优选为5质量%以上、更优选为10质量%以上、进一步优选为12质量%以上、特别优选为15质量%以上、最优选为20质量%以上,并且优选为90质量%以下、更优选为80质量%以下、进一步优选为70质量%以下、特别优选为60质量%以下。从同样的方面出发,相对于具有2价低聚芴作为重复单元的聚合物,2价低聚芴的优选摩尔分数优选为1%以上、更优选为2%以上、进一步优选为3%以上、特别优选为4%以上、最优选为5%以上,并且优选为50%以下、更优选小于50%、进一步优选为40%以下、更进一步优选为35%以下、特别优选为30%以下、最优选为20%以下。
另一方面,在使用r1和r2为具有或不具有取代基的亚甲基的2价低聚芴的情况下,即使相对于树脂组合物整体的质量以任意的含有比例含有2价低聚芴,也具有相位差的波长分散性小、能够表现出平坦分散性的倾向。特别是通过提高含有比例,具有能够在维持平坦分散性的同时降低相位差的倾向,因而该组合物也能够用作在全波长区域不易表现出相位差的宽带域零双折射材料。其优选含有比例相对于树脂组合物整体的质量优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,相对于树脂组合物整体所含有的重复单元,2价低聚芴的优选摩尔分数优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。
此外,从同样的方面出发,2价低聚芴的优选含有比例相对于含有2价低聚芴作为重复单元的聚合物的质量优选为30质量%以上、更优选为40质量%以上、进一步优选为50质量%以上、特别优选为60质量%以上,并且优选为95质量%以下、更优选为90质量%以下、进一步优选为85质量%以下、特别优选为80质量%以下。从同样的方面出发,2价低聚芴的优选摩尔分数相对于含有2价低聚芴作为重复单元的聚合物优选为10%以上、更优选为15%以上、进一步优选为20%以上、特别优选为25%以上,并且优选为80%以下、更优选为75%以下、进一步优选为70%以下、特别优选为65%以下。
<3-4.相位差比>
本发明的树脂组合物在假设为相位差膜用途的情况下,优选在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比、即相位差比满足下式(2)。
re450/re550≦1.0(2)
此处,“本发明的树脂组合物的相位差比满足上述式(2)”是指,在成型为拉伸膜时,在下述测定条件,在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比满足上述式(2)。
特别是在本发明的树脂组合物中,在相位差膜中的显示出逆波长分散性的相位差膜、即假设为逆波长分散膜用途的情况下,优选相位差比满足下式(2’)。
0.5≦re450/re550<1.0(2’)
从可以适当地用作相位差膜的方面出发,优选本发明的树脂组合物为包含上述具有2价低聚芴作为重复单元的聚合物,在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比满足上述式(2)。特别是在本发明的树脂组合物含有具有2价低聚芴作为重复单元的聚合物、且满足上述式(2)的情况下,2价低聚芴的含有比例具有至少显示出充分的逆波长分散性的倾向。
相位差比按下述方法测定。利用热压机对树脂组合物进行压制,制作膜。将该膜切成规定的尺寸,进行自由端单向拉伸,制作拉伸膜。使用相位差测定装置(王子计测机器社制造kobra-wpr),对于该拉伸膜在波长450nm的相位差(re450)与在波长550nm的相位差(re550)进行测定。在相对于拉伸方向的相位差比(re450/re550)满足上述式(2)的情况下,该树脂组合物显示出作为相位差膜有用的波长分散性。此外,在相位差比(re450/re550)满足上述式(2’)的情况下,该树脂组合物显示出作为逆波长分散膜有用的逆波长分散性。详细测定条件在下文叙述。
本发明的树脂组合物在假定为相位差膜中的逆波长分散膜用途的情况下,相位差比(re450/re550)的上限优选为1.0以下、更优选小于1.0、进一步优选为0.95以下、更进一步优选为0.93以下、特别优选为0.91以下。此外,相位差比(re450/re550)的下限优选为0以上、更优选为0.50以上,进一步优选超过0.50、个进一步优选为0.70以上、特别优选为0.75以上、最优选为0.80以上。
只要相位差比(re450/re550)的值为上述范围内,则波长越长,越可表现出相位差,在可见区域的各波长下能够得到理想的相位差特性。例如,作为使得在相互垂直方向振动的偏振光的相位进行1/4波长(90°)变化的1/4λ板,可使用由具有这样的波长分散性的本发明树脂组合物得到的相位差膜,将其与偏振片粘贴,从而制作圆偏振片等,能够实现在全部波长下具有防止外光反射功能的黑色性优异的圆偏振片和图像显示装置。另一方面,在相位差比(re450/re550)的值为上述范围外的情况下,波长所致的脱色(色抜け)变大,具有在圆偏振片或图像显示装置中产生着色问题的倾向。
此外,本发明的树脂组合物在假定为逆波长分散膜用途的情况下,优选在波长630nm测定的相位差(re630)与在波长550nm处测定的相位差(re550)之比、即相位差比’满足下式(25)。
1.0≦re630/re550(25)
此处,“本发明的树脂组合物的相位差比’满足上述式(25)”是指,在成型为拉伸膜时,在下述测定条件下,在波长630nm测定的相位差(re630)与在波长550nm处测定的相位差(re550)之比满足上述式(25)。
相位差比’按下述方法测定。利用热压机对树脂组合物进行压制,制作膜。将该膜切成规定的尺寸,进行自由端单向拉伸,制作拉伸膜。使用相位差测定装置(王子计测机器社制造kobra-wpr),对于该拉伸膜在波长630nm的相位差(re630)与在波长550nm的相位差(re550)进行测定。在相对于拉伸方向的相位差比’(re630/re550)满足上述式(25)的情况下,该树脂组合物显示出作为相位差膜有用的波长分散性。
在本发明的树脂组合物中,在假设为相位差膜中的逆波长分散膜用途的情况下,相位差比’(re630/re550)的上限优选为1.25以下、更优选为1.20以下、特别优选为1.15以下。此外,相位差比(re630/re550)的下限优选为1.00以上、更优选为1.01以上、进一步优选为1.02以上、特别优选为1.03以上。
相位差比’(re630/re550)的值为上述范围内时,波长越长,越可表现出相位差,在可见区域的各波长下能够得到理想的相位差特性。例如,作为1/4λ板可以使用由具有这样的波长分散性的本发明树脂组合物得到的相位差膜,将其与偏振片粘贴,从而制作圆偏振片等,能够实现在全部波长下具有防止外光反射功能的黑色性优异的圆偏振片和图像显示装置。另一方面,在相位差比’(re630/re550)的值为上述范围外的情况下,波长所致的脱色(色抜け)变大,具有在圆偏振片或图像显示装置中产生着色问题的倾向。特别是在不论波长如何均可得到防止外光反射功能的方面出发,优选相位差比(re450/re550)与相位差比’(re630/re550)两者的值均为上述范围内。
如此,关于用于使相位差比(re450/re550)、相位差比’(re630/re550)为上述范围内的具体方法并无任何限定,例如可以举出作为2价低聚芴使用规定量的下述2价低聚芴的方法:位于两末端的芴的9位碳原子为2价基团的2价低聚芴、或者与位于两末端的芴的9位碳原子分别键合的r1和r2为2价基团并且r1与r2的至少任意一个的碳原子数为2以上。这种情况下的2价低聚芴,也可以进一步合用与位于两末端的芴的9位碳原子分别键合的碳原子数为1的r1和r2为2价基团的2价低聚芴。
此外,在假定为对图像显示装置的漏色进行校正的逆波长分散膜用途的情况下,为了根据装置进行漏色的校正,设定最佳的相位差比(re450/re550)即可,在上限小于1.0时,下限不特别设定。
另一方面,在本发明的树脂组合物中,在假设为相位差的波长分散性小的平坦分散材料的情况下,优选相位差比满足下式(23)。
0.9<re450/re550<1.1(23)
此处,“本发明的树脂组合物中的相位差比满足上述式(23)”是指,在成型为拉伸膜时,在下述测定条件下,在波长450nm处测定的相位差(re450)与在波长550nm处测定的相位差(re550)之比满足上述式(23)。
在本发明的树脂组合物中,在假设为相位差的波长分散性小的平坦分散材料的情况下,相位差比(re450/re550)优选为0.93以上、更优选为0.95以上、特别优选为0.98以上,并且优选为1.08以下、更优选为1.06以下、特别优选为1.05以下。
相位差比(re450/re550)的值为上述范围内的情况下,通过使用本发明的树脂组合物,能够得到对va模式液晶显示装置的脱色进行校正的相位差膜,能够实现波长所致的脱色少的液晶显示装置。进一步地,通过满足后述<3-12.双折射>中记载的条件,能够在可见区域的各波长下得到理想的相位差特性,能够制成宽带域零双折射材料。此外,作为液晶显示装置的偏振片保护膜,通过将本发明的宽带域零双折射材料与偏振片贴合,能够实现波长所致的脱色少的偏振片和图像显示装置。
此外,在本发明的树脂组合物中,在假设为平坦分散材料的情况下,优选在波长630nm测定的相位差(re630)与在波长550nm处测定的相位差(re550)之比、即相位差比’满足下式(26)。
0.97<re630/re550<1.02(26)
此处,“本发明的树脂组合物的相位差比’满足上述式(26)”是指,在成型为拉伸膜时,在下述测定条件下,在波长630nm测定的相位差(re630)与在波长550nm处测定的相位差(re550)之比满足上述式(26)。
在本发明的树脂组合物中,在假设为平坦分散材料的情况下,相位差比’(re630/re550)的上限优选为1.02以下、更优选为1.01以下、特别优选为1.00以下。此外,相位差比(re630/re550)的下限优选为0.97以上、更优选为0.98以上、特别优选为0.99以上。
相位差比’(re630/re550)的值为上述范围内的情况下,通过使用本发明的树脂组合物,能够得到对va模式液晶显示装置的脱色进行校正的相位差膜,能够实现波长所致的脱色少的液晶显示装置。进一步地,通过满足后述<3-12.双折射>中记载的条件,能够在可见区域的波长下得到理想的相位差特性,能够制成宽带域零双折射材料。此外,作为液晶显示装置的偏振片保护膜,通过将本发明的宽带域零双折射材料与偏振片贴合,能够实现波长所致的脱色少的偏振片和图像显示装置。进一步地,特别优选相位差比(re450/re550)与相位差比’(re630/re550)这两者的值均为上述范围内。
如此,关于用于使相位差比(re450/re550)、相位差比’(re630/re550)为上述范围内的具体方法并无任何限定,例如可以举出作为2价低聚芴使用规定量的下述2价低聚芴的方法:与位于两末端的芴的9位碳原子分别键合的碳原子数为1的r1和r2为2价基团的2价低聚芴。这种情况下,也可以进一步合用位于两末端的芴的9位碳原子为2价基团的2价低聚芴、或者与位于两末端的芴的9位碳原子分别键合的碳原子数为2以上的r1和r2为2价基团的2价低聚芴。
<3-5.芴骨架的质量>
相对于本发明的树脂组合物的质量,芴骨架的质量优选为5%以上、更优选为8%以上、进一步优选为10%以上,并且优选为70%以下、更优选为50%以下、进一步优选为30%以下。若为该范围,则具有可得到逆波长分散性、正双折射各向异性之类的所期望的光学特性的倾向。若低于该范围,则具有不会表现出逆波长分散性、或者逆波长分散性不足的可能性。此外,在高于该范围的情况下,折射率各向异性可能为负,进而具有树脂变脆等的机械强度降低的可能性。
另一方面,在使用r1和r2为具有或不具有取代基的亚甲基的2价低聚芴的情况下,关于相对于本发明树脂组合物质量的芴骨架质量,即使相对于树脂组合物整体的质量以任意质量含有,也具有能够表现出相位差的波长分散性小的平坦分散性的倾向。特别是通过提高含有比例,具有能够在维持平坦分散性的同时降低相位差的倾向,因而该组合物也能够用作在全波长区域不易表现出相位差的宽带域零双折射材料。其优选含有比例相对于树脂组合物整体的质量优选为20质量%以上、更优选为25质量%以上、进一步优选为30质量%以上、特别优选为35质量%以上,并且优选为90质量%以下、更优选为85质量%以下、进一步优选为80质量%以下、特别优选为75质量%以下。
此处的芴骨架表示的是上述通式(1)中的包括形成芴环的13个碳原子与取代基r4~r9的结构。其中,此处所说的芴骨架是指不仅包含在上述通式(1)所表示的具有2价低聚芴作为重复单元的聚合物中、而且包含在树脂组合物中的总量。
<3-6.芴比例>
另一方面,含有具有芴作为重复单元的聚合物的树脂组合物中,通过具有芳香环的芴环在主链进行取向,具有表现出所期望的光学特性的倾向。例如,在芴环大致垂直于主链进行取向的情况下,显示出逆波长分散性;在芴环相对于主链倾斜45度左右进行取向的情况下,显示出平坦分散性。因此,为了有效地表现出逆波长分散性、平坦分散性、宽带域零双折射等所期望的光学物性,优选提高重复单元中的芴环的比例。在本说明书中,将其称为芴比例,按下式(27)进行定义。此处,芴环的分子量为13个碳原子的原子量的总和,氢原子不包含在该分子量中,并且,即使在具有取代基的情况下,取代基也不包含在该分子量中。此外,芴环的分子量的总和是指具有芴的重复单元所含有的全部芴环的分子量的合计值,例如,在具有2个芴环的情况下,为2个芴环的分子量;同样地,在具有3个芴环的情况下,为3个芴环的分子量。另一方面,具有芴的重复单元的分子量是指该重复单元本身的分子量。
芴比例(%)=芴环的分子量的总和/具有芴的重复单元的分子量×100(27)
在本发明中,通过使用特定的2价低聚芴,能够提高重复单元中的芴环的比例,因而具有即使减少其含有比例也能够表现出所期望的光学物性的倾向。从这方面考虑,芴比例优选为30%以上、更优选为40%以上、进一步优选为50%以上、进而优选为60%以上,并且通常为90%以下。
<3-7.玻璃化转变温度>
本发明的树脂组合物的玻璃化转变温度优选为90℃以上、更优选为100℃以上、进一步优选为110℃以上、特别优选为120℃以上,并且优选为170℃以下、更优选为160℃以下、进一步优选为150℃以下。若低于该范围,则在使用环境下,光学物性可能会由设计值发生变化,可能不满足实用上所需要的耐热性。此外,若高于该范围,则树脂组合物的熔融加工性降低,可能得不到良好的外观及尺寸精度高的成型体。进而,耐热性会过高,而另一方面,机械强度会降低,因而认为树脂组合物会变脆,加工性或成型体的处理性可能会变差。
<3-8.熔融粘度>
本发明的树脂组合物的熔融粘度在测定温度240℃、剪切速度91.2sec-1的条件下优选为500pa·s以上、更优选为800pa·s以上、进一步优选为1000pa·s以上,并且优选为5000pa·s以下、更优选为4500pa·s以下、进一步优选为4000pa·s以下。若低于该范围,则可能得不到可耐实用的机械强度。此外,在后述的熔融制膜法中,可能会处于适当的熔融粘度范围之外。若高于该范围,则与上述玻璃化转变温度过高的情况同样地具有成型性恶化的可能性。
<3-9.分子量>
本发明的树脂组合物的分子量可利用比浓粘度来表示。如后述实施例一节中记载的那样,本发明的树脂组合物的比浓粘度如下测定:使用二氯甲烷作为溶剂,将高分子浓度精密调整为0.6g/dl,在温度20.0℃±0.1℃下使用乌氏粘度管进行测定。本发明的树脂组合物的比浓粘度没有特别限制,优选为0.30dl/g以上、更优选为0.35dl/g以上。比浓粘度的上限优选为1.20dl/g以下、更优选为0.60dl/g以下、进一步优选为0.50dl/g以下。
<3-10.金属含有比例>
本发明的树脂组合物在含有大量的金属和金属离子时,在聚合或加工时可能会着色、或者可能易于发生热分解。重要的是尽可能降低例如在制造树脂组合物时所用的催化剂的残留物、或者在树脂组合物的原料中污染的金属成分、或者由反应装置等中溶出的金属等。由于na、k、cs、fe的影响特别显著,因而本发明的聚碳酸酯树脂组合物中,na、k、cs、fe的含有比例的合计优选为3质量ppm以下、更优选为1质量ppm以下、进一步优选为0.8质量ppm以下、特别优选为0.5质量ppm以下。树脂组合物中的金属量可在利用湿灰化等方法回收树脂组合物中的金属后使用原子发光、原子吸光、icp等方法进行测定。
<3-11.光弹性系数>
本发明的树脂组合物的光弹性系数优选为45×10-12pa-1以下。进一步优选为40×10-12pa-1以下、特别优选为35×10-12pa-1以下,并且通常为5×10-12pa-1以上。光弹性系数增高时,在用于大型成型品的情况下、或者在对成型品进行弯折的情况下,在发生应力的部分,材料的双折射发生变化,光学物性的均匀性可能会受损。
<3-12.双折射>
本发明的树脂组合物中,在假设为显示出逆波长分散性的相位差膜、或假设为显示出平坦分散性的相位差膜用途的情况下,在成膜时,在550nm处的双折射优选为0.001以上。如下文所述,为了将使用本发明的树脂组合物进行成型的膜的厚度设计得非常薄,优选双折射高。因而,550nm处的双折射更优选为0.002以上,并且通常为0.005以下。550nm处的双折射小于0.001的情况下,需要使膜的厚度过大,具有增加成膜材料的用量、从厚度·透明性·相位差的方面考虑难以控制均质性的倾向。因此,在550nm处的双折射小于0.001的情况下,可能无法适合于要求精密性·薄型·均质性的器械中。
双折射为相位差除以膜厚度得到的值,因而可通过使用相位差测定装置(王子计测机器社制造kobra-wpr)测定膜的相位差并测定膜厚度来求得。
另一方面,在本发明的树脂组合物中,在假设为宽带域零双折射材料的情况下,在制成膜时,优选550nm处的双折射为0.0005以下。如上所述,为了使用本发明的树脂组合物来设计具有宽带域零双折射的偏振片保护膜,优选双折射较低。因而,550nm处的双折射更优选为0.0002以下、特别优选为0.0001以下,并且通常为0.00001以上。550nm处的双折射超过0.0005的情况下,由于双折射不够小,因而若膜的厚度较厚,则有产生脱色的可能性。
该光学膜特别是在用于液晶显示装置的偏振片的保护膜时表现出极为优异的特性。其中,其并不限于偏振片的保护膜,也可用于相位差膜、塑料电池(プラセル)基板膜、防反射膜、亮度上升膜、光盘的保护膜、扩散膜等其它用途中。
<3-13.折射率>
本发明的树脂组合物中,在假设为光学透镜等宽带域零双折射材料的情况下,在589nm的折射率优选为1.54以上。为了使用本发明的树脂组合物设计光学透镜,为使透镜较薄,也优选折射率高。因而,589nm的折射率更优选为1.56以上、特别优选为1.58以上,通常为1.65以下。
<3-14.阿贝值>
本发明的树脂组合物中,在假设为摄像系光学透镜等宽带域零双折射材料的情况下,优选阿贝值为35以下。为了使用本发明的树脂组合物设计摄像系光学透镜,优选阿贝值较低。因而,阿贝值更优选为30以下、特别优选为25以下,通常为15以上。
<3-15.芴环的取向>
本发明的树脂组合物中,源于芴环取向的740cm-1的吸收在拉伸方向与垂直方向的强度比优选为1.2以上、更优选为1.3以上、进一步优选为1.4以上,并且通常为2.0以下。将本发明的树脂组合物用作逆波长分散膜用途的情况下,在源于芴环取向的740cm-1的吸收在拉伸方向与垂直方向的强度比高时,即使该树脂组合物中所含有的具有芴环的重复单元的比例减少,也具有显示出逆波长分散性的倾向。需要说明的是,上述强度比可按照下述方法进行测定。
首先由本发明的树脂组合物制作拉伸膜,实施偏光atr分析。在该分析结果中,源于羰基取向的1245cm-1的吸收在拉伸方向与垂直方向的强度比(2色比:拉伸方向的强度/垂直方向的强度)为1.2以上,确认到主链在拉伸方向取向。接着计算出源于芴环取向的740cm-1的吸收在拉伸方向与垂直方向的强度比。
<3-16.主链与芴所成的角度>
关于本发明的树脂组合物,2价低聚芴的特定构象(conformation)不以歪扭构象为稳定构象的情况下,在反式构象的主链与芴环所成的角度为50°以上、优选为60°以上、更优选为70°以上时,预计可表现出逆波长分散性。
2价低聚芴的特定构象(构型)的能量计算和该构象中的芴环与主链所成的角度的计算可如下来计算出。
对于am1法,软件使用美国wavefunction社制造的pcspartanpro1.0.5(windows(注册商标)32位版本)。需要说明的是,聚焦判定值等与计算精度相关的输入值全部使用该软件的默认值。
此处,关于2价低聚芴,在聚碳酸酯树脂组合物的情况下,对于将重复单元的两末端进行碳酸甲酯化而成的结构进行计算;在聚酯或聚酯碳酸酯树脂组合物的情况下,对于将重复单元的两末端进行甲基酯化而成的结构进行计算。
使用am1法,对于在各单体中存在的2个侧链的两者为反式构象、以及两者为两种歪扭构象的构象异构体的能量差进行计算。此外,对于反式构象与歪扭构象(两种歪扭构象中稳定构象),进行主链与芴环所成角度的计算。
需要说明的是,主链与芴环所成的角度如下确定。首先,将两末端的甲基的碳原子彼此连接而成的直线作为主链方向,将穿过芴的3位、6位、9位碳原子的平面作为芴平面。此时,尽管与主链方向交叉的芴平面上的直线是无限存在的,但与主链方向的角度最小的芴平面上的直线是唯一确定的。将该角度作为主链与芴环所成的角度。
<4.低聚芴单体>
上述通式(1)所表示的具有2价低聚芴作为重复单元的聚合物例如可通过下述通式(20)所表示的低聚芴单体的聚合等方法来制造。
[化53]
(式中,r3~r9和n与上述通式(1)的相同。a3和a4各自独立地表示聚合反应性基团。)
<4-1.聚合反应性基团>
下面举出a3和a4中的“聚合反应性基团”的具体结构,但并不限于这些,可以举出:羟基甲基、2-羟基乙基、3-羟基丙基、羟基丁基、2,2-二甲基-3-羟基丙基、2-甲氧基甲基-2-甲基亚丙基、4-羟基苯基、4-羟基-3-甲基苯基、4-(2-羟基乙氧基)苯基、(4-(羟基甲基)环己烷-1-基)甲基等羟基;甲氧基羰基、乙氧基羰基、苯氧基羰基、乙氧基羰基甲基、2-(乙氧基羰基)乙基、2-(甲氧基羰基)丙基等酯基;2-羟基乙氧基羰基、2-(2-羟基乙氧基)羰基乙基、2-(2-羟基乙氧基)羰基丙基、2-(4-羟基丁氧基)羰基乙基、2-[[4-(羟基甲基)环己烷-1-基]甲氧基]羰基乙基等羟基酯基;羧基、羧基甲基、羧基乙基等羧基;氨基甲基、2-氨基乙基、3-氨基丙基等氨基;丙烯酰氧基甲基、甲基丙烯酰氧基甲基、2-(丙烯酰氧基)乙基、3-(丙烯酰氧基)丙基等丙烯酰基;2,3-环氧丙基、2,3-环氧丙氧基甲基、2-(2,3-环氧丙氧基)乙基等环氧基等。
需要说明的是,上述通式(20)所表示的低聚芴单体可以作为具有2价低聚芴作为重复单元的聚合物的原料使用,聚合反应性基团优选仅为a3和a4这2处,在用于制造各种树脂组合物的聚合条件下,作为聚合反应性基团发挥作用的取代基优选不包含在r3~r9中。
需要说明的是,a3和a4可以相同,也可以不同。在不同的情况下,作为a3和a4的组合,例如可以举出羟基甲基与乙氧基羰基、2-(2-羟基乙氧基)羰基与羧基、2-(2-羟基乙氧基)羰基乙基与羧基乙基等的组合。
它们之中,优选a3和a4相同的情况,a3和a4相同的情况具有能够以较短工序实施该单体的制造的倾向,更优选在作为优选聚合物的聚酯、聚碳酸酯或者聚酯碳酸酯中使用的羟基、酯基或者羟基酯基的情况。a3和a4为羟基的情况下,形成下述通式(10a)
[化54]
(式中,r1~r9与上述通式(1)的r1~r9相同,n表示1~5的整数值。)所表示的具有低聚芴的二羟基化合物,该二羟基化合物是能够通用于聚酯、聚碳酸酯、聚酯碳酸酯的单体,该聚酯、聚碳酸酯、聚酯碳酸酯是由于光学性能良好而优选的聚合物。
关于上述羟基中的羟基甲基(式(10a)中,在r1和r2为亚甲基的情况下,为-r1-oh基和-r2-oh基),由于其容易导入且可得到高玻璃化转变温度的树脂组合物因而特别优选。羟基甲基能够提高单体分子中的芴比例,而且尽管在具有经9位与主链连接的芴骨架,但是出人意料的是负折射率各向异性仍较小,因而在树脂组合物中,即使上述2价低聚芴的质量比和/或摩尔分数为较高范围,也具有正折射率各向异性。因此具有能够得到如下树脂组合物的倾向:该聚合物具有正折射率各向异性与高折射率且光弹性系数小。进一步地,由于具有波长分散小的倾向,因而适于平坦分散相位差膜。此外,关于上述羟基中的羟基丙基,由于在添加量为少量的情况下也能够发挥出作为相位差膜的特性,因而特别优选。即,在树脂组合物的折射率各向异性为正且550nm处双折射为0附近时,逆波长分散性增大,但由于来源于具有羟基丙基的低聚芴单体的结构单元的折射率各向异性为较大的负值,因而在相对于除此以外的具有正折射率各向异性的结构单元为更少量的情况下,就能够使树脂组合物的折射率各向异性为正且能够使双折射为0附近。
a3和a4为酯基的情况下,形成下述通式(10b)
[化55]
(式中,r1~r9与上述通式(1)中的r1~r9相同,r17表示碳原子数1~10的有机取代基,n表示1~5的整数值。其中,r17在左右可以相同、也可以不同。)所表示的具有低聚芴的二酯化合物,该二酯化合物是能够通用于聚酯、聚酯碳酸酯的单体,该聚酯、聚酯碳酸酯是由于光学性能良好而优选的聚合物。
a3和a4为酯基的情况下,r17中的“碳原子数1~10的有机取代基”的具体结构如下面所举出下述结构,但并不限于这些,可以举出:甲基、乙基、正丙基、正丁基、正戊基、正己基、正癸基等直链状的烷基;异丙基、2-甲基丙基、2,2-二甲基丙基、2-乙基己基等包含支链的烷基;环丙基、环戊基、环己基、环辛基等环状的烷基;苯基、1-萘基、2-萘基等芳基;2-吡啶基、2-噻吩基、2-呋喃基等杂芳基;苄基、2-苯基乙基、对甲氧基苄基等芳烷基等。它们之中,在r17为甲基或乙基的情况下,通过将通过与二羟基化合物的酯交换而产生的低沸点的醇除去,能够有效地合成聚酯和聚酯碳酸酯,因而特别优选。另一方面,r17为芳基的情况下,由于酯交换反应容易进行,因而通过将上述二酯化合物与二羟基化合物、碳酸二酯一次性添加装入到反应器中,能够一步合成作为优选聚合物的聚酯碳酸酯,因而是优选的。特别是苯基的分子量小,在聚酯碳酸酯合成后能够以苯酚的形式进行蒸馏去除,r17为苯基的情况是特别优选的。此外,在r17为芳基的情况下,从聚合时的反应性的方面出发,作为碳酸二酯优选使用后述的二芳基碳酸酯类;从能够容易地除去副产物的方面出发,更优选r17的芳基与二芳基碳酸酯类中的芳基相同。
上述通式(20)所表示的低聚芴单体中,在a3和a4为酯基的情况下,上述酯基中的2-(甲氧基羰基)乙基、2-(乙氧基羰基)乙基或2-(甲氧基羰基)丙基能够分别使用可工业获得的丙烯酸甲酯、丙烯酸乙酯、甲基丙烯酸甲酯而容易地导入,在制成树脂组合物时的柔软性高,并且在少量添加时即可显示出较高的逆波长分散性,因而是特别优选的。另一方面,关于苯氧基羰基烷基,由于酯基的活性提高,酯交换反应容易地进行,因而通过使上述二酯化合物与二羟基化合物、碳酸二酯在同一条件下发生反应,能够一步合成作为优选聚合物的聚酯碳酸酯,因此苯氧基羰基烷基是优选的。特别是对于2-(苯氧基羰基)乙基和2-(苯氧基羰基)丙基来说,由于可采用使用丙烯酸苯酯以及甲基丙烯酸苯酯的导入法、或基于由其它丙烯酸酯类以及甲基丙烯酸酯类出发的酯交换的导入法,因而是特别优选的。
a3和a4为羟基酯基的情况下,形成下述通式(10c)
[化56]
(式中,r1~r10与上述通式(1)和上述通式(3)中的r1~r10相同,n表示1~5的整数值。其中,r10在左右可以相同、也可以不同。)所表示的具有低聚芴的二羟基酯化合物,该二羟基酯化合物是能够通用于聚酯、聚酯碳酸酯的单体,该聚酯、聚酯碳酸酯是由于光学性能良好而优选的聚合物。上述羟基酯基中的2-(2-羟基乙氧基)羰基乙基、2-(4-羟基丁氧基)羰基乙基、2-[[4-(羟基甲基)环己烷-1-基]甲氧基]羰基乙基能够通过与相应的丙烯酸衍生物的反应以及酯交换反应而容易地导入,利用通常的聚碳酸酯制造工序制成树脂组合物时,具有可制成柔软性高、并且少量添加时即可显示出较高的逆波长分散性的聚酯碳酸酯的倾向,因而是特别优选的。
需要说明的是,上述通式(10c)所表示的具有低聚芴的二羟基酯化合物同时具有上述通式(1)所表示的低聚芴与上述通式(3)所表示的2价有机基团。作为r10,优选可采用<2-3.有机基团的具体例>中说明的在上述通式(3)所表示的2价有机基团中作为r10示例出的基团。
<4-2.低聚芴单体的具体例>
作为上述通式(20)所表示的低聚芴单体的具体例,可以举出下述[l]组中示出的结构。
[化57]
[l]
[化58]
[化59]
[化60]
[化61]
<4-3.低聚芴二醇>
上述通式(10a)所表示的低聚芴之中,特别优选的具有羟基甲基的低聚芴单体为下述通式(19)中示出的二羟基化合物(下文中称为低聚芴二醇。)。
[化62]
(式中,r3为具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基或氰基,r4~r9中,相邻的至少2个基团可以相互键合形成环。n表示1~5的整数值。)
作为r3~r9和n,可以应用通式(1)中优选示例出的r3~r9和n。
在使用该低聚芴二醇得到树脂组合物时,在2价低聚芴中,由于与位于两末端的芴的9位碳原子分别键合的r1和r2的碳原子数为1,因而具有能够简便地得到可表现出平坦分散性的树脂组合物的倾向。
上述通式(19)中,优选r4~r9全部为氢原子,这种情况下,能够由工业成本低的芴衍生出,并且能够提高芴比例,芴环彼此间不易产生空间位阻,容易得到源于芴环的所期望的光学特性;更优选r3为具有或不具有取代基的碳原子数1~10的亚烷基、且r4~r9全部为氢原子,这种情况下,由于主链不包含芳香环,因而可认为光弹性较小;进一步优选r3为亚甲基、亚乙基、正亚丙基、正亚丁基或2,2-二甲基亚丙基并且r4~r9全部为氢原子,特别优选r3为亚甲基或亚乙基并且r4~r9全部为氢原子的双(9-羟基甲基芴-9-基)甲烷或双(9-羟基甲基芴-9-基)乙烷。r3为长链状基团的情况下,树脂组合物的玻璃化转变温度可能会降低。使用上述通式(19)所表示的具有羟基甲基的低聚芴单体得到的树脂组合物与使用其它芴单体得到的树脂组合物不同,尽管具有经9位与主链连接的芴环,但是具有如下令人吃惊的特征:在几乎所有共聚比下具有正折射率各向异性,显示出了接近于平坦的波长分散性。据认为,这是由于,羟基甲基优先采取弯曲结构,芴环并未相对于拉伸方向垂直取向而是倾斜取向。此外,由于可通过改变共聚比来控制双折射的值,因而在具有羟基甲基的低聚芴单体的比例高的区域,能够作为宽带域的零双折射材料使用。
作为上述通式(19)所表示的低聚芴二醇的具体例,可以举出双(9-羟基甲基芴-9-基)甲烷、1,2-双(9-羟基甲基芴-9-基)乙烷、1,3-双(9-羟基甲基芴-9-基)丙烷、1,3-双(9-羟基甲基芴-9-基)-2,2-二甲基丙烷、1,4-双(9-羟基甲基芴-9-基)丁烷、1,4-双(9-羟基甲基芴-9-基)苯、1,3-双(9-羟基甲基芴-9-基)苯、1,4-双[(9-羟基甲基芴-9-基)甲基]苯、9,9-双[(9-羟基甲基芴-9-基)甲基]芴、9,9-双[2-(9-羟基甲基芴-9-基)乙基]芴、双[9-[(9-羟基甲基芴-9-基)甲基]芴-9-基]甲烷、1,2-双[9-[2-(9-羟基甲基芴-9-基)乙基]芴-9-基]乙烷等。
<4-4.低聚芴二芳基酯>
上述通式(10b)之中,特别优选的具有芳基酯基的低聚芴单体为下述通式(10d)所表示的化合物。
[化63]
通式(10d)中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团。
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基。
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、具有或不具有取代基的氨基、具有取代基的硫原子、卤原子、硝基和氰基。其中,r4~r9中,相邻的至少2个基团可以相互键合形成环。
ar1为具有或不具有取代基的碳原子数4~10的芳基,n表示1~5的整数值。其中,ar1在左右可以相同、也可以不同。)所表示的二芳基酯化合物(下文中称为低聚芴二芳基酯。)
作为r1~r9和n,可以应用通式(1)中优选示例出的r1~r9和n。
此外,ar1为具有或不具有取代基的碳原子数4~10的芳基,其碳原子数为4以上、优选为6以上,并且为10以下、优选为8以下。若为该范围内,则具有能够通过蒸馏将在聚碳酸酯或聚酯碳酸酯制造时副生的芳基醇进行蒸馏去除的倾向,因而能够提高所制造的聚碳酸酯或聚酯碳酸酯的聚合度。此外,作为该芳基可以具有的取代基,可以举出甲基、乙基、氯原子、溴原子或者苯基等。
作为ar1的具体例,可以举出苯基、甲苯基、氯苯基、萘基、间甲苯基、联苯基等,它们之中,从工业成本低、分子量较小的方面出发,优选苯基、甲苯基、氯苯基或者间甲苯基,更优选在聚合后能够以苯酚的形式蒸馏去除的苯基。
上述通式(10d)中,优选r4~r9全部为氢原子,这种情况下,能够由工业成本低的芴衍生出,并且能够提高芴比例,芴环彼此间不易产生空间位阻,容易得到源于芴环的所期望的光学特性;更优选r1、r2和r3各自独立地为具有或不具有取代基的碳原子数1~10的亚烷基并且r4~r9全部为氢原子,这种情况下,由于主链不包含芳香环,因而可认为光弹性较小;进一步优选ar1为苯基、甲苯基、氯苯基、间甲苯基、萘基或者联苯基,且r1、r2和r3各自独立地为亚甲基、亚乙基、正亚丙基、正亚丁基或2,2-二甲基亚丙基,并且r4~r9全部为氢原子,这种情况下,工业成本低、分子量比较小;特别优选ar1为苯基并且r1、r2和r3为亚甲基或亚乙基、且r4~r9全部为氢原子的双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷或者1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷,这种情况下,在聚酯或聚酯碳酸酯的合成后能够以苯酚的形式蒸馏去除。r3为长链状基团的情况下,树脂组合物的玻璃化转变温度可能会降低。上述通式(10d)所表示的具有二芳基酯基的低聚芴单体中,酯基的活性提高,酯交换反应容易进行,因而通过将二羟基化合物、碳酸二酯一次性添加装入到反应器中,能够一步合成作为优选聚合物的聚酯碳酸酯。
作为上述通式(10d)所表示的低聚芴二芳基酯的具体例,可以举出[9-(2-苯氧基羰基乙基)芴-9-基]甲烷、1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷、[9-(苯氧基羰基甲基)芴-9-基]甲烷、1,2-双[9-(苯氧基羰基甲基)芴-9-基]乙烷、[9-(2-苯氧基羰基丙基)芴-9-基]甲烷、1,2-双[9-(2-苯氧基羰基丙基)芴-9-基]乙烷、1,3-双[9-(2-苯氧基羰基乙基)芴-9-基]丙烷、1,3-双[9-(2-苯氧基羰基乙基)芴-9-基]-2,2-二甲基丙烷、1,4-双[9-(2-苯氧基羰基乙基)芴-9-基]丁烷、1,4-双[9-(2-苯氧基羰基乙基)芴-9-基]苯、1,3-双[9-(2-苯氧基羰基乙基)芴-9-基]苯、1,4-双[[9-(2-苯氧基羰基乙基)芴-9-基]甲基]苯、9,9-双[[9-(2-苯氧基羰基乙基)芴-9-基]甲基]芴、9,9-双[[9-(2-苯氧基羰基乙基)芴-9-基]乙基]芴、双[[[9-(2-苯氧基羰基乙基)芴-9-基]甲基]芴-9-基]甲烷、1,2-双[[[9-(2-苯氧基羰基乙基)芴-9-基]乙基]芴-9-基]乙烷等。
<4-5.低聚芴单体的物性值>
本发明的低聚芴单体中的氯含有比例以cl换算质量计优选为100质量ppm以下。进一步优选为10质量ppm以下。氯成分的含有比例多的情况下,会使聚合反应中所用的催化剂失活,聚合无法进行至所期望的分子量,反应呈不稳定化,具有生产率变差的可能性。并且在所得到的聚合物中也有氯成分残存,可能使聚合物的热稳定性降低。
本发明的低聚芴二醇中的单羟基体的含有比例优选为全部单体质量的10质量%以下。进一步优选为2质量%以下。在单羟基体经聚合反应掺入到聚合物中时会形成末端封链基团,因而若单羟基体增多,则聚合进行不到所期望的分子量、或者聚合物中的低聚物等低分子成分的残存量增多,可能会使所得到的聚合物的机械强度或耐热性降低。此外可认为还有低分子成分从成型体中渗出等使制品的品质降低的可能性。需要说明的是,单羟基体是指低聚芴二醇的末端羟基中的任意一个为聚合反应性基团以外的基团。
本发明的低聚芴二酯中的低聚芴单酯体的含有比例优选为全部单体质量的10质量%以下。进一步优选为2质量%以下。在低聚芴单酯体经聚合反应掺入到聚合物中时会形成末端封链基团,因而若低聚芴单酯体增多,则聚合进行不到所期望的分子量、或者聚合物中的低聚物等低分子成分的残存量增多,可能会使所得到的聚合物的机械强度或耐热性降低。此外认为还有低分子成分从成型体中渗出等使制品的品质降低的可能性。需要说明的是,单酯体是指低聚芴二酯的末端酯基中的任意一个为聚合反应性基团以外的基团。
本发明的低聚芴二醇中,可能会含有来源于下述工序的钠或钾等长周期型周期表第1族金属或钙等第2族金属,该工序为在碱存在下使甲醛类发生作用来进行羟基甲基化的工序;这些金属的含有比例优选为500质量ppm以下、更优选为200质量ppm以下、进一步优选为50质量ppm以下、特别优选为10质量ppm以下。若金属成分多,则在聚合反应或树脂加工时,聚合物可能变得易着色。此外,所含有的金属成分显示出催化剂作用或催化剂失活作用,也可能会使聚合呈不稳定化。
本发明的低聚芴二芳基酯中,可能会含有由下述工序带来的钛、铜、铁等过渡金属、钠、钾等长周期型周期表第1族、镁、钙等第2族金属、锌、镉等第12族金属、或锡等第14族金属,该工序为酯交换反应催化剂存在下使碳酸二芳基类发生作用来进行酯交换的工序;这些金属的含有比例优选为500质量ppm以下、更优选为200质量ppm以下、进一步优选为50质量ppm以下、特别优选为10质量ppm以下。若金属成分多,则在聚合反应或树脂加工时,聚合物可能变得易着色。此外,所含有的金属成分显示出催化剂作用或催化剂失活作用,也可能会使聚合呈不稳定化。
本发明低聚芴单体的10质量%四氢呋喃溶液的色调优选为50以下。进一步优选为10以下。低聚芴单体的吸收端延伸至靠近可见光的区域,具有在因聚合或树脂加工而暴露于高温时容易着色的性质。为了得到色调良好的聚合物,优选聚合反应中所用的低聚芴单体尽可能着色少。由于色调与浓度成比例,因而可以为在不同浓度下进行测定、并标准化为10质量%浓度而得到的值。此处,低聚芴单体的色调(apha值)可如下测定:按照jis-k0071-1(1998年),将kishidachemical社制造色度标准液(1000度)稀释,将所制作的稀释液与低聚芴二醇装入到内径20mm的比色管中并进行比较,由此来测定该色调。
本发明的低聚芴单体在热重测定中的5%失重温度优选为230℃以上、更优选为250℃以上。进一步特别优选为270℃以上。芴是非常富含电子的结构,键合在芴环上的取代基的反应性高,容易产生热分解。若在聚合反应中使用热分解温度低的低聚芴单体,则在聚合时会产生热分解、聚合不到所期望的分子量、或者所得到的聚合物可能会着色。
<5.有机基团的导入方法>
作为在本发明的树脂组合物中导入上述通式(3)所表示的2价有机基团作为重复单元的方法,从所得到的树脂组合物的透明性、均匀性的方面出发,优选下述方法:
1.将上述式(10a)所表示的具有低聚芴的二羟基化合物与具有上述通式(3)所表示的有机基团的下式(21)所表示的二羟基化合物共聚的方法;
2.将上述式(10b)所表示的具有低聚芴的二酯化合物利用具有上述通式(3)所表示的有机基团的下式(21)所表示的二羟基化合物进行酯交换后,与具有上述通式(3)所表示的有机基团的下式(21)所表示的二羟基化合物进行共聚,通过两步导入的方法;
3.将上述式(10d)所表示的具有低聚芴的二芳基酯化合物与具有上述通式(3)所表示的有机基团的下式(21)所表示的二羟基化合物共聚的方法;
4.将上述式(10a)所表示的具有低聚芴的二羟基化合物、具有上述通式(3)所表示的有机基团的下式(28)所表示的二羧酸化合物、以及具有上述通式(3)所表示的有机基团的下式(21)所表示的二羟基化合物共聚的方法。
ho-r10-oh(21)
hocor10-cooh(28)
(式中,r10与上述通式(3)中的r10相同。)
此处,上述通式(3)所表示的2价有机基团可以使用单一种类的基团,也可将两种以上不同的有机基团组合使用。组合使用两种以上不同的有机基团可通过使用两种以上不同的上述通式(21)所表示的二羟基化合物和/或上述通式(28)所表示的二羧酸化合物来达成。
<6.聚合物的制造方法>
如上所述,作为聚合物优选聚酯、聚碳酸酯、聚酯碳酸酯,聚碳酸酯相比于聚酯通常具有充分的玻璃化转变温度、耐水解性优异,出于该原因特别优选聚碳酸酯。另一方面,聚酯的柔软性通常优于聚碳酸酯,出于该原因特别优选聚酯。聚酯碳酸酯的玻璃化转变温度与耐水解性以及柔软性的平衡优异,因而特别优选。
需要说明的是,聚碳酸酯和聚酯碳酸酯可通过<8.聚碳酸酯的聚合方法>等方法进行制造。另外,聚酯也可利用相同的方法进行制造,具体地说,可通过<9.聚酯的聚合方法>等方法来制造。
<7.聚碳酸酯树脂组合物>
本发明的树脂组合物中,从玻璃化转变温度和耐水解性的方面出发,优选上述聚合物为聚碳酸酯。下文中,有时将聚合物为聚碳酸酯的树脂组合物简称为“聚碳酸酯树脂组合物”。其中,2价低聚芴优选含有经下述[y]组中示出的碳酸酯键和/或酯键连接而成的重复单元结构,该重复单元结构用于构成具有耐热性与熔融加工性、机械强度的平衡优异的倾向的聚酯、聚碳酸酯或聚酯碳酸酯。
[化64]
[y]
(上述[y]组中示出的各连接基团中,z表示2价低聚芴、上述通式(3)所表示的2价有机基团和任意的重复单元连接的部位。此外,在连接基团为非对称的情况下,连接基团相对于2价低聚芴可以按任意方向连接。)
本发明的聚碳酸酯树脂组合物表示含有聚碳酸酯作为聚合物的树脂组合物,可以仅由聚碳酸酯构成,也可以共存有聚碳酸酯以外的聚合物。
本发明的聚碳酸酯树脂组合物可以含有2价低聚芴经任意的连接基团连接而成的聚合物,可以含有2价低聚芴与上述通式(3)所表示的2价有机基团经任意的连接基团连接而成的共聚物,也可以含有2价低聚芴、上述通式(3)所表示的2价有机基团以及任意的重复单元经任意的连接基团连接而成的共聚物。为了控制为膜成型时所需要的光学特性和物性范围,也可以使用2价低聚芴、上述通式(3)所表示的2价有机基团以及任意的重复单元经上述[y]组中示出的至少一种连接基团连接而成的共聚物。
在本发明的聚碳酸酯树脂组合物中可以共存有2价低聚芴经任意的连接基团连接而成的聚合物以外的聚合物,可以共存有2价低聚芴经任意的连接基团连接而成的聚合物、以及上述通式(3)所表示的2价有机基团经任意的连接基团连接而成的聚合物,也可以共存有2价低聚芴经任意的连接基团连接而成的聚合物、上述通式(3)所表示的2价有机基团经任意的连接基团连接而成的聚合物、以及任意的重复单元经任意的连接基团连接而成的聚合物。为了控制为膜成型时所需要的光学特性和物性范围,可以共存有经上述[y]组中示出的至少一种连接基团连接而成的具有2价低聚芴作为重复单元的聚合物、具有上述通式(3)所表示的2价有机基团作为重复单元的聚合物、以及具有任意重复单元的聚合物。
此处,上述通式(3)所表示的2价有机基团可以使用单一种类的基团,也可以将两种以上的有机基团的结构单元组合使用。
<7-1.包含低聚芴的重复单元结构>
为了提供充分高的玻璃化转变点和所期望的光学特性,本发明的聚碳酸酯树脂组合物优选含有具有碳酸酯键作为上述连接基团的、具有下述通式(22)所示重复单元结构的聚合物。
[化65]
(式中,r1和r2各自独立地为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,或者为选自由具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基以及具有或不具有取代基的碳原子数6~10的亚芳烷基组成的组中的2个以上基团经氧原子、具有或不具有取代基的硫原子、具有或不具有取代基的氮原子或羰基连接而成的基团;
r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、卤原子、硝基或氰基,r4~r9中,相邻的至少2个基团可以相互键合形成环。
n表示1~5的整数值。)
需要说明的是,作为r1~r9和n,可以应用在通式(1)中优选地示例出的r1~r9和n。
此外,在本发明的聚碳酸酯树脂组合物中,优选含有聚酯碳酸酯聚合物,该聚酯碳酸酯聚合物具有碳酸酯键与酯键这两者作为上述连接基团且含有下述通式(24)所表示的重复单元结构,下述通式(24)所表示的重复单元结构具有玻璃化转变温度与耐水解性、柔软性的平衡优异的倾向。此处,下述通式(24)所表示的聚酯碳酸酯的重复单元结构为含有低聚芴骨架的重复单元结构,因而具有以较少的摩尔分数和/或质量比得到逆波长分散性的倾向,从而优选。
[化66]
(式中,r1~r10和n与上述通式(22)中的r1~r10和n相同。)
<7-2.含有有机基团的重复单元结构的具体例>
本发明的聚碳酸酯树脂组合物中,为了将物性和光学特性调整在优选的范围内,具有2价低聚芴作为重复单元的聚合物可以具有上述通式(3)所表示的2价有机基团作为重复单元,具有2价低聚芴作为重复单元的聚合物也可以与具有上述通式(3)所表示的2价有机基团作为重复单元的聚合物共存。此处,在具有上述通式(3)所表示的2价有机基团作为重复单元时,优选以具有高耐水解性的、经下述通式(12)所表示的碳酸酯键连接而成的重复单元结构的形态具有。
[化67]
(式中,r10表示具有或不具有取代基的碳原子数2~20的亚烷基、具有或不具有取代基的碳原子数4~20的亚芳香基、具有或不具有取代基的碳原子数6~20的亚芳烷基、具有或不具有取代基的碳原子数2~100的亚烷基醚基、具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团、或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团。)
上述通式(12)所表示的重复单元结构中,r10优选的是因在主链不具有芳香环或者在主链大量含有芳香环以外的部分结构因而具有能够达成光学膜所要求的低光弹性系数的倾向的、含有具有或不具有取代基的碳原子数2~20的亚烷基、具有或不具有取代基的碳原子数2~100的亚烷基醚基、具有或不具有取代基的具有碳原子数4~20的脂环结构的有机基团、或者具有或不具有取代基的具有碳原子数4~20的杂环结构的有机基团的重复单元结构。更优选为选自下述重复单元结构中的至少一种重复单元结构:
具有可赋予高透明性与玻璃化转变温度、吸水性、低光弹性系数的倾向的、下述通式(13)所表示的重复单元结构;
[化68]
具有可赋予适度的疏水性与柔软性、低光弹性系数的倾向的、下述通式(14)所表示的重复单元结构,
[化69]
(式中,r11表示具有或不具有取代基的碳原子数0~18的直链状亚烷基。);
具有可赋予高透明性与玻璃化转变温度的倾向的、下述通式(15)所表示的重复单元结构
[化70]
(式中,r12表示具有或不具有取代基的碳原子数4~20的环亚烷基。);
具有可赋予柔软性与吸水性、低光弹性系数的倾向的、下述通式(16)所表示的重复单元结构
[化71]
(式中,r13表示具有或不具有取代基的碳原子数2~10的亚烷基,p为1~40的整数。);
具有可赋予高透明性与玻璃化转变温度的倾向的、下述通式(17)所表示的重复单元结构
[化72]
(式中,r14表示具有或不具有取代基的碳原子数2~10的亚烷基,r15表示具有或不具有取代基的碳原子数12~30的亚芳香基。);以及
具有可赋予高透明性与玻璃化转变温度的倾向的、下述通式(18)所表示的重复单元结构
[化73]
(式中,r16表示具有或不具有取代基的具有碳原子数2~20的乙缩醛环的基团。)。
进一步优选为上述通式(13)所表示的重复单元结构,其可通过提供高透明性与玻璃化转变温度、吸水性、低光弹性系数而赋予作为相位差膜的优异物性。
需要说明的是,作为上述式(14)中的r11,可以采用上述式(5)中优选示例出的r11。同样地,作为上述式(15)中的r12,可以采用上述式(6)中优选示例出的r12;作为上述式(16)中的r13,可以采用上述式(7)中优选示例出的r13。此外,作为上述式(17)中的r14和r15,可以各自独立地采用上述式(8)中优选示例出的r14和r15;作为上述式(18)中的r16,可以采用上述式(9)中优选示例出的r16。
本发明的聚碳酸酯树脂组合物中,关于2价低聚芴与上述通式(3)所表示的2价有机基团,只要在表现出上述光学物性的范围内,即可在聚碳酸酯树脂组合物中以任意质量含有。
<8.聚碳酸酯的聚合方法>
作为本发明的聚碳酸酯树脂组合物的制造方法,优选包括将二羟基化合物与下述通式(11)所表示的碳酸二酯熔融缩聚的方法(熔融聚合法)。作为通常的聚碳酸酯的制造方法已知的另一种方法为界面聚合法,但该方法中所能够使用的单体被限定在芳香族二羟基化合物,因而优选使用熔融法,该熔融法能够应用于也包括具有醇性羟基的二羟基化合物在内的更宽泛的结构。此外,界面法还需要使用毒性强的光气或二氯甲烷、氯苯等含氯溶剂,具有环境负荷也会增高的倾向。
[化74]
(式中,a1和a2分别为取代或无取代的碳原子数1~18的脂肪族烃基或取代或者无取代的芳香族烃基,a1与a2可以相同,也可以不同。)
本发明的聚碳酸酯树脂组合物之中,作为含有具有优选的上述通式(22)所示重复单元结构的聚合物的树脂组合物的制造方法,优选包括将上述通式(10a)所表示的具有低聚芴的二羟基化合物与上述通式(11)所表示的碳酸二酯熔融缩聚的方法(熔融聚合法)。此时也可以合用上述通式(10a)所表示的具有低聚芴的二羟基化合物以外的二羟基化合物。
此外,在本发明的聚碳酸酯树脂组合物之中,作为含有具有优选的上述通式(24)所示重复单元结构的聚合物的树脂组合物的制造方法,可以举出经前工序与后工序的两步进行制造的方法(熔融聚合法),在该前工序中,将上述通式(10b)所表示的低聚芴二酯化合物利用具有上述通式(3)所表示的有机基团的上述式(21)所表示的二羟基化合物进行酯交换;在后工序中,对于经前工序生成的生成物与上述通式(11)所表示的碳酸二酯进行熔融缩聚(其中,在进行该后工序时,也可以合用具有上述通式(3)所表示的有机基团的上述式(21)所表示的二羟基化合物。);或者可以举出将上述通式(10d)所表示的具有低聚芴的二芳基酯化合物、具有上述通式(3)所表示的有机基团的上述式(21)所表示的二羟基化合物以及上述通式(11)所表示的碳酸二酯一步熔融缩聚的方法(熔融聚合法)。
<8-1.碳酸二酯等>
作为该熔融聚合法中使用的碳酸二酯,通常可以举出上述通式(11)所表示的物质。作为上述式(11)所表示的碳酸二酯,例如可以举出:以碳酸二苯酯、碳酸二苄酯、双(氯苯基)碳酸酯、碳酸间甲苯酯、碳酸二萘酯、双(二苯基)碳酸酯等为代表的二芳基碳酸酯类;以碳酸二甲酯、碳酸二乙酯、碳酸二丁酯、碳酸二环己酯等为代表的碳酸二烷基酯类。其中优选使用二芳基碳酸酯类,特别优选使用碳酸二苯酯。这些碳酸二酯可以单独使用一种,也可将2种以上混合使用。
碳酸二酯相对于反应中所用的全部二羟基化合物优选以0.90以上的摩尔分数进行使用,更优选为0.96以上、进一步优选为0.98以上,并且优选为1.10以下、更优选为1.05以下、进一步优选为1.03以下。此外,在导入二羧酸结构的情况下,相对于从全部二羟基化合物的摩尔数中减去全部二羧酸的摩尔数所得到的二羟基化合物的摩尔数优选以0.90以上的摩尔分数进行使用,更优选为0.96以上、进一步优选为0.98以上,并且优选为1.10以下、更优选为1.05以下、进一步优选为1.03以下。该摩尔比例若小于上述下限值,则所制造的聚碳酸酯的末端羟基增加,聚碳酸酯的热稳定性变差、或得不到所期望的高分子量体。此外,该摩尔比例若大于上述上限值,则在同一条件下酯交换反应的速度降低、难以制造所期望分子量的聚碳酸酯,不仅如此,所制造的聚碳酸酯中的残存碳酸二酯量还会增加,该残存碳酸二酯在坯膜(原反)制膜时或拉伸时挥发,具有招致膜的缺陷的可能性。
本发明的聚碳酸酯树脂组合物中含有的聚碳酸酯为具有来源于二羟基化合物的重复单元经碳酸酯键连接而成的结构的聚合物,在本发明中,除了碳酸酯键的一部分被二羧酸结构取代而成的聚酯碳酸酯以外,还可以含有具有碳酸酯键的聚氨酯等。
<8-2.聚酯碳酸酯>
通过下述等方法得到聚酯碳酸酯:将聚合中所用的碳酸二酯的一部分置换为上述通式(28)所表示的二羧酸化合物和/或上述通式(20)所表示的低聚芴单体的聚合反应性基团a3和a4为羧基的二羧酸化合物,作为聚合中所用的二羟基化合物的一部分使用二羟基酯和/或二羟基酯低聚物。此处可以使用的二羟基酯和/或二羟基酯低聚物可通过二羧酸化合物与二羟基化合物的反应来合成。作为上述通式(28)所表示的二羧酸化合物,可以举出:对苯二甲酸、邻苯二甲酸、间苯二甲酸、4,4’-二苯基二羧酸、4,4’-二苯基醚二羧酸、4,4’-二苯甲酮二羧酸、4,4’-二苯氧基乙烷二羧酸、4,4’-二苯砜二羧酸、2,6-萘二羧酸等芳香族二羧酸;1,2-环己烷二羧酸、1,3-环己烷二羧酸、1,4-环己烷二羧酸等脂环式二羧酸;丙二酸、琥珀酸、戊二酸、己二酸、庚二酸、辛二酸、壬二酸、癸二酸等脂肪族二羧酸等,从所得到的聚酯碳酸酯的耐热性或热稳定性的方面出发,优选芳香族二羧酸,特别是从处理或获得的容易性的方面出发,优选对苯二甲酸或间苯二甲酸,其中适宜为对苯二甲酸。这些二羧酸成分本身作为二羧酸,可以当作上述聚酯碳酸酯的原料,根据制造法的不同,也可以以甲基酯体、苯基酯体等二羧酸酯或二酰卤等二羧酸衍生物为原料。需要说明的是,作为二羧酸化合物使用上述通式(20)所表示的低聚芴单体的情况下,作为二羟基化合物,并非必须使用上述通式(10a)所表示的具有低聚芴的二羟基化合物,从削减制造成本的方面出发,优选不使用上述通式(10a)所表示的具有低聚芴的二羟基化合物。
此外,在聚合中所用的上述通式(20)所表示的低聚芴单体的聚合反应性基团a3和/或a4为羟基酯基、即为具有酯骨架的羟基的情况下,或者a3和a4为羟基和羧基的情况下,均可得到聚酯碳酸酯。作为具有酯骨架的羟基的具体例,可以举出2-羟基乙氧基羰基、2-(2-羟基乙氧基)羰基乙基、2-(2-羟基乙氧基)羰基丙基等。此外,作为a3和a4为羟基和羧基的具体例,可以举出羟基甲基与乙氧基羰基、2-(2-羟基乙氧基)羰基与羧基、2-(2-羟基乙氧基)羰基乙基与羧基乙基等。
上述聚酯碳酸酯中,关于来源于全部二羧酸化合物的重复单元结构的含有比例,在设来源于全部二羟基化合物的重复单元结构与来源于全部羧酸化合物的结构单元合计为100摩尔%的情况下,来源于全部二羧酸化合物的重复单元结构的含有比例通常为45摩尔%以下、优选为30摩尔%以下、更优选为20摩尔%以下、进一步优选为0摩尔%。此处,来源于二羧酸化合物的重复单元结构的含有比例中的二羧酸化合物是指聚合中所用的全部二羧酸化合物,表示上述通式(28)所表示的二羧酸化合物或上述通式(20)所表示的低聚芴单体的聚合反应性基团a3和a4为羧基的二羧酸化合物。来源于二羧酸化合物的重复单元结构的含有比例若高于上述上限值,则聚合性降低,聚合有可能进行不到所期望的分子量。
<8-3.聚合催化剂>
作为熔融聚合中的聚合催化剂(酯交换催化剂),例如使用长周期型周期表第1族和/或第2族的金属化合物。酯交换催化剂(下文中有时简单称为催化剂、聚合催化剂)能够对反应速度或进行缩聚得到的聚碳酸酯树脂组合物的品质带来非常大的影响。
作为所使用的催化剂,只要可满足所制造的聚碳酸酯树脂组合物的透明性、色调、耐热性、耐候性以及机械强度就没有限定。例如可以举出长周期型周期表中的1族和/或2族(下文中简单记为“1族”、“2族”。)的金属化合物。
作为上述的1族金属化合物,例如可以举出:氢氧化钠、氢氧化钾、氢氧化锂、氢氧化铯、碳酸氢钠、碳酸氢钾、碳酸氢锂、碳酸氢铯、碳酸钠、碳酸钾、碳酸锂、碳酸铯、乙酸钠、乙酸钾、乙酸锂、乙酸铯、硬脂酸钠、硬脂酸钾、硬脂酸锂、硬脂酸铯、硼氢化钠、硼氢化钾、硼氢化锂、硼氢化铯、苯基化硼钠、苯基化硼钾、苯基化硼锂、苯基化硼铯、苯甲酸钠、苯甲酸钾、苯甲酸锂、苯甲酸铯、磷酸氢二钠、磷酸氢二钾、磷酸氢二锂、磷酸氢二铯、苯基磷酸二钠、苯基磷酸氢二钾、苯基磷酸二锂、苯基磷酸二铯;钠、钾、锂、铯的醇盐、酚盐;双酚a的2钠盐、2钾盐、2锂盐和2铯盐等。其中,从聚合活性与所得到的聚碳酸酯树脂组合物的色调的方面出发,优选锂化合物。
作为上述的2族金属化合物,例如可以举出:氢氧化钙、氢氧化钡、氢氧化镁、氢氧化锶、碳酸氢钙、碳酸氢钡、碳酸氢镁、碳酸氢锶、碳酸钙、碳酸钡、碳酸镁、碳酸锶、乙酸钙、乙酸钡、乙酸镁、乙酸锶、硬脂酸钙、硬脂酸钡、硬脂酸镁和硬脂酸锶等。其中优选镁化合物、钙化合物或钡化合物,从聚合活性与所得到的聚碳酸酯树脂组合物的色调的方面出发,进一步优选镁化合物和/或钙化合物,最优选钙化合物。
也可以与长周期型周期表第1族和第2族金属化合物一起辅助性地合用碱性硼化合物、碱性磷化合物、碱性铵化合物、胺系化合物等碱性化合物,但特别优选仅使用长周期型周期表第1族和/或第2族的金属化合物。
作为上述的碱性磷化合物,例如可以举出三乙基膦、三正丙基膦、三异丙基膦、三正丁基膦、三苯基膦、三丁基膦和季鏻盐等。
作为上述的碱性铵化合物,例如可以举出四甲基氢氧化铵、四乙基氢氧化铵、四丙基氢氧化铵、四丁基氢氧化铵、三甲基乙基氢氧化铵、三甲基苄基氢氧化铵、三甲基苯基氢氧化铵、三乙基甲基氢氧化铵、三乙基苄基氢氧化铵、三乙基苯基氢氧化铵、三丁基苄基氢氧化铵、三丁基苯基氢氧化铵、四苯基氢氧化铵、苄基三苯基氢氧化铵、甲基三苯基氢氧化铵和丁基三苯基氢氧化铵等。
作为上述的胺系化合物,例如可以举出4-氨基吡啶、2-氨基吡啶、n,n-二甲基-4-氨基吡啶、4-二乙基氨基吡啶、2-羟基吡啶、2-甲氧基吡啶、4-甲氧基吡啶、2-二甲氨基咪唑、2-甲氧基咪唑、咪唑、2-巯基咪唑、2-甲基咪唑、氨基喹啉和胍等。
关于上述聚合催化剂的用量,在使用长周期型周期表第1族与第2族金属化合物的情况下,相对于反应中所用的全部二羟基化合物1摩尔,以金属换算量计在通常为0.1μmol~100μmol的范围内使用,优选为0.5μmol~50μmol的范围内、进一步优选为1μmol~25μmol的范围内。聚合催化剂的用量若过少,则可能得不到制造所期望分子量的聚碳酸酯时所需要的聚合活性;另一方面,若聚合催化剂的用量过多,则所得到的聚合物的色调变差、产生副产物,流动性的降低或凝胶的产生增多,可能难以制造目标品质的聚碳酸酯。
其中,在使用包含选自由长周期型周期表中的2族组成的组和锂中的至少一种金属的化合物的情况下,特别是在使用镁化合物和/或钙化合物的情况下,以金属换算量计,在每1摩尔的上述全部二羟基化合物中优选为0.1μmol以上、更优选为0.3μmol以上、特别优选为0.5μmol以上。并且,作为上限,优选为20μmol以下、更优选为10μmol以下、进一步优选为5μmol以下、特别优选为3μmol以下。
催化剂量过少时,聚合速度缓慢,因而具有为了得到所期望分子量的聚碳酸酯树脂组合物,不得不提高聚合温度的倾向。因而所得到的聚碳酸酯树脂组合物的色调变差的可能性增高,并且未反应的原料在聚合途中挥发,二羟基化合物与碳酸二酯的摩尔比例被破坏,有可能达不到所期望的分子量。另一方面,聚合催化剂的用量过多时,有可能并发不优选的副反应,招致所得到的聚碳酸酯树脂组合物的色调的恶化或成型加工时的树脂的着色。
其中,若1族金属中的钠、钾或铯在聚碳酸酯树脂组合物中大量含有,则可能会对色调带来不良影响。并且,这些金属并不仅来源于所使用的催化剂,也可能由原料或反应装置中混入。不论来源如何,聚碳酸酯树脂组合物中这些金属的化合物的总量以金属量计优选为1重量ppm以下、进而更优选为0.5重量ppm以下。
此外,在导入二羧酸结构的情况下,也可以在与上述碱性化合物合用或不进行合用的情况下使用钛化合物、锡化合物、锗化合物、锑化合物、锆化合物、铅化合物、锇化合物等酯交换催化剂。相对于反应中所用的全部二羟基化合物1mol,这些酯交换催化剂的用量以金属换算量计通常为10μmol以上、优选为20μmol以上、更优选为50μmol以上,并且通常为1mmol以下、优选为800μmol以下、更优选为500μmol以下。
<8-4.聚合法>
作为将本发明的聚碳酸酯树脂组合物中含有的聚碳酸酯利用熔融聚合法进行制造的方法,使二羟基化合物和必要时的二羧酸化合物在聚合催化剂的存在下与碳酸二酯发生反应。聚合通常利用两步以上的多步工序实施,关于聚合反应器,可使用1个反应器改变条件利用两步以上工序进行实施,也可使用2个以上的反应器并改变各自的条件利用两步以上工序来实施,从生产效率的方面出发,使用2个以上、优选3个以上、进一步优选3~5个、特别优选4个反应器来实施。聚合反应可以为分批式、连续式、或者分批式与连续式的组合中的任一种方式,从生产效率与品质稳定性的方面出发,优选连续式。
在用于得到本发明的聚碳酸酯树脂组合物中所含有的聚碳酸酯的熔融聚合反应中,控制温度与反应体体系内压力的平衡这一点是重要的。若使温度和压力中的任意一者过快地变化,则未反应的单体馏出到反应体系外,二羟基化合物与碳酸二酯的摩尔比例发生变化,可能得不到所期望的高分子。
具体地说,关于第1步的反应,作为聚合反应器的内温的最高温度,通常为130℃以上、优选为140℃以上、更优选为150℃以上,并且通常为250℃以下、优选为240℃以下、更优选为230℃以下。此外,压力通常为110kpa以上、优选为70kpa以上、更优选为30kpa以上,并且通常为5kpa以下、优选为3kpa以下、更优选为1kpa以下(绝对压力)的压力下。另外,反应时间通常为0.1小时以上、优选为0.5小时以上,并且通常为10小时以下、优选为3小时以下,一边将所产生的碳酸二酯来源的单羟基化合物(使用碳酸二苯酯作为碳酸二酯的情况下,所谓单羟基化合物表示苯酚。)蒸馏去除到反应体系外一边实施反应。
在第2步以后,将反应体系的压力从第1步的压力缓慢地下降,接着将所产生的单羟基化合物排除到反应体系外的同时,最终使反应体系的压力(绝对压力)为5kpa以下、优选为3kpa,使内温的最高温度通常为210℃以上、优选为220℃以上,并且通常为270℃以下、优选为260℃以下,进行通常为0.1小时以上、优选为0.5小时以上、更优选为1小时以上,并且通常为10小时以下、优选为6小时以下、更优选为3小时以下。
特别是为了抑制本发明聚碳酸酯树脂组合物的着色或热劣化,得到色调、耐光性良好的聚碳酸酯树脂组合物,优选全部反应阶段中的内温的最高温度为270℃以下、特别优选为260℃以下。
<8-5.造粒>
本发明的聚碳酸酯树脂组合物在如上所述缩聚后,通常可进行冷却固化,并利用旋转式切割器等进行造粒。造粒的方法没有限定,可以举出下述方法:从最终的聚合反应器中以熔融状态抽出,以线料的形态使其冷却固化并进行造粒的方法;从最终的聚合反应器中以熔融状态将树脂供给到单螺杆或双螺杆的挤出机中,熔融挤出后,使其冷却固化并进行造粒的方法;或者从最终的聚合反应器中以熔融状态抽出,以线料的形态使其冷却固化,暂且进行造粒后再次将树脂供给到单螺杆或双螺杆的挤出机中,熔融挤出后使其冷却固化并进行造粒的方法;等等。如下文所述,如果在聚碳酸酯中大量含有副生的碳酸二酯来源的单羟基化合物,则在加工成相位差膜之后,会招致环境变化所致的光学特性的变化,因而本发明的聚碳酸酯树脂组合物优选使用挤出机除去碳酸二酯来源的单羟基化合物,其中优选从最终的聚合反应器中将树脂以熔融状态供给到具有单个或2个以上排气口的单螺杆或双螺杆的挤出机中,一边使排气口减压除去单羟基化合物一边进行熔融挤出,之后使其冷却固化并进行造粒的方法。
<8-6.碳酸二酯来源的单羟基化合物含量>
在熔融聚合法中,由于在聚合反应中从碳酸二酯中副生苯酚等单羟基化合物,因而该单羟基化合物残存在本发明的聚碳酸酯树脂组合物中,在进行膜的成膜时或拉伸时发生挥发,成为异味的原因、或招致膜的缺陷。此外,在本发明的聚碳酸酯树脂组合物被加工成相位差膜之后,残存在该膜中的碳酸二酯来源的单羟基化合物会由于环境变化而使相位差膜的光学特性发生变化,因而本发明的聚碳酸酯树脂组合物所含有的碳酸二酯来源的单羟基化合物优选为1500质量ppm以下。进一步优选为1000质量ppm以下。关于下限,为了解决上述问题,下限低为佳,但在熔融聚合法中,难以使残存在高分子中的单羟基化合物为零、为将其除去需要过多的劳力,因而其通常为1质量ppm。为了降低残存在本发明的聚碳酸酯树脂组合物中的碳酸二酯来源的单羟基化合物,如上所述将高分子利用挤出机进行脱挥处理、使聚合最终阶段的压力为3kpa以下、优选为2kpa以下是有效的,但若过分降低压力,则分子量急剧上升,可能难以进行反应的控制,因而优选使高分子的末端基团浓度呈羟基过剩或芳基过剩,使末端基团失去平衡来进行制造。其中,从热稳定性的方面出发,羟基末端浓度优选为50mol/ton以下、特别优选为30mol/ton以下。羟基末端浓度可利用1h-nmr等进行定量。羟基末端浓度可利用碳酸二酯与全部二羟基化合物的投料摩尔比来调节。
<9.聚酯的聚合方法>
通过下式方法等得到聚酯:将聚合中所用的碳酸二酯置换为上述通式(28)所表示的二羧酸化合物和/或上述通式(20)所表示的低聚芴单体的聚合反应性基团a3和a4为羧基的二羧酸化合物;作为聚合中所用的二羟基化合物的一部分置换成上述通式(20)所表示的低聚芴单体的聚合反应性基团a3和a4为羟基的二羟基化合物。
优选的二羧酸、聚合催化剂、聚合条件等与<8.聚碳酸酯的聚合方法>中记载的方法相同。
<10.添加剂>
在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中可以含有任意的添加剂。同样地,在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中所含有的聚合物中也可以含有任意的添加剂。
<10-1.热稳定剂>
为了防止成型时等的分子量的降低或色调的恶化,在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中可以混配热稳定剂。同样地,出于同样的理由,在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中所含有的聚合物中也可以混配热稳定剂。
作为该热稳定剂,可以举出通常已知的受阻酚系热稳定剂和/或磷系热稳定剂。
作为受阻酚系化合物,具体地说,可以举出2,6-二叔丁基苯酚、2,4-二叔丁基苯酚、2-叔丁基-4-甲氧基苯酚、2-叔丁基-4,6-二甲基苯酚、2,6-二叔丁基-4-甲基苯酚、2,6-二叔丁基-4-乙基苯酚、2,5-二叔丁基对苯二酚、正十八烷基-3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯、2-叔丁基-6-(3’-叔丁基-5’-甲基-2’-羟基苄基)-4-甲基丙烯酸苯酯、2,2’-亚甲基-双(4-甲基-6-叔丁基苯酚)、2,2’-亚甲基-双(6-环己基-4-甲基苯酚)、2,2’-亚乙基-双(2,4-二叔丁基苯酚)、四[亚甲基-3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯]甲烷、1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯等。其中可以举出四[亚甲基-3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯]-甲烷、正十八烷基-3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯、1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯。
作为磷系化合物,可以举出亚磷酸、磷酸、亚膦酸、膦酸和它们的酯等,具体地说,可以举出亚磷酸三苯酯、亚磷酸三(壬基苯基)酯、亚磷酸三(2,4-二叔丁基苯基)酯、亚磷酸三癸酯、亚磷酸三辛酯、亚磷酸三(十八烷基)酯、亚磷酸二癸基单苯基酯、亚磷酸二辛基单苯基酯、亚磷酸二异丙基单苯酯、亚磷酸单丁基二苯酯、亚磷酸单癸基二苯酯、亚磷酸单辛基二苯酯、双(2,6-二叔丁基-4-甲基苯基)季戊四醇二亚磷酸酯、2,2-亚甲基双(4,6-二叔丁基苯基)辛基亚磷酸酯、双(壬基苯基)季戊四醇二亚磷酸酯、双(2,4-二叔丁基苯基)季戊四醇二亚磷酸酯、二硬脂基季戊四醇二亚磷酸酯、磷酸三丁酯、磷酸三乙酯、磷酸三甲酯、磷酸三苯酯、磷酸二苯基单邻联苯酯、磷酸二丁酯、磷酸二辛酯、磷酸二异丙酯、4,4’-亚联苯基二次膦酸四(2,4-二叔丁基苯基)酯、苯膦酸二甲酯、苯膦酸二乙酯、苯膦酸二丙酯等。这些热稳定剂可以单独使用一种,也可将2种以上合用。
关于该热稳定剂,除了在熔融聚合时所添加的添加量以外,还可以进一步追加混配。即,在混配适当量的热稳定剂得到本发明的树脂组合物或本发明的聚碳酸酯树脂组合物之后,若进一步混配热稳定剂,则可避免雾度的上升、着色以及耐热性的降低,若可进一步混配较多的热稳定剂,则能够防止色调的恶化。此外,关于热稳定剂,例如在熔融挤出法等使用挤出机进行成膜的情况下,可以在挤出机中添加上述热稳定剂等进行成膜,也可以预先使用挤出机在树脂组合物或聚碳酸酯树脂组合物中添加上述热稳定剂等,制成颗粒等形状进行使用。
在设本发明的树脂组合物或本发明的聚碳酸酯树脂组合物为100质量份的情况下,这些热稳定剂的混合量优选为0.0001质量份以上、更优选为0.0005质量份以上、进一步优选为0.001质量份以上,并且优选为1质量份以下、更优选为0.5质量份以下、进一步优选为0.2质量份以下。
<10-2.抗氧化剂>
此外,在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中,出于抗氧化的目的,还可以混配通常已知的抗氧化剂。作为该抗氧化剂,例如可以举出季戊四醇四(3-巯基丙酸酯)、季戊四醇四(3-月桂基硫代丙酸酯)、甘油-3-硬脂基硫代丙酸酯、三甘醇-双[3-(3-叔丁基-5-甲基-4-羟基苯基)丙酸酯]、1,6-己二醇-双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、季戊四醇-四[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、十八烷基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯、1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯、n,n-六亚甲基双(3,5-二叔丁基-4-羟基-氢化肉桂酰胺)、3,5-二叔丁基-4-羟基-苄基膦酸酯-二乙基酯、三(3,5-二叔丁基-4-羟基苄基)异氰脲酸酯、4,4’-亚联苯基二次膦酸四(2,4-二叔丁基苯基)酯、3,9-双{1,1-二甲基-2-[β-(3-叔丁基-4-羟基-5-甲基苯基)丙酰基氧基]乙基}-2,4,8,10-四氧杂螺环(5,5)十一烷等的1种或2种以上。在设本发明的树脂组合物或本发明的聚碳酸酯树脂组合物为100质量份的情况下,这些抗氧化剂的混合量优选为0.0001质量份以上,并且优选为0.5质量份。
进一步地,在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物,也可以在无损于本发明目的的范围内含有通常使用的成核剂、阻燃剂、无机填充剂、冲击改良剂、发泡剂、染颜料等。
上述的添加剂可以将上述成分同时或以任意的顺序利用转鼓混合机、v型混合机、诺塔混合机(nautamixer)、班伯里混炼机、开炼机、挤出机等混合机混合在本发明的树脂组合物或本发明的聚碳酸酯树脂组合物中,进行制造,其中,从提高分散性的方面出发,优选利用挤出机、特别优选利用双螺杆挤出机进行混炼。
<11.用途>
本发明中所涉及的树脂组合物或聚碳酸酯树脂组合物的光弹性系数小、耐热性和成型性也优异,进而具有兼具着色少、透明性高的倾向,因而将它们成型得到的成型体在膜或透镜、光学棱镜之类的光学部件中是适合的。例如,本发明中涉及的膜可用作各种显示屏(液晶显示装置、有机el显示装置、等离子体显示装置、fed电场放出显示装置、sed表面电场显示装置)的可视角补偿用、防外光反射用、色调补偿用、向线性偏振光的圆偏振光转换用等的相位差膜。此外,本发明中所涉及的透镜、光学棱镜还可用于菲涅耳透镜、取物镜等光学透镜或光学棱镜中。
<11-1.膜>
本发明的树脂组合物和本发明的聚碳酸酯树脂组合物可适当地作为膜使用。可通过对本发明的树脂组合物或本发明的聚碳酸酯树脂组合物进行成膜来得到膜。
<11-2.膜制造法>
作为使用本发明的树脂组合物或本发明的聚碳酸酯树脂组合物进行坯膜(原反フィルム)的成膜的方法,有使本发明的树脂组合物或本发明的聚碳酸酯树脂组合物溶解在溶剂中并进行浇注后除去溶剂的流延法、不使用溶剂而进行熔融制膜的方法,具体地说,有使用t模的熔融挤出法、压延成型法、热压法、共挤出法、共熔融法、多层挤出、吹胀成型法等,没有特别限定,其中由于流延法可能会产生残存溶剂所致的问题,因而优选熔融制膜法,其中从之后进行拉伸处理的容易性的方面考虑,优选使用t模的熔融挤出法。
在熔融制膜法进行坯膜的成型的情况下,成型温度优选为265℃以下、更优选为260℃以下、特别优选为258℃以下。成型温度过高时,所得到的坯膜中的由异物或气泡产生所致的缺陷可能会增加、或者坯膜可能着色。其中,在成型温度过低时,本发明的树脂组合物或本发明的聚碳酸酯树脂组合物的粘度过高,难以进行坯膜的成型,可能难以制造厚度均匀的坯膜,因而成型温度的下限通常为200℃以上、优选为210℃以上、更优选为220℃以上。此处,坯膜的成型温度是指熔融制膜法中的成型时的温度,通常为对挤出熔融树脂的模口温度进行测定得到的值。
坯膜的厚度并无限制,但若过厚,则容易产生厚度不均(厚み斑),若过薄,则可能招致拉伸时的断裂,因而厚度通常为50μm以上、优选为70μm以上,并且通常为200μm以下、优选为120μm以下。此外,在坯膜具有厚度不均时,可能招致相位差膜的相位差不均,因而作为相位差膜使用的部分的厚度优选为设定厚度±3μm以下、更优选为设定厚度±2μm以下、特别优选为设定厚度±1μm以下。
<11-3.膜物性>
本发明的膜的内部雾度优选为3%以下、更优选为1.5%以下。相位差膜的内部雾度若大于上述上限值,则会产生光的散射,在与例如偏振元件进行层积时,可能会成为去偏振产生的原因。内部雾度的下限值没有特别限定,通常为0.2%以上。测定样品使用的是事先进行了雾度测定的带粘合剂透明膜贴合在试样膜的两面而制作出的消除了外部雾度的影响的状态的样品,测定值使用的是带粘合剂透明膜的雾度值的差分。
本发明的膜的b*值优选为3以下。膜的b*值若过大,则会产生着色等问题。本发明的膜的b*值更优选为2以下、特别优选为1以下。
关于本发明的膜,不论厚度如何,该膜本身的全光线透过率优选为80%以上,该透过率更优选为90%以上。透过率为上述下限以上时,可得到着色少的膜,在与偏振片贴合时,形成偏光度及透过率高的圆偏振片,在用于图像显示装置时,能够实现高的显示品位。需要说明的是,本发明的膜的全光线透过率的上限没有特别限制,但通常为99%以下。
本发明的膜优选在后述的弯曲测试中不发生脆性破坏。发生脆性破坏的膜在进行成膜时或拉伸时容易发生膜的断裂,可能使制造的成品率恶化。为了制成不发生脆性破坏的膜,将本发明的树脂组合物或聚碳酸酯树脂组合物的分子量或熔融粘度、玻璃化转变温度设计成上述的优选范围是很重要的。此外,通过在树脂组合物或聚碳酸酯树脂组合物中共聚或共混能够赋予柔软性的成分来调整膜的物性的方法也是有效的。
<11-4.拉伸膜的制造法>
如此得到的坯膜可通过沿至少一个方向进行拉伸来制成本发明的拉伸膜。关于该拉伸的方法,可单独使用自由端拉伸、固定端拉伸、自由端收缩、固定端收缩等各种拉伸方法,也可同时或者逐次使用这些方法。此外,关于拉伸方向,可在水平方向·垂直方向·厚度方向、对角方向等各种方向或维度来进行拉伸,没有特别限定。优选可以举出横单向拉伸方法、纵横同时双向拉伸方法、纵横逐次双向拉伸方法等。作为进行拉伸的手段,可以使用拉幅机拉伸机、双向拉伸机等任意适当的拉伸机。
拉伸温度可根据目的酌情选择适当的值。优选的是,拉伸在相对于坯膜(即作为坯膜的制膜材料的树脂组合物)的玻璃化转变温度(tg)通常为tg-20℃以上、优选为tg-10℃以上、更优选为tg-5℃以上,并且通常为tg+30℃以下、优选为tg+20℃以下、更优选为tg+10℃以下的范围来进行。通过选择这样的条件,相位差值容易均匀,并且膜不易浑浊。具体地说,上述拉伸温度通常为90℃以上、优选为100℃以上,并且通常为210℃以下、优选为200℃以下、更优选为180℃以下。
拉伸倍率根据目的酌情选择,设未拉伸的情况为1倍时,拉伸倍率优选为1.1倍以上、更优选为1.5倍以上、进一步优选为1.8倍以上、特别优选为2倍以上,并且优选为6倍以下、更优选为4倍以下、进一步优选为3倍以下、特别优选为2.5倍以下。拉伸倍率若过大,则不仅可能招致拉伸时的断裂,而且在高温条件下长期使用所致的光学特性的变动可能会增大;若其过小,则可能无法在所期望的厚度下赋予所谋求的光学特性。
拉伸速度也根据目的酌情选择,以下式所表示的应变速率计通常为50%以上、优选为100%以上、更优选为200%以上、特别优选为250%以上,并且通常为2000%以下、优选为1500%以下、更优选为1000%以下、特别优选为500%以下。拉伸速度过大时,可能会招致拉伸时的断裂,或者在高温条件下长期使用所致的光学特性的变动可能会增大。另外,若拉伸速度过小,则不仅生产率会降低,而且为了得到所期望的相位差,有时必须使拉伸倍率过大。
应变速率(%/分钟)=拉伸速度(mm/分钟)/坯膜的长度(mm)×100
此外,在拉伸后可利用加热炉进行热固定处理,也可控制拉幅机的幅度或调整辊圆周速度来进行缓和工序。通过进行该处理,能够抑制在高温条件下长期使用所致的光学特性的变动。
本发明的拉伸膜可通过对这样的拉伸工序中的处理条件进行酌情选择·调整来制作。
<11-5.拉伸膜物性>
本发明的拉伸膜优选为在波长450nm处测定的相位差(re450)和在波长550nm处测定的相位差(re550)之比满足下式(2)的相位差膜。
re450/re550≦1.0(2)
re450/re550优选为0.50以上1.00以下,更优选大于0.5且小于1.00,进一步与优选为0.70以上0.95以下,更进一步优选为0.75以上0.93以下,特别优选为0.80以上0.91以下。re450/re550的值为上述范围时,波长越长越可表现出相位差,能够在可见区域的各波长中得到理想的相位差特性。例如可通过将作为1/4λ板的具有这样的波长依赖性的本发明的相位差膜与偏振片贴合来制作圆偏振片等,能够实现在所有波长下具有防止外光反射功能的黑色性优异的圆偏振片和图像显示装置。另一方面,在re450/re550的值为上述范围外的情况下,波长所致的脱色增大,在圆偏振片或图像显示装置中产生着色的问题。
此外,本发明的拉伸膜优选满足<3-4.相位差比>中记载的物性值。同样地,优选满足<3-11.光弹性系数>中记载的物性值。同样地,优选满足<3-12.双折射>中记载的物性值。
本发明的拉伸膜的厚度通常优选为150μm以下、更优选为100μm以下、进一步优选为60μm以下。相位差膜的厚度若过厚,为了制造相同面积的膜可能需要更多的成膜材料而没有效率,或者使用该膜得到的制品的厚度可能会变厚,同时难以进行均匀性的控制,可能无法适合于要求精密性·薄型·均质性的机器。作为本发明相位差膜的厚度的下限优选为5μm以上、更优选为10μm以上。相位差膜的厚度若过薄,则难以进行膜的处理,在制造中会产生褶皱,或者难以与保护膜等其它膜或片等进行贴合。
本发明的拉伸膜的双折射优选为0.001以上。为了将后述使用本发明的树脂组合物进行成型的膜的厚度设计得非常薄,优选双折射高。因而,双折射更优选为0.002以上。双折射小于0.001的情况下,需要使膜的厚度过大,因而成膜材料的用量增加,从厚度·透明性·相位差的方面考虑,难以进行均质性的控制。因此,在双折射小于0.001的情况下,可能无法适合于要求精密性·薄型·均质性的机器。
<11-6.吸水率>
本发明的膜的饱和吸水率优选大于1.0质量%。若饱和吸水率大于1.0质量%,则在将该膜与其它膜等贴合时,具有能够容易地确保粘接性的倾向。例如,在与偏振片贴合时,由于膜为亲水性的,因而水的接触角也较低,容易自由地设计接合剂,可得到较高的粘接设计。在饱和吸水率为1.0质量%以下的情况下,膜为疏水性的,水的接触角也较高,难以进行粘接性的设计。并且膜容易带电而卷入异物等,在组装到圆偏振片、图像显示装置中时,具有会产生外观缺陷变多的问题的倾向。另一方面,若饱和吸水率大于2.0质量%,则湿度环境下的光学特性的耐久性有变差的倾向,因而不优选。本发明的膜的饱和吸水率优选大于1.0质量%、更优选为1.1质量%以上,并且优选为2.0质量%以下、更优选为1.5质量%以下。
另一方面,根据膜或使用膜的图像显示装置的使用条件的不同,饱和吸水率也可以为1.0质量%以下。
<11-7.器件用途等>
本发明中所涉及的膜可用作各种显示屏(液晶显示装置、有机el显示装置、等离子体显示装置、fed电场放出显示装置、sed表面电场显示装置)的可视角补偿用、防外光反射用、色调补偿用、线性偏振光向圆偏振光转换用等的相位差膜。
本发明中所涉及的树脂组合物和聚碳酸酯树脂组合物的光弹性系数小、耐热性和成型性也优异,进而具有兼具着色少、透明性高的倾向,因而还可用于其它光学膜或光盘、光学棱镜、取物镜等。
本发明中所涉及的拉伸膜的用途没有特别限制,由于在可见区域的各波长中具备理想的相位差特性,光弹性系数小、耐热性和成型性也优异,进而具有兼具着色少、透明性高的倾向,因而适用于1/4λ板、圆偏振片、图像显示装置等。
例如,通过使本发明的拉伸膜满足上述的<3-4.相位差比>中记载的条件,能够作为1/4λ板使用。可通过将如此制作的本发明的1/4λ板与偏振片贴合来制作圆偏振片等,能够制成在所有波长下具有防止外光反射功能的黑色性优异的圆偏振片,进而在应用于图像显示装置的情况下,能够实现黑色的再现性极为良好的图像显示装置。作为上述偏振片,可以采用公知的各种结构的偏振片。例如可以使用如下得到的偏振片等:通过现有公知的方法使碘或二色性染料等二色性物质吸附于各种膜、进行染色,通过交联、拉伸、干燥来制备偏振元件,在所制得的偏振元件上层积保护膜,得到偏振片。
此外,通过使本发明的拉伸膜满足上述的<3-4.相位差比>中记载的条件,能够得到对va模式液晶显示装置的脱色进行校正的相位差膜,能够实现波长所致的脱色少的液晶显示装置。进一步地,通过使其满足上述的<1-13.双折射>中记载的条件,能够在可见区域的波长中得到理想的相位差特性,能够制成宽带域零双折射材料。并且,作为液晶显示装置的偏振片保护膜,通过将本发明的宽带域零双折射材料与偏振片贴合,能够实现波长所致的脱色少的偏振片和图像显示装置。
<12.低聚芴单体的制造方法>
本发明所使用的低聚芴单体(20)的制造方法并无任何限定,例如可通过下式中示出的制造法a、制造法b或制造法c等方法进行制造。
[化75]
此处,各结构式中,r3为直接键合、具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基;
r4~r9各自独立地为氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数1~10的酰基、具有或不具有取代基的碳原子数1~10的烷氧基、具有或不具有取代基的碳原子数1~10的芳氧基、卤原子、硝基或氰基,r4~r9中,相邻的至少2个基团可以相互键合形成环。
a3和a4各自独立地表示聚合反应性基团。n表示1~5的整数值。
此处,各结构式中的r3~r9的优选取代基与上述通式(1)中的r3~r9相同。此外,各结构式中的n的优选值与上述通式(1)中的n相同。
<12-1.制造法a>
制造法a为下述方法:将芴类(i)作为原料,合成9位单取代芴类(iiia)和(iiib)之后,通过对这些单取代体进行交联来制造低聚芴单体(20)。此处,通式(iiia)与(iiib)可以相同,也可以不同。需要说明的是,也可以不将芴类(i)作为原料来合成9位单取代芴类(iiia)和/或(iiib)。
例如,作为9-芴羧酸酯的合成方法,已知有使用芴的方法(j.chem.soc.,1949,2623.)和使用二苯乙醇酸的方法(can.j.chem.,1956,34,991.)。还已知有将9-芴羧酸酯利用亚烷基进行交联的方法(anal.chem.,1960,32,554.)。可参考这些方法得到芴的单取代体,之后通过进行交联反应来制造低聚芴单体(20)。
<12-2.制造法b>
制造法b为下述方法:将芴类(i)作为原料,转换为9-羟基甲基芴类(iv),之后通过脱水合成烯烃体(v),使烯烃体(v)与芴基阴离子发生反应,从而来制造r3为亚甲基的低聚芴化合物(iia)。需要说明的是,无取代的9-羟基甲基芴可作为试剂购入。也可由此处得到的低聚芴化合物(iia)按照制造法c的工序(ii)导入聚合反应性基团a3和a4来制成低聚芴单体(20)。
例如已知有将9-羟基甲基芴转换成9-亚甲基-9h-芴(ジベンゾフルバン)后通过阴离子聚合来合成低聚芴(iia)的混合物的方法(j.am.chem.soc.,123,2001,9182-9183.)。可以参考这些方法来制造低聚芴(iia)。
<12-3.制造法c>
制造法c为下述方法:通过进行原料芴类(i)的交联反应(工序(i))来合成低聚芴化合物(ii),其后通过导入羟基、酯基、羧基等聚合反应性基团(工序(ii))来制造低聚芴单体(20)。
[化76]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。a3和a4各自独立地表示聚合反应性基团。)
下面将制造法c分成工序(i)低聚芴化合物(ii)的制造法和工序(ii)低聚芴单体(20)的制造法进行记载。
<12-4.工序(i):低聚芴化合物(ii)的制造方法>
[化77]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。)。下面将工序(i)中的低聚芴化合物(ii)的制造方法分成r3和n的数值的情况进行记载。
<12-4-1.r3为直接键合且n=1的情况:9,9’-联芴的制造方法>
9,9’-联芴的合成法已知有多种,可由芴酮或9-溴芴进行合成(j.chem.res.,2004,760;tetrahedronlett.,2007,48,6669.)。需要说明的是,9,9’-联芴可作为试剂购入。
<12-4-2.工序(ia):r3为亚甲基且n=1~5的情况的制造方法>
下述通式(iia)所表示的具有亚甲基交联的低聚芴化合物可由芴类(i)和甲醛类在碱存在下按照下式所表示的反应进行制造。
[化78]
(式中,r4~r9和n与式(1)中的r4~r9和n含义相同。)
<12-4-2-1.甲醛类>
工序(ia)中使用的甲醛类只要为能够向反应体系中供给甲醛的物质就没有特别限定,可以举出气态的甲醛、甲醛水溶液、甲醛聚合而成的多聚甲醛、三氧杂环已烷等。它们之中,出于工业成本低且由于为粉末状因而操作性容易、能够精确称量的原因,特别优选使用多聚甲醛。
(理论量的定义)
在制造目标n数的低聚芴化合物(iia)的情况下,甲醛类相对于原料芴类(i)的理论量(摩尔比)由n/(n+1)表示。
(优选不超过理论量的理由)
相对于芴类(i)使用超过理论量的甲醛类的情况下,具有会生成目标n数以上的低聚芴化合物(iia)的倾向。由于n数越增加溶解性越会降低,因而在目的物中存在有目标n数以上的低聚芴化合物(iia)的情况下,可知具有精制负荷增大的倾向。因此,通常优选甲醛类的用量为与目标n数对应的理论量的n/(n+1)倍摩尔以下。
(优选不大大低于理论量的理由)
此外,若甲醛类的用量大大低于理论量的n/(n+1),则目标n数以下的低聚芴化合物(iia)为主生成物或者原料芴类(i)未反应而有残留,因而可知具有收率大幅降低的倾向。
由此,关于甲醛类的最佳用量,具体地说,在n=1的情况下,相对于芴类(i)通常为0.1倍摩尔以上、优选为0.3倍摩尔以上、进一步优选为0.38倍摩尔以上,并且通常为0.5倍摩尔以下、优选为0.46倍摩尔以下、进一步优选为0.42倍摩尔以下。
此外,在n=2的情况下,通常为0.5倍摩尔以上、优选为0.55倍摩尔以上、进一步优选为0.6倍摩尔以上,并且通常为0.66倍摩尔以下、优选为0.65倍摩尔以下、进一步优选为0.63倍摩尔以下。由此可知,根据甲醛类的用量,主生成物的结构与生成物的比例有很大变化,通过在限定甲醛类用量的条件下进行使用,能够以高收率得到目标n数的低聚芴化合物(iia)。
<12-4-2-2.碱>
作为工序(ia)中使用的碱,使用氢氧化锂、氢氧化钠、氢氧化钾等碱金属氢氧化物;氢氧化钙、氢氧化钡等碱土金属的氢氧化物;碳酸钠、碳酸氢钠、碳酸钾等碱金属的碳酸盐;碳酸镁、碳酸钙等碱土金属的碳酸盐;磷酸钠、磷酸氢钠、磷酸钾等磷酸的碱金属盐;正丁基锂、叔丁基锂等有机锂盐;甲醇钠、乙醇钠、叔丁醇钾等碱金属的醇盐;氢化钠、氢化钾等氢化碱金属盐;三乙胺、二氮杂双环十一碳烯等叔胺;四甲基氢氧化铵、四丁基氢氧化铵等氢氧化季铵等。它们可以单独使用一种,也可以合用两种以上。
它们之中,优选在该反应中具有充分碱性的碱金属的醇盐,更优选工业成本低的甲醇钠和乙醇钠。此处的碱金属的醇盐可以使用粉状的物质,也可以使用醇溶液等液态物质。并且可以使碱金属与醇发生反应来制备。
碱的用量相对于原料芴类(i)并无特别上限,但若用量过多,则在搅拌或反应后的精制负荷会增大,因而通常为芴类(i)的10倍摩尔以下、优选为5倍摩尔以下、进一步优选为1倍摩尔以下。另一方面,碱的用量若过少,则反应的进行迟缓,因而作为下限,通常相对于原料芴类(i)为0.01倍摩尔以上、优选为0.1倍摩尔以上、进一步优选为0.2倍摩尔以上。
<12-4-2-3.溶剂>
工序(ia)优选使用溶剂进行。作为能够使用的溶剂的具体例,可以举出:作为烷基腈系溶剂的乙腈、丙腈等;作为醚系溶剂的二乙醚、四氢呋喃、1,4-二氧六环、甲基环戊基醚、叔丁基甲醚等;作为卤素系溶剂的1,2-二氯乙烷、二氯甲烷、氯仿、1,1,2,2-四氯乙烷等;作为卤素系芳香族烃的氯苯、1,2-二氯苯等;作为酰胺系溶剂的n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等;作为亚砜系溶剂的二甲基亚砜、环丁砜等;作为环式脂肪族烃的环戊烷、环己烷、环庚烷、环辛烷等单环式脂肪族烃,为其衍生物的甲基环戊烷、乙基环戊烷、甲基环己烷、乙基环己烷、1,2-二甲基环己烷、1,3-二甲基环己烷、1,4-二甲基环己烷、异丙基环己烷、正丙基环己烷、叔丁基环己烷、正丁基环己烷、异丁基环己烷、1,2,4-三甲基环己烷、1,3,5-三甲基环己烷等,十氢化萘等多环式脂肪族烃;正戊烷、正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷等非环式脂肪族烃;作为芳香族烃的甲苯、对二甲苯、邻二甲苯、间二甲苯等;作为醇系溶剂的甲醇、乙醇、异丙醇、正丁醇、叔丁醇、己醇、辛醇、环己醇等。
其中,出于由芴类(i)生成的阴离子的溶解性高、具有反应进行良好的倾向的原因,优选极性溶剂的酰胺系溶剂、或者亚砜系溶剂。其中,在制造n=1或2的低聚芴化合物(iia)的情况下,特别优选n,n-二甲基甲酰胺。这是由于,n=1或2的低聚芴在n,n-二甲基甲酰胺中的溶解性低,在目的物生成后迅速析出,抑制其以上的反应的进行,具有目的物的选择性增高的倾向。
这些溶剂可以单独使用一种,也可以两种以上混合使用。
对于工序(ia)中制造的低聚芴化合物(iia),可知n值越大,在溶剂中的溶解性越会减小,通过生成的目的物的迅速析出,可抑制其以上的反应的进行。因此,溶剂的用量优选根据n的值进行适当调整。特别是在制造n=1或2的低聚芴化合物(iia)的情况下,为了提高目的物的选择性,不过量使用溶剂为佳。例如,在使用作为最优选溶剂的n,n-二甲基甲酰胺的情况下,所使用的溶剂量的上限通常为原料芴类(i)的10倍体积量、优选为7倍体积量、进一步优选为4倍体积量的量。另一方面,若溶剂的用量过少,则搅拌困难、同时反应的进行缓慢,因而作为下限,通常使用原料芴类(i)的1倍体积量、优选为2倍体积量、进一步优选为3倍体积量的量。
<12-4-2-4.反应形式>
在进行工序(ia)时,反应的形式可以采用分批型反应、流通型反应、或它们组合而成的形式,对其形式没有特别限制。
<12-4-2-5.反应条件>
工序(ia)可根据目标n值的低聚芴化合物(iia)进行酌情调整。为了抑制反应进行至目标n值以上,优选尽可能在低温下进行反应。另一方面,若温度过低,则可能得不到充分的反应速度。
因此,在使用作为最佳溶剂的n,n-二甲基甲酰胺和作为最佳碱的乙醇钠的情况下,n=1和2的具体反应温度,通常在上限为30℃、优选为20℃、更优选为10℃的温度下进行实施。另一方面,在下限为-50℃、优选为-20℃、更优选为0℃进行实施。
关于工序(ia)中的一般反应时间,通常下限为30分钟、优选为60分钟、进一步优选为2小时,上限不特别限定,但通常为20小时、优选为10小时、进一步优选为5小时。
<12-4-2-6.目的物的分离·精制>
在反应终止后,可将反应液添加到稀盐酸等酸性水中,或者将稀盐酸等酸性水添加到反应液中,使目的物低聚芴化合物(iia)析出,由此来进行分离。
此外,在反应终止后,也可将目的物低聚芴化合物(iia)可溶性的溶剂与水添加到反应液中,进行提取。利用溶剂提取出的目的物可通过将溶剂浓缩的方法、或者添加不良溶剂的方法等进行分离。其中,由于低聚芴化合物(iia)在室温下具有在溶剂中的溶解性非常低的倾向,因而通常优选与酸性水接触使其析出的方法。
所得到的低聚芴化合物(iia)可直接作为工序(ii)的原料使用,也可在进行精制后用于工序(ii)中。作为精制法,可以采用通常的精制法、例如再结晶或再沉淀法、提取精制、柱色谱等,并无限制。
<12-4-3.工序(ib):r3为直接键合以外且n=1的情况下的二芴化合物(iib)的制造方法>
下述通式(iib)所表示的二芴化合物可将芴类(i)作为原料,在烷基化剂(viiia)和碱的存在下按照下述工序(ib)所表示的反应进行制造。
[化79]
上述式中的二芴化合物以结构式(iib)表示。(式中,r3为具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,r4~r9和n与式(1)中的r4~r9和n含义相同。x表示离去基团。作为离去基团的示例,可以举出卤原子(不包括氟原子。)、甲磺酰基、或甲苯磺酰基等。)
二芴化合物(iib)的制造法中,使用正丁基锂作为碱,在产生芴类(i)的阴离子后,使其与烷基化剂(viiia)偶联的方法是广为人知的,已知有r3为亚乙基的情况或r3为亚丙基等的制造法(organometallics,2008,27,3924;j.molec.cat.a:chem.,2004,214,187.)。此外,除了亚烷基以外还有利用亚二甲苯基进行交联的报告例(j.am.chem.soc.,2007,129,8458.)。但是,在利用这些使用正丁基锂的方法进行工业制造时,在安全方面、成本方面均有非常困难的倾向。此外,作为二芴化合物(iib)的制造方法,已知有使芴与乙二醇在碱存在下在高温下进行脱水缩合的方法(j.org.chem.,1965,30,2540.)。
作为工序(ib)中使用的烷基化剂,可以举出:二碘甲烷、1,2-二碘乙烷、1,3-二碘丙烷、1,4-二碘丁烷、1,5-二碘戊烷、1,6-二碘己烷、二溴甲烷、1,2-二溴乙烷、1,3-二溴丙烷、1,4-二溴丁烷、1,5-二溴戊烷、1,6-二溴己烷、二氯甲烷、1,2-二氯乙烷、1,3-二氯丙烷、1,4-二氯丁烷、1,5-二氯戊烷、1,6-二氯己烷、1-溴-3-氯丙烷等直链状的烷基二卤化物(不包括氟原子);2,2-二甲基-1,3-二氯丙烷等包含支链的烷基二卤化物(不包括氟原子);1,4-双(溴甲基)苯、1,3-双(溴甲基)苯等芳烷基二卤化物(不包括氟原子);乙二醇二甲磺酸酯、乙二醇二甲苯磺酸酯、丙二醇二甲磺酸酯、四亚甲基二醇二甲磺酸酯等二醇的二磺酸酯等。
<12-4-4.工序(ic):r3为直接键合以外且为n=2以上的情况的低聚芴化合物(iic)的制造方法>
下述通式(iid)所表示的低聚芴化合物可将低聚芴化合物(iic)作为原料,在烷基化剂(viiia)与碱存在下在溶剂中按照下述工序(ic)所表示的反应进行制造。
[化80]
上述式中的低聚芴化合物由结构式(iid)所表示。(式中,r3为具有或不具有取代基的碳原子数1~10的亚烷基、具有或不具有取代基的碳原子数4~10的亚芳香基、或者具有或不具有取代基的碳原子数6~10的亚芳烷基,r4~r9和n与式(1)中的r4~r9和n含义相同。x表示离去基团。作为离去基团的示例,可以举出卤原子(不包括氟原子。)、甲磺酰基、或甲苯磺酰基等。)
<12-5.低聚芴单体(20)的制造方法>
下面,将下式所表示的工序(ii)中的低聚芴单体(20)的制造方法按a3和a4的种类分别进行记载。
[化81]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。a3和a4各自独立地表示聚合反应性基团。)
<12-5-1.工序(iia):通式(20)中,a3和a4为羟基甲基的情况的制造方法>
a3和a4为羟基甲基的低聚芴二醇(19)可在碱存在下按照下述工序(iia)所表示的反应由低聚芴化合物(ii)和甲醛类进行制造。
[化82]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。)
<12-5-1-1.甲醛类>
工序(iia)中使用的甲醛类只要为能够向反应体系中供给甲醛的物质就没有特别限定,可以举出气态的甲醛、甲醛水溶液、甲醛聚合而成的多聚甲醛、三氧杂环已烷等。它们之中,出于工业成本低且由于为粉末状因而操作性容易、能够精确称量的原因,特别优选使用多聚甲醛。
关于甲醛类的用量,相对于原料低聚芴化合物(ii),其上限没有特别限制,但若其用量过多,则具有反应后的精制负荷增大的倾向,因而通常相对于低聚芴化合物(ii)为20倍摩尔以下、优选为10倍摩尔以下、进一步优选为5倍摩尔以下。关于下限,由于相对于原料以理论量计为2倍摩尔,因而下限通常为2倍摩尔以上。由于不会残留有加速反应的进行的原料或中间体,因而甲醛类相对于原料低聚芴化合物(ii)稍微过量使用也没有任何问题。此时甲醛类的优选用量相对于原料低聚芴化合物(ii)为2.1倍摩尔以上、进一步优选为2.2倍摩尔以上。
<12-5-1-2.碱>
作为碱,使用氢氧化锂、氢氧化钠、氢氧化钾等碱金属氢氧化物;氢氧化钙、氢氧化钡等碱土金属的氢氧化物;碳酸钠、碳酸氢钠、碳酸钾等碱金属的碳酸盐;碳酸镁、碳酸钙等碱土金属的碳酸盐;磷酸钠、磷酸氢钠、磷酸钾等磷酸的碱金属盐;正丁基锂、叔丁基锂等有机锂盐;甲醇钠、乙醇钠、叔丁醇钾等碱金属的醇盐;氢化钠、氢化钾等氢化碱金属盐;三乙胺、二氮杂双环十一碳烯等叔胺;四甲基氢氧化铵、四丁基氢氧化铵等氢氧化季铵等。它们可以单独使用一种,也可以合用两种以上。
它们之中,优选在该反应中具有充分碱性的碱金属的醇盐,更优选工业成本低的甲醇钠或乙醇钠。此处的碱金属的醇盐可以使用粉状的物质,也可以使用醇溶液等液态物质。并且可以使碱金属与醇发生反应来制备。
碱的用量相对于原料低聚芴化合物(ii)过量使用时,可只有进行低聚芴二醇(19)的分解反应的倾向。因此,其相对于低聚芴化合物(ii)为1倍摩尔以下、优选为0.5倍摩尔以下、进一步优选为0.2倍摩尔以下为佳。另一方面,若碱的用量过少,则反应的进行迟缓,因而作为下限,通常相对于原料低聚芴化合物(ii)为0.01倍摩尔以上、优选为0.05倍摩尔以上。
<12-5-1-3.溶剂>
工序(iia)优选使用溶剂来进行。
具体地说,可使用的溶剂可以举出:作为烷基腈系溶剂的乙腈、丙腈等;作为酮系溶剂的丙酮、甲基乙基酮、甲基异丁基酮等;作为酯系溶剂的乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸苯酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丙酸苯酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸甲酯、乳酸甲酯、乳酸乙酯等直链状的酯类,γ-丁内酯、己内酯等环状酯类,乙二醇单甲基醚乙酸酯、乙二醇单乙基醚乙酸酯、乙二醇单丁醚乙酸酯、丙二醇-1-单甲醚乙酸酯、丙二醇-1-单乙醚乙酸酯等醚酯类等;作为醚系溶剂的二乙醚、四氢呋喃、1,4-二氧六环、甲基环戊基醚、叔丁基甲醚等;作为卤素系溶剂的1,2-二氯乙烷、二氯甲烷、氯仿、1,1,2,2-四氯乙烷等;作为卤素系芳香族烃的氯苯、1,2-二氯苯等;作为酰胺系溶剂的n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等;作为亚砜系溶剂的二甲基亚砜、环丁砜等;作为环式脂肪族烃的环戊烷、环己烷、环庚烷、环辛烷等单环式脂肪族烃,为其衍生物的甲基环戊烷、乙基环戊烷、甲基环己烷、乙基环己烷、1,2-二甲基环己烷、1,3-二甲基环己烷、1,4-二甲基环己烷、异丙基环己烷、正丙基环己烷、叔丁基环己烷、正丁基环己烷、异丁基环己烷、1,2,4-三甲基环己烷、1,3,5-三甲基环己烷等,十氢化萘等多环式脂肪族烃;正戊烷、正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷等非环式脂肪族烃;作为芳香族烃的甲苯、对二甲苯、邻二甲苯、间二甲苯等;作为醇系溶剂的甲醇、乙醇、异丙醇、正丁醇、叔丁醇、己醇、辛醇、环己醇等。
其中,出于由低聚芴化合物(ii)生成的阴离子的溶解性高、具有反应进行良好的倾向的原因,优选极性溶剂的酰胺系溶剂或者亚砜系溶剂,特别优选n,n-二甲基甲酰胺。
这些溶剂可以单独使用一种,也可以两种以上混合使用。关于溶剂的用量,上限没有特别限制,然而若考虑到每一反应器中的目的物的生成效率,则通常使用原料低聚芴化合物(ii)的10倍体积量、优选为7倍体积量、进一步优选为4倍体积量的量。另一方面,若溶剂的用量过少,则变得搅拌困难,同时具有反应的进行缓慢的倾向,因而作为下限,通常使用原料低聚芴化合物(ii)的1倍体积量、优选为2倍体积量、进一步优选为3倍体积量的量。
<12-5-1-4.反应形式>
在进行工序(iia)时,反应的形式可以采用分批型反应、流通型反应、或它们组合而成的形式,对其形式没有特别限制。
在分批式情况下的将反应试剂投入到反应器中的投入方法中,在反应开始时将碱通过一次性添加而进行填料的情况下,由于已知分解反应容易进行,因而优选在加入原料低聚芴化合物(ii)、甲醛类以及溶剂后分多次、每次少量添加碱的方法。
<12-5-1-5.反应条件>
工序(iia)的温度若过低,则得不到充分的反应速度,反之若温度过高,则可知有进行分解反应的倾向,温度管理是极为重要的。因此,在使用作为最佳溶剂的n,n-二甲基甲酰胺和作为最佳碱的乙醇钠的情况下,通常在下限为-50℃、上限为30℃进行实施。具体地说,在r3为亚甲基且n=1的情况下,在反应温度的上限优选为20℃、更优选为10℃的条件下进行实施。另一方面,在下限优选为-20℃、更优选为0℃以上的条件下进行实施。在r3为亚乙基且n=1的情况下,在反应温度的上限优选为25℃、更优选为20℃的条件下实施。另一方面,在下限优选为0℃、更优选为10℃以上的条件下进行实施。此外,在r3为亚甲基且n=2的情况下,在反应温度的上限优选为25℃、更优选为20℃的条件下实施。另一方面,在下限优选为0℃、更优选为10℃以上的条件下进行实施。
<12-5-1-6.目的物的分离·精制>
在反应终止后,可将反应液添加到稀盐酸等酸性水中,或者将稀盐酸等酸性水添加到反应液中,使目的物低聚芴二醇(19)析出,由此来进行分离。
此外,在反应终止后,也可将可溶性的溶剂与水添加到反应液中,对目的物低聚芴二醇(19)进行提取。利用溶剂提取出的目的物可通过将溶剂浓缩的方法、或者添加不良溶剂的方法等进行分离。
所得到的低聚芴二醇(19)可作为聚合物原料直接用于聚合中,也可在进行精制后进行聚合。作为精制法,可以采用通常的精制法、例如再结晶或再沉淀法、提取精制、柱色谱等,并无限制。
金属成分的存在在聚合反应中多产生问题,可以使长周期型周期表第1族与第2族金属在单体中的含有比例为500质量ppm以下、更优选为200质量ppm以下、进一步优选为50质量ppm以下、特别优选为10质量ppm以下。为了去除金属成分,分液操作通常是非常有效的,但由于目的物低聚芴二醇(19)仅溶解在n,n-二甲基甲酰胺或四氢呋喃等高极性溶剂中,因而利用2层体系进行的分液操作非常困难。另一方面,对于反应停止后的析出物,即使进行水洗、利用任意溶剂进行的热悬浮洗涤等通常的精制处理,也难以充分除去所混入的金属成分,在析出物中可能残存有数百质量ppm数量级的金属成分。作为用于除去金属成分的良好的精制法,将含有杂质的反应析出物溶解在n,n-二甲基甲酰胺、四氢呋喃等溶解性较高的溶剂中之后添加水使其析出的方法作为简便且有效的无机盐除去法而优选。
<12-5-2.工序(iib):通式(20)中,a3和a4为羟基、酯基、羧基、具有氨基的基团的情况的制造方法(基于迈克尔(michael)加成反应的制造法)>
下述通式(vii)所表示的低聚芴衍生物可在碱存在下按照下述工序(iib)所表示的反应由低聚芴化合物(ii)和吸电子基团取代烯烃(vi)进行制造。
[化83]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基。ewg表示吸电子基团。)
<12-5-2-1.吸电子基团取代烯烃>
作为反应试剂的吸电子基团取代烯烃由工序(iib)中的通式(vi)所表示,通式(vi)中,ri、rii和riii各自独立地表示氢原子、或者碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、具有或不具有取代基的碳原子数6~10的芳烷基。具体地说,可以举出:甲基、乙基、正丙基、异丙基、环己基等(可以为直链也可以为支链)烷基;苯基、1-萘基、2-萘基、2-噻吩基等芳基;苄基、2-苯基乙基、对甲氧基苄基等芳烷基。此外,通式(vi)中,吸电子基团取代烯烃的ewg表示吸电子基团。具体地说,可以举出硝基、氰基、甲酰基、羧基、具有碳原子数1~10的有机取代基的酮基或酯基。酮基或酯基所具有的有机取代基具体可以举出:甲基、乙基、正丙基、异丙基、正丁基等链状的(可以为直链也可以为支链)烷基;环己基等环状的烷基;羟基乙基、羟基丙基等羟基烷基;苯基、1-萘基、2-萘基、2-噻吩基等芳基;苄基、2-苯基乙基、对甲氧基苄基等芳烷基等。这些取代基可在不阻碍工序(iib)中的反应的范围内进一步被任意的取代基所取代。
作为吸电子基团取代烯烃(vi),可以举出:丙烯酸甲酯、丙烯酸乙酯、丙烯酸苯酯、丙烯酸烯丙酯、丙烯酸缩水甘油酯、丙烯酸-2-羟基乙酯、丙烯酸-4-羟基丁酯、1,4-环己烷二甲醇单丙烯酸酯等丙烯酸酯类;甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸苯酯、甲基丙烯酸烯丙酯、甲基丙烯酸酸缩水甘油酯、甲基丙烯酸-2-羟基乙酯等甲基丙烯酸酯类;2-乙基丙烯酸甲酯、2-苯基丙烯酸甲酯等α-取代不饱和酯类;肉桂酸甲酯、肉桂酸乙酯、丁烯酸甲酯、丁烯酸乙酯等β-取代不饱和酯类;β-硝基苯乙烯等共轭硝基烯烃类;丙烯腈等α,β-不饱和腈类;丙烯醛、异丁烯醛、巴豆醛等α,β-不饱和醛类。其中优选能够直接导入聚合反应性基团的下述通式(vi-1)所表示的不饱和羧酸酯
[化84]
(式中,r17表示碳原子数1~10的有机取代基,riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基。),更优选包含在其中的丙烯酸酯类、甲基丙烯酸酯类或α-取代不饱和酯类,从反应速度与反应选择性的方面出发,进一步优选riii为氢原子或甲基的丙烯酸酯类或甲基丙烯酸酯类。由于r17为更小的基团时,工业成本低并且蒸馏精制容易、反应性也高,因而特别优选丙烯酸甲酯、甲基丙烯酸甲酯、丙烯酸乙酯、甲基丙烯酸乙酯、丙烯酸苯酯、或者甲基丙烯酸苯酯。
另一方面,关于酯基的有机取代基,在为丙烯酸-2-羟基乙酯、丙烯酸-4-羟基丁酯、1,4-环己烷二甲醇单丙烯酸酯等具有羟基烷基的酯类的情况下,由于可利用一步来得到聚酯碳酸酯、聚酯的原料,因而特别优选。
也可以使用2种以上不同的吸电子基团取代烯烃(vi),但从精制简便性的方面考虑,优选使用一种吸电子基团取代烯烃(vi)。
此时,在作为吸电子基团取代烯烃(vi)使用酯类的情况下,低聚芴衍生物(vii)为下述的低聚芴二酯(20b)。特别是在所使用的酯类为具有羟基烷基的酯类的情况下,低聚芴衍生物(vii)为下述的低聚芴二羟基酯(20d)。
[化85]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,riv和rv表示碳原子数1~10的有机取代基。)
吸电子基团取代烯烃(vi)的聚合活性高,因而在以高浓度存在时,具有容易由于光、热、酸·碱等外部刺激而发生聚合的倾向。此时,由于伴随有大量放热,因而可能会非常危险。因此,从安全性的方面出发,吸电子基团取代烯烃(vi)的用量不过分地过剩使用为佳。通常相对于原料低聚芴(ii)为10倍摩尔以下、优选为5倍摩尔以下、进一步优选为3倍摩尔以下。关于下限,由于相对于原料以理论量计为2倍摩尔,因而下限通常为2倍摩尔以上。为了不会残留有加速反应进行的原料或中间体,吸电子基团取代烯烃(vi)的用量相对于原料低聚芴(ii)为2.2倍摩尔以上、进一步优选为2.5倍摩尔以上。
<12-5-2-2.碱>
作为碱,使用氢氧化锂、氢氧化钠、氢氧化钾等碱金属氢氧化物;氢氧化钙、氢氧化钡等碱土金属的氢氧化物;碳酸钠、碳酸氢钠、碳酸钾等碱金属的碳酸盐;碳酸镁、碳酸钙等碱土金属的碳酸盐;磷酸钠、磷酸氢钠、磷酸钾等磷酸的碱金属盐;正丁基锂、叔丁基锂等有机锂盐;甲醇钠、乙醇钠、叔丁醇钾等碱金属的醇盐;氢化钠、氢化钾等氢化碱金属盐;三乙胺、二氮杂双环十一碳烯等叔胺;四甲基氢氧化铵、四丁基氢氧化铵、苄基三甲基氢氧化铵等氢氧化季铵。它们可以单独使用一种,也可以合用两种以上。
在r3为亚甲基的情况与除此以外的情况下,具有低聚芴(ii)的反应性有较大差异的倾向。因此,分成r3为亚甲基的情况与除此以外的情况进行记载。
在r3为亚甲基的情况下,低聚芴(ii)在溶剂中在碱存在下容易进行分解反应。因此,出于在利用有机层与水层的2层体系进行反应的情况下能够抑制分解反应等副反应的原因,优选使用水溶性的无机碱。其中从成本、反应性的方面考虑,优选碱金属的氢氧化物,进一步特别优选氢氧化钠或氢氧化钾。
此外,关于水溶液的浓度,在使用特别优选的氢氧化钠水溶液的情况下,由于若浓度地则反应速度会显著降低,因而特别优选使用通常为5wt/vol%以上、优选为10wt/vol%以上、更优选为25wt/vol%以上的水溶液。
在r3为亚甲基以外的情况下,即使利用有机层与水层的2层体系也可进行反应,但在使用溶解于有机层中的有机碱进行反应的情况下,由于反应进行得迅速而优选使用有机碱。其中优选在该反应中具有充分碱性的碱金属的醇盐,更优选工业成本低的甲醇钠或乙醇钠。此处的碱金属的醇盐可以使用粉状的物质,也可以使用醇溶液等液态物质。并且可以使碱金属与醇发生反应来制备。
在r3为亚甲基的情况下,关于碱的用量,相对于原料低聚芴(ii),其上限没有特别限制,但在碱的用量过多时,搅拌或反应后的精制负荷可能会增大,因而在使用作为特别优选的碱的25wt/vol%以上的氢氧化钠水溶液的情况下,通常相对于低聚芴(ii)为20倍体积量以下、优选为10倍体积量以下、进一步优选为5倍体积量以下。在碱量过少时,反应速度显著降低,因而碱相对于原料低聚芴(ii)通常为0.2倍体积量以上。优选为0.5倍体积量以上、更优选为1倍体积量以上。
在r3为亚甲基以外的情况下,关于碱的用量,相对于原料低聚芴(ii),其上限没有特别限制,但在碱的用量过多时,搅拌或反应后的精制负荷可能会增大,因而在使用作为特别优选的碱的甲醇钠或乙醇钠的情况下,通常相对于低聚芴(ii)为5倍摩尔以下、优选为2倍摩尔以下、进一步优选为1倍摩尔以下、特别优选为0.5倍摩尔以下。在碱量过少时,反应速度显著降降低,因而碱相对于原料低聚芴(ii)通常为0.005倍摩尔以上。优选的是,0.01倍摩尔以上、更优选为0.05倍摩尔以上、特别优选为0.1倍摩尔以上。
<12-5-2-3.相转移催化剂>
在工序(iib)中,在利用有机层与水层的2层体系进行反应的情况下,为了提高反应速度,优选使用相转移催化剂。
作为相转移催化剂,可以举出:四甲基氯化铵、四丁基溴化铵、甲基三辛基氯化铵、甲基三癸基氯化铵、苄基三甲基氯化铵、三辛基甲基氯化铵、四丁基碘化铵、乙酰基三甲基溴化铵、苄基三乙基氯化铵等季铵盐的卤化物(不包括氟原子);n,n-二甲基吡咯烷鎓氯化物、n-乙基-n-甲基吡咯烷鎓碘化物、n-丁基-n-甲基吡咯烷鎓溴化物、n-苄基-n-甲基吡咯烷鎓氯化物、n-乙基-n-甲基吡咯烷鎓溴化物等季吡咯烷鎓盐的卤化物(不包括氟原子);n-丁基-n-甲基吗啉鎓溴化物、n-丁基-n-甲基吗啉鎓碘化物、n-烯丙基-n-甲基吗啉鎓溴化物等季吗啉鎓盐的卤化物(不包括氟原子);n-甲基-n-苄基哌啶鎓氯化物、n-甲基-n-苄基哌啶鎓溴化物、n,n-二甲基哌啶鎓碘化物、n-甲基-n-乙基哌啶鎓乙酸酯、n-甲基-n-乙基哌啶鎓碘化物等季哌啶鎓盐的卤化物(不包括氟);冠醚类等。优选季铵盐,进一步优选苄基三甲基氯化铵、或者苄基三乙基氯化铵。
这些相转移催化剂可以单独使用一种,也可以合用两种以上。
相转移催化剂的用量相对于原料低聚芴(ii)过多时,具有酯的水解或逐级michael反应等副反应显著进行的倾向,并且从成本的方面出发,通常相对于低聚芴(ii)也为5倍摩尔以下、优选为2倍摩尔以下、进一步优选为1倍摩尔以下。相转移催化剂的用量过少时,有反应速度显著降低的倾向,因而相转移催化剂的用量相对于原料低聚芴通常为0.01倍摩尔以上。优选为0.1倍摩尔以上、更优选为0.5倍摩尔以上。
<12-5-2-4.溶剂>
工序(iib)优选使用溶剂进行。
具体地说,可使用的溶剂可以举出:作为烷基腈系溶剂的乙腈、丙腈等;作为酮系溶剂的丙酮、甲基乙基酮、甲基异丁基酮等;作为酯系溶剂的乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸苯酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丙酸苯酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸甲酯、乳酸甲酯、乳酸乙酯等直链状的酯类,γ-丁内酯、己内酯等环状酯类,乙二醇单甲基醚乙酸酯、乙二醇单乙基醚乙酸酯、乙二醇单丁醚乙酸酯、丙二醇-1-单甲醚乙酸酯、丙二醇-1-单乙醚乙酸酯等醚酯类等;作为醚系溶剂的二乙醚、四氢呋喃、1,4-二氧六环、甲基环戊基醚、叔丁基甲醚等;作为卤素系溶剂的1,2-二氯乙烷、二氯甲烷、氯仿、1,1,2,2-四氯乙烷等;作为卤素系芳香族烃的氯苯、1,2-二氯苯等;作为酰胺系溶剂的n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等;作为亚砜系溶剂的二甲基亚砜、环丁砜等;作为环式脂肪族烃的环戊烷、环己烷、环庚烷、环辛烷等单环式脂肪族烃,为其衍生物的甲基环戊烷、乙基环戊烷、甲基环己烷、乙基环己烷、1,2-二甲基环己烷、1,3-二甲基环己烷、1,4-二甲基环己烷、异丙基环己烷、正丙基环己烷、叔丁基环己烷、正丁基环己烷、异丁基环己烷、1,2,4-三甲基环己烷、1,3,5-三甲基环己烷等,十氢化萘等多环式脂肪族烃;正戊烷、正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷等非环式脂肪族烃;作为芳香族烃的甲苯、对二甲苯、邻二甲苯、间二甲苯等;作为芳香族杂环的吡啶等;作为醇系溶剂的甲醇、乙醇、异丙醇、正丁醇、叔丁醇、己醇、辛醇、环己醇等。
在r3为亚甲基的情况下,通过使用与水发生相分离的溶剂,可知具有能够抑制低聚芴(ii)的分解反应等副反应的倾向。进一步地,在使用可较好地溶解原料低聚芴(ii)的溶剂的情况下,出于具有反应进行良好的倾向,优选使用原料低聚芴(ii)的溶解度为0.5质量%以上的溶剂,更优选使用该溶解度为1.0质量%以上、特别优选使用该溶解度为1.5质量%以上的溶剂。具体地说,优选卤素系脂肪族烃、卤素系芳香族烃、芳香族烃、或者醚系溶剂,特别优选二氯甲烷、氯苯、氯仿、1,2-二氯苯、四氢呋喃、1,4-二氧六环、或者甲基环戊基醚。
这些溶剂可以单独使用一种,也可以两种以上混合使用。
关于溶剂的用量,上限没有特别限制,然而若考虑到每一反应器中的目的物的生成效率,则通常使用原料低聚芴(ii)的20倍体积量、优选为15倍体积量、进一步优选为10倍体积量的量。另一方面,若溶剂的用量过少,则试剂的溶解性变差、搅拌变难,同时反应的进行缓慢,因而作为下限,通常使用原料低聚芴(ii)的1倍体积量、优选为2倍体积量、进一步优选为4倍体积量的量。
r3为亚甲基以外的情况下,可知具有有机碱和低聚芴(ii)的溶解性对反应速度带来较大影响的倾向,为了确保该溶解性,优选使用具有一定值以上的介电常数的溶剂。作为可较好地溶解有机碱和低聚芴(ii)的溶剂,优选芳香族杂环、烷基腈系溶剂、酰胺系溶剂、亚砜系溶剂,特别优选吡啶、乙腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、环丁砜。
这些溶剂可以单独使用一种,也可以两种以上混合使用。
关于溶剂的用量,上限没有特别限制,然而若考虑到每一反应器中的目的物的生成效率,则通常使用原料低聚芴(ii)的20倍体积量、优选为15倍体积量、进一步优选为10倍体积量的量。另一方面,若溶剂的用量过少,则试剂的溶解性变差、搅拌变难,同时具有反应的进行缓慢的倾向,因而作为下限,通常使用原料低聚芴(ii)的1倍体积量、优选为2倍体积量、进一步优选为4倍体积量的量。
<12-5-2-5.反应形式>
在进行工序(iib)时,反应的形式可以采用分批型反应、流通型反应、或它们组合而成的形式,对其形式没有特别限制。
在分批式的情况下的将反应试剂投入到反应器中的投入方法中,在反应开始时将吸电子基团取代烯烃(vi)通过一次性添加投料的情况下,由于吸电子基团取代烯烃(vi)以高浓度存在,因而副聚合反应容易进行。从而优选在添加原料低聚芴(ii)、相转移催化剂、溶剂和碱之后每次少量逐次添加吸电子基团取代烯烃(vi)。
<12-5-2-6.反应条件>
工序(iib)中的温度若过低,则得不到充分的反应速度,反之若温度过高,则具有吸电子基团取代烯烃(vi)容易进行聚合反应的倾向,因而温度管理是极为重要的。因此,作为反应温度,具体地说,通常在下限为0℃、优选为10℃、更优选为15℃进行实施。另一方面,通常在上限为40℃、优选为30℃、更优选为20℃进行实施。
关于工序(iib)中的一般反应时间,通常下限为2小时、优选为4小时、进一步优选为6小时,上限没有特别限定,通常为30小时、优选为20小时、进一步优选为10小时。
<12-5-2-7.目的物的分离·精制>
在反应终止后,可采用下述方法使目的物低聚芴衍生物(vii)析出,从而进行目的物低聚芴衍生物(vii)的分离,所述方法为通过过滤将副生的金属卤化物以及残存的无机碱从反应液中除去之后进行溶剂浓缩的方法、或者添加目的物的不良溶剂的方法;等等。
此外,在反应终止后中,也可以向反应液中添加酸性水与可溶解目的物低聚芴衍生物(vii)的溶剂来进行提取。利用溶剂提取出的目的物可通过对溶剂进行浓缩的方法、或添加不良溶剂的方法等进行分离。
作为在提取时能够使用的溶剂,只要可溶解目的物低聚芴衍生物(vii)即可,没有特别限制,但适于使用甲苯、二甲苯等芳香族烃化合物、二氯甲烷、氯仿等卤素系溶剂等中的1种或2种以上。
此处得到的低聚芴衍生物(vii)之中,在ewg为具有碳原子数1~10的有机取代基的酯基的情况下,该低聚芴衍生物可以直接以聚酯或以聚酯碳酸酯原料单体的形式使用,或者也可以以聚碳酸酯原料单体的前体的形式进行使用等,但在进行精制后使用为佳。作为精制法,可以采用通常的精制法、例如再结晶或再沉淀法、提取精制、柱色谱等,并无限制。此外还可将低聚芴二酯(20b)溶解在适当的溶剂中,利用活性炭进行处理。此时能够使用的溶剂与提取时能够使用的溶剂相同。
在此处得到的低聚芴衍生物(vii)的ewg为羧基的情况下,可以直接以聚酯或以聚酯碳酸酯原料单体的形式使用,或者也可以以聚碳酸酯原料单体的前体的形式进行使用。此外可通过酯化反应转换成a3和a4为酯基的低聚芴二酯(20b)。
在此处得到的低聚芴衍生物(vii)的ewg为硝基或氰基的情况下,可通过利用氢气氛下的钯碳等方法进行的氢化或利用氢化锂铝等还原剂进行的氢化物还原来制造a3和a4为具有氨基的基团的低聚芴单体(20)。此外,在ewg为氰基的情况下,可通过专利文献7和8等方法转换为低聚芴二酯(20b)。
在此处得到的低聚芴衍生物(vii)的ewg为醛基的情况下,可通过利用氢气氛下的钯碳等方法进行的氢化或利用硼氢化钠等还原剂进行的氢化物还原来制造a3和a4为具有羟基的基团的低聚芴二醇(20c)。
在此处得到的低聚芴衍生物(vii)的ewg为具有碳原子数1~10的有机取代基的酮基的情况下,可通过利用氢气氛下的钯碳等方法进行的氢化或利用硼氢化钠等还原剂进行的氢化物还原来制造a3和a4为具有羟基的基团的低聚芴单体(20)。
<12-5-3.工序(iic):通式(20)中,a3和a4为具有羟基的基团的情况的制造方法(通过低聚芴二酯(20b)的还原反应进行的低聚芴二醇(20c)的制造法)>
下述通式(20c)所表示的低聚芴二醇可在还原剂存在下按下述工序(iic)由低聚芴二酯(20b)进行制造。
[化86]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,riv表示碳原子数1~10的有机取代基。)
关于通过酯的还原进行的二醇的合成,其合成法是广为人知的,在美国专利申请公开第2012/0170118号说明书中,通过使用还原剂氢化铝锂的还原反应进行制造。作为使用其它金属氢化物的方法,可以举出二异丁基氢化铝、双(2-甲氧基乙氧基)氢化铝钠等。此外,利用在催化剂中使用钌、铑、钯、铂的接触氢化反应进行的酯还原也是广为人知的方法。
<12-5-4.工序(iid):通式(20)中,a3和a4为具有羟基酯基的基团的情况的制造方法(利用低聚芴二酯(20b)的酯交换反应进行的低聚芴二羟基酯(20d)的制造法)>
下述通式(20d)所表示的低聚芴二羟基酯可在碱存在下按照下述工序(iid)由低聚芴二酯(20b)和二醇(viii)进行制造。
[化87]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,riv表示碳原子数1~10的有机取代基,rv表示碳原子数1~10的有机取代基。)
<12-5-4-1.二醇>
工序(iid)中使用的二醇(viii)表示碳原子数1~10的二醇。具体地说,可以举出:乙二醇、新戊二醇、1,4-丁二醇、1,6-己二醇等链状(可以为直链、也可以为支链)的亚烷基二醇;环己烷二甲醇等环状的亚烷基二醇;二甘醇、三甘醇、四甘醇等低聚乙二醇;异山梨醇等仲二醇;间苯二酚等包含芳香族的二醇等。这些二醇可以在不阻碍该反应的范围内被任意的取代基所取代。
其中,从反应速度与成本的方面出发,优选烷撑二醇或低聚乙二醇,特别优选乙二醇。
在工序(iid)中,也可以使用2种以上不同的二醇(viii),但从精制简便性的方面考虑,通常使用一种二醇(viii)。
关于二醇(viii)的用量,由于由原料低聚芴二酯(20b)的酯基的有机取代基生成的醇与所加入的二醇(viii)具有形成竞争反应的倾向,因而二醇(viii)的用量越多,反应的进行越迅速。此外,在二醇(viii)的用量多时,下述通式(ix)所示的低聚芴能够抑制经二醇交联而成的副产物的生成。由于该自身酯交换生成物(ix)本身作为含有聚酯碳酸酯的聚碳酸酯原料、或聚酯原料发挥作用,因而可认为,即使其在低聚芴二羟基酯(20d)中含有,作为聚碳酸酯原料、聚酯、聚酯碳酸酯原料也没有大的问题。但是,从聚碳酸酯、聚酯、聚酯碳酸酯的品质的方面出发,自身酯交换生成物(ix)的含有比例相对于生成物低聚芴二羟基酯(20d)通常为0.1倍摩尔以下、优选为0.05倍摩尔以下、更优选为0.03倍摩尔以下。
[化88]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,rv表示碳原子数1~10的有机取代基。)
因此,二醇(viii)的用量相对于低聚芴二酯(20b)通常为3倍摩尔以上、优选为10倍摩尔以上、进一步优选为50倍摩尔以上。
二醇(viii)可以在投料时一次性添加,也可以随着反应的进行分次添加。通式(ix)所示的自身酯交换生成物可由于二醇(viii)的添加而转换成低聚芴二羟基酯(20d)。
<12-5-4-2.碱>
作为工序(iid)中使用的碱,使用氢氧化锂、氢氧化钠、氢氧化钾等碱金属氢氧化物;氢氧化钙、氢氧化钡等碱土金属的氢氧化物;碳酸钠、碳酸氢钠、碳酸钾等碱金属的碳酸盐;碳酸镁、碳酸钙等碱土金属的碳酸盐;磷酸钠、磷酸氢钠、磷酸钾等磷酸的碱金属盐;正丁基锂、叔丁基锂等有机锂盐;甲醇钠、乙醇钠、叔丁醇钾等碱金属的醇盐;氢化钠、氢化钾等氢化碱金属盐;四甲基氢氧化铵、四丁基氢氧化铵等氢氧化季铵。
它们可以单独使用一种,也可以合用两种以上。
其中,从成本、反应性的方面考虑,优选碱金属的醇盐,进一步特别优选甲醇钠或乙醇钠。
关于碱的用量,相对于原料低聚芴二酯(20b),并无特别上限,但若用量过多,则具有搅拌或反应后的精制负荷增大的倾向,因而通常为芴的10倍摩尔以下、优选为5倍摩尔以下、进一步优选为1倍摩尔以下。
另一方面,碱的用量过少时,具有反应的进行缓慢的倾向,因而作为下限,通常相对于原料芴为0.01倍摩尔以上、优选为0.05倍摩尔以上、进一步优选为0.1倍摩尔以上。
<12-5-4-3.溶剂>
工序(iid)在无溶剂下也可进行,但在原料低聚芴二酯(20b)在反应试剂二醇(viii)中的溶解性低、反应性低的情况下,也可以使用溶剂来进行。
具体地说,可使用的溶剂可以举出:作为烷基腈系溶剂的乙腈、丙腈等;作为醚系溶剂的二乙醚、四氢呋喃、1,4-二氧六环、甲基环戊基醚、叔丁基甲醚、二乙二醇二甲醚、三乙二醇二甲醚等;作为卤素系溶剂的1,2-二氯乙烷、二氯甲烷、氯仿、1,1,2,2-四氯乙烷等;作为卤素系芳香族烃的氯苯、1,2-二氯苯等;作为酰胺系溶剂的n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等;作为亚砜系溶剂的二甲基亚砜、环丁砜等;作为环式脂肪族烃的环戊烷、环己烷、环庚烷、环辛烷等单环式脂肪族烃,作为其衍生物的甲基环戊烷、乙基环戊烷、甲基环己烷、乙基环己烷、1,2-二甲基环己烷、1,3-二甲基环己烷、1,4-二甲基环己烷、异丙基环己烷、正丙基环己烷、叔丁基环己烷、正丁基环己烷、异丁基环己烷、1,2,4-三甲基环己烷、1,3,5-三甲基环己烷等,十氢化萘等多环式脂肪族烃;正戊烷、正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷等非环式脂肪族烃;作为芳香族烃的甲苯、对二甲苯、邻二甲苯、间二甲苯等。
其中,在使用原料低聚芴二酯(20b)与二醇(viii)这两者的溶解性高的溶剂的情况下,出于具有反应进行良好的倾向的原因,优选醚系溶剂;出于能够进行高温下的反应的原因,特别优选二乙二醇二甲醚、或者三乙二醇二甲醚。
这些溶剂可以单独使用一种,也可以两种以上混合使用。
关于溶剂的用量,其上限没有特别限制,然而若考虑到每一反应器中的目的物的生成效率,则通常使用原料低聚芴二酯(20b)的20倍体积量、优选为15倍体积量、进一步优选为10倍体积量的量。另一方面,若溶剂的用量过少,则试剂的溶解性变差、搅拌变难,同时具有反应的进行缓慢的倾向,因而作为下限,通常使用原料低聚芴二酯(20b)的1倍体积量、优选为2倍体积量、进一步优选为4倍体积量的量。
<12-5-4-4.反应形式>
在进行工序(iid)时,反应的形式可以采用分批型反应、流通型反应、或它们组合而成的形式,对其形式没有特别限制。
<12-5-4-5.反应条件>
在溶剂或反应试剂二醇(viii)中含有水分的情况下,进行酯的水解,作为副产物,根据所含有的水含量,具有会生成以下所示的二羧酸(x)或羟基羧酸(xi)的倾向。
[化89]
(式中,r3~r9和n与式(1)中的r3~r9和n含义相同。ri、rii和riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,rv表示碳原子数1~10的有机取代基。)
因此,优选溶剂或反应试剂二醇(viii)使用无水物,或者在反应前利用甲苯、二甲苯等不参与反应且与水共沸的溶剂进行共沸脱水,之后进行反应。
需要说明的是,二羧酸(x)或羟基羧酸(xi)也可作为含有聚酯碳酸酯的聚碳酸酯原料、或聚酯原料进行使用。
工序(iid)的温度若过低,则具有得不到充分的反应速度的倾向,因而作为反应温度,具体地说,通常在下限为20℃、优选为50℃、更优选为80℃进行实施。另一方面,通常在上限为150℃、优选为120℃、更优选为100℃进行实施。
关于工序(iid)中的一般反应时间,通常下限为2小时、优选为4小时、进一步优选为6小时,上限没有特别限定,通常为30小时、优选为20小时、进一步优选为10小时。
<12-5-4-6.目的物的分离·精制>
在反应终止后,可采用下述方法使目的物低聚芴二羟基酯(20d)析出,从而进行目的物低聚芴二羟基酯(20d)的分离,所述方法为通过过滤将副生的金属卤化物以及残存的无机碱等不溶物从反应液中除去之后进行溶剂浓缩的方法、或者添加目的物的不良溶剂的方法;等等。
此外,在反应终止后,也可以向反应液中添加酸性水与可溶解目的物低聚芴二羟基酯(20d)的溶剂来进行提取。利用溶剂提取出的目的物可通过对溶剂进行浓缩的方法、或添加不良溶剂的方法等进行分离。
此外,通过利用碳酸钠、碳酸钾等水溶液对利用溶剂提取出目的物进行清洗,可除去副产物羧酸。
作为在提取时能够使用的溶剂,只要可溶解目的物低聚芴二羟基酯(20d)即可,没有特别限制,但适于使用乙酸乙酯等酯系溶剂、甲苯、二甲苯等芳香族烃化合物、二氯甲烷、氯仿等卤素系溶剂等中的1种或2种以上。
低聚芴二羟基酯(20d)可以作为含有聚酯碳酸酯的聚碳酸酯原料、或聚酯原料直接用于聚合中,也可以在进行精制后用于接下来的工序中。作为精制法,可以采用通常的精制法、例如再结晶或再沉淀法、提取精制等,并无限制。此外还可将低聚芴二羟基酯(20d)溶解在适当的溶剂中,利用活性炭进行处理。此时能够使用的溶剂与提取时能够使用的溶剂相同。
<12-5-5.通式(20)中,a3和a4为具有羟基的基团的情况的制造方法(利用低聚芴化合物(ii)的烷基化、水解反应进行的低聚芴二醇(10a)的制造法)>
低聚芴二醇(10a)可通过下述方法进行制造,所述方法为:进行通过低聚芴化合物(ii)与烷基化剂(viiib)和(viiic)的烷基化反应来合成具有离去基团的低聚芴(ix)的工序(工序(iie))以及接下来的水解反应(工序(iif))的方法;或者进行通过低聚芴化合物(ii)与烷基化剂(xa)和(xb)的烷基化反应来合成具有保护基团的低聚芴(xi)的工序(工序(iig))以及接下来的水解反应(工序(iih))的方法;等等。
[化90]
(式中,r1~r9和n与式(1)中的r1~r9和n含义相同。x表示离去基团。作为离去基团的示例,可以举出卤原子(不包括氟原子。)、甲磺酰基、或甲苯磺酰基等。t表示保护基团。作为保护基团的示例,可以举出甲氧基甲基、2-甲氧基乙氧基甲基、四氢吡喃基、叔丁氧基羰基、苄氧基羰基、三甲基甲硅烷基、或者叔丁基二甲基甲硅烷基等。)
芴类的烷基化反应是广为人知的,例如,有报告提出了9,9-双(溴己基)芴、9,9-双(碘己基)芴等9,9-双(卤烷基)芴(j.org.chem.,2010,75,2714.)。根据这些技术思想,能够将低聚芴(ii)作为原料来合成具有离去基团的低聚芴。此外,关于卤素的水解,一直也有多例报告(bull.koreanchem.soc.,2008,29.2491.),因而可由该路线合成低聚芴二醇(10a)。
作为工序(iie)中使用的烷基化剂,可以举出:二碘甲烷、1,2-二碘乙烷、1,3-二碘丙烷、1,4-二碘丁烷、1,5-二碘戊烷、1,6-二碘己烷、二溴甲烷、1,2-二溴乙烷、1,3-二溴丙烷、1,4-二溴丁烷、1,5-二溴戊烷、1,6-二溴己烷、二氯甲烷、1,2-二氯乙烷、1,3-二氯丙烷、1,4-二氯丁烷、1,5-二氯戊烷,1,6-二氯己烷、1-溴-3-氯丙烷等直链状的烷基二卤化物(不包括氟原子);2,2-二甲基-1,3-二氯丙烷等包含支链的烷基二卤化物(不包括氟原子);1,4-双(溴甲基)苯、1,3-双(溴甲基)苯等芳烷基二卤化物(不包括氟原子);乙二醇二甲磺酸酯、乙二醇二甲苯磺酸酯、丙二醇二甲磺酸酯、四亚甲基二醇二甲磺酸酯等二醇的二磺酸酯等。
作为工序(iig)中使用的烷基化剂,可以举出3-溴丙醇、2-溴丙醇、3-氯-2,2-二甲基-1-丙醇等卤代烷基醇的保护体等。
<12-5-6.通式(10d)的低聚芴二芳基酯的制造方法(通过在低聚芴二酯化合物(10b)合成后进行酯交换反应来进行的低聚芴二芳基酯化合物(10d)的制造法)>
低聚芴二芳基酯化合物(10d)可通过经由合成低聚芴二酯化合物(10b)的工序(工序(iij)、工序(iik)、或者工序(iil))以及接下来的与二芳基碳酸酯类(11a)的酯交换反应(工序(iim))的方法进行制造。
[化91]
(式中,r1~r9和n与式(1)中的r1~r9和n含义相同。x表示离去基团。)
作为离去基团的示例,可以举出卤原子(不包括氟原子。)、甲磺酰基、或甲苯磺酰基等。riii表示氢原子、具有或不具有取代基的碳原子数1~10的烷基、具有或不具有取代基的碳原子数4~10的芳基、或者具有或不具有取代基的碳原子数6~10的芳烷基,r17表示碳原子数1~10的有机取代基,ar1表示具有或不具有取代基的碳原子数4~10的芳基。)
<12-5-6-1-1.工序(iij):通式(10b)中,r1和r2为直接键合的情况的制造方法>
r1和r2为直接键合的低聚芴二酯(10b)按照<12-1.制造法a>所记载的方法由9位单取代芴类(iiic)和(iiid)进行制造。
<12-5-6-1-2.工序(iik):通式(10b)中,r1和r2为具有或不具有取代基的亚乙基的情况的制造方法>
r1和r2为具有或不具有取代基的亚乙基的低聚芴二酯(10b)可在碱存在下,按照<12-5-2:工序(iib)>记载的方法由低聚芴化合物(ii)和不饱和羧酸酯(vi-1)进行制造。
<12-5-6-1-3.工序(iil):通式(10b)中,r1和r2为直接键合以外的情况的制造方法>
r1和r2为直接键合以外的低聚芴二酯(10b)可通过在碱存在下,按照与<12-5-5>中作为工序(iie)或(iig)所记载的方法相同的方法对低聚芴(ii)与烷基化剂(xc)和(xd)进行烷基化反应来进行制造。
作为工序(iil)中使用的烷基化剂,可以举出:氯乙酸甲酯、氯乙酸乙酯、氯乙酸丙酯、氯乙酸正丁酯、氯乙酸叔丁酯、溴乙酸甲酯、溴乙酸乙酯、溴乙酸叔丁酯、碘乙酸甲酯、碘乙酸乙酯、碘乙酸叔丁酯、氯丙酸甲酯、氯丙酸乙酯、氯丙酸叔丁酯、溴丙酸甲酯、溴丙酸乙酯、溴丙酸叔丁酯、碘丙酸甲酯、碘丙酸乙酯、碘丙酸叔丁酯等卤代链烷烃酸烷基酯;4-氯甲基苯甲酸甲酯、4-溴甲基苯甲酸甲酯、4-氯甲基苯甲酸乙酯、4-溴甲基苯甲酸乙酯、3-氯甲基苯甲酸甲酯、3-溴甲基苯甲酸甲酯等卤代烷基苯甲酸烷基酯等。
<12-5-6-2.通式(10d)的低聚芴二芳基酯的制造方法(利用酯交换反应进行的低聚芴二芳基酯化合物(10d)的制造法)>
ar1为具有或不具有取代基的碳原子数4~10的芳基的低聚芴二芳基酯化合物(10d)可在酯交换催化剂存在下按照工序(iim)所表示的反应由低聚芴二酯化合物(10b)和二芳基碳酸酯类(11a)进行制造。
<12-5-6-2-1.二芳基碳酸酯类>
作为反应试剂的二芳基碳酸酯类可以举出碳酸二苯酯、碳酸二苄酯、双(氯苯基)碳酸酯、碳酸间甲苯酯、碳酸二萘酯、碳酸双(联苯)酯等。其中优选低成本且能够工业获得的碳酸二苯酯。
这些二芳基碳酸酯可以单独使用一种,也可将2种以上混合使用。
关于二芳基碳酸酯类的用量,相对于原料低聚芴二酯(10b)并无特别上限,但若用量过多,则具有反应后的精制负荷增大的倾向,因而通常为低聚芴二酯的20倍摩尔以下、优选为10倍摩尔以下、进一步优选为5倍摩尔以下。
另一方面,碱的用量若过少,则原料低聚芴二酯(10b)或作为中间体的如下所示的低聚芴单芳基酯(10e)可能会有残留,因而作为下限,通常相对于原料低聚芴二酯(10b)为1倍摩尔以上、优选为1.5倍摩尔以上、进一步优选为2倍摩尔以上。
[化92]
(式中,r1~r9和n与式(1)中的r1~r9和n含义相同。r17表示碳原子数1~10的有机取代基,ar1表示具有或不具有取代基的碳原子数4~10的芳基。)
<12-5-6-2-2.酯交换反应催化剂>
作为酯交换反应催化剂,可以举出:四丁氧基钛、四异丁氧基钛、四甲氧基钛、四异丙氧基钛、四乙氧基钛、四(2-乙基己氧基)钛、四(十八烷基)原钛酸酯、四苯氧基钛、乙酰丙酮钛(iv)、二异丙氧基双(乙酰丙酮)钛(iv)等钛化合物;碳酸锂、二丁基氨基锂、乙酰丙酮锂、苯氧基钠、苯氧基钾等碱金属化合物;乙酰丙酮镉、碳酸镉等镉化合物;乙酰丙酮锆、二茂锆等锆化合物;硫化铅、氢氧化铅、铅酸盐、锌酸盐、碳酸铅、乙酸铅、四丁基铅、四苯基铅、三苯基铅、二甲氧基铅、二苯氧基铅等铅化合物;乙酸铜、双乙酰丙酮铜、油酸铜、丁基铜、二甲氧基铜、氯化铜等铜化合物;氢氧化铁、碳酸铁、三乙酰氧基铁、三甲氧基铁、三苯氧基铁等铁化合物;双乙酰丙酮锌、二乙酰氧基锌、二甲氧基锌、二乙氧基锌、二苯氧基锌等锌化合物;二正丁基氧化锡、二苯基氧化锡、二正辛基氧化锡、二甲氧基二正丁基锡、二正丁基锡二丙烯酸酯、二正丁基锡二甲基丙烯酸酯、二正丁基锡二月桂酸酯、四甲氧基锡、四苯氧基锡、四丁基-1,3-二乙酰氧基二锡氧烷等有机锡化合物;乙酸铝、甲氧基铝、乙氧基铝、苯氧基铝等铝化合物;二氯化钒、三氯化钒、四氯化钒、硫酸钒等钒化合物;苯氧基四苯基磷鎓等鏻盐等。它们可以单独使用一种,也可以合用两种以上。
它们之中,出于工业成本低、具有反应操作上的优越性的原因,优选使用鏻盐、锂化合物、锆化合物、有机锡化合物或者钛化合物等,其中特别优选有机锡化合物或钛化合物。
关于酯交换反应催化剂的用量,相对于原料低聚芴二酯(20b)并无特别上限,但若用量过多,则反应后的精制负荷会增大,因而通常为芴的20摩尔%以下、优选为10摩尔%以下、进一步优选为5摩尔%以下。
另一方面,酯交换反应催化剂的用量过少时,反应时间可能会过长,因而作为下限,通常相对于原料低聚芴二酯为0.1摩尔%以上、优选为0.5摩尔%以上、进一步优选为1摩尔%以上。
<12-5-6-2-3.溶剂>
在工序(iim)中可以使用反应溶剂,但优选不使用反应溶剂,仅利用原料低聚芴二酯(10b)、二芳基碳酸酯类以及酯交换反应催化剂进行反应。但是,在原料低聚芴二酯(10b)、二芳基碳酸酯类常温为固体、难以搅拌的情况下,也可以使用反应溶剂。在使用反应溶剂的情况下,只要为能够适当地溶解和/或分散上述原料低聚芴二酯(10b)、二芳基碳酸酯类以及酯交换反应催化剂的溶剂,其种类为任意的。
具体地说,能够使用的溶剂可以举出:作为烷基腈系溶剂的乙腈、丙腈等;作为酮系溶剂的丙酮、甲基乙基酮、甲基异丁基酮等;作为醚系溶剂的二乙醚、四氢呋喃、1,4-二氧六环、甲基环戊基醚、叔丁基甲醚等;作为卤素系溶剂的1,2-二氯乙烷、二氯甲烷、氯仿、1,1,2,2-四氯乙烷等;作为卤素系芳香族烃的氯苯、1,2-二氯苯等;作为酰胺系溶剂的n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等;作为亚砜系溶剂的二甲基亚砜、环丁砜等;作为环式脂肪族烃的环戊烷、环己烷、环庚烷、环辛烷等单环式脂肪族烃,作为其衍生物的甲基环戊烷、乙基环戊烷、甲基环己烷、乙基环己烷、1,2-二甲基环己烷、1,3-二甲基环己烷、1,4-二甲基环己烷、异丙基环己烷、正丙基环己烷、叔丁基环己烷、正丁基环己烷、异丁基环己烷、1,2,4-三甲基环己烷、1,3,5-三甲基环己烷等,十氢化萘等多环式脂肪族烃;正戊烷、正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷等非环式脂肪族烃;作为芳香族烃的甲苯、对二甲苯、邻二甲苯、间二甲苯、1,3,5-三甲基苯、1,2,4-三甲基苯、1,2,3,4-四氢萘等;作为芳香族杂环的吡啶等。
由于该反应优选在通常为100℃以上的高温下进行,因而在上述溶剂中,优选沸点为100℃以上的溶剂的氯苯、1,2-二氯苯、三氯苯、甲苯、对二甲苯、邻二甲苯、间二甲苯、1,3,5-三甲基苯、1,2,4-三甲基苯、1,2,3,4-四氢萘、十氢化萘、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜或者环丁砜,出于能够适当地溶解原料低聚芴二酯(10b)、沸点为130℃以上、能够进行更高温度下的反应的原因,特别优选1,2-二氯苯、二甲苯、1,3,5-三甲基苯、1,2,4-三甲基苯、或1,2,3,4-四氢萘、十氢化萘。
这些溶剂可以单独使用一种,也可以两种以上混合使用。
关于溶剂的用量,上限没有特别限制,然而若考虑到每一反应器中的目的物的生成效率,则通常使用原料低聚芴二酯(10b)的15倍体积量、优选为10倍体积量、进一步优选为5倍体积量的量。另一方面,若溶剂的用量过少,则试剂的溶解性变差、搅拌变难,同时反应的进行缓慢,因而作为下限,通常使用以原料低聚芴二酯(10b)的浓度计的1倍体积量、优选为2倍体积量、进一步优选为4倍体积量的量。
<12-5-6-2-4.反应形式>
在进行工序(iim)时,反应的形式可以采用分批型反应、流通型反应、或它们组合而成的形式,对其形式没有特别限制。
<12-5-6-2-5.反应条件>
在工序(iim)中,若温度过低,则具有得不到充分的反应速度的倾向,因而通常在下限为50℃、优选为70℃、更优选为100℃进行实施。另一方面,在上限通常为250℃、优选为200℃、更优选为180℃进行实施。
关于工序(iim)中的一般反应时间,通常下限为1小时、优选为2小时、进一步优选为3小时,上限没有特别限定,通常为30小时、优选为20小时、进一步优选为10小时。
在工序(iim)中,为了使平衡向生成物侧偏移,可在减压下蒸馏除去副产物同时进行反应。进行减压的情况下,在压力通常为20kpa以下、优选为10kpa以下、更优选为5kpa以下进行实施。另一方面,若真空度过高,则可能升华为作为试剂使用的二芳基碳酸酯类,因而在通常为0.1kpa以上、优选为0.5kpa以上、更优选为1.0kpa以上进行实施。
<10-5-6-2-6.目的物的分离·精制>
在反应终止后,可向反应液中添加不良溶剂,使目的物低聚芴二芳基酯(10d)析出,由此来进行分离。
此外,在反应终止后,也可将可溶性的溶剂与水添加到反应液中,对目的物低聚芴二芳基酯(10d)进行提取。利用溶剂提取出的目的物可通过对溶剂进行浓缩的方法、或者添加不良溶剂的方法等进行分离。
所得到的低聚芴二芳基酯(10d)也可作为含有聚酯碳酸酯的聚碳酸酯原料、或聚酯原料直接用于聚合中。作为精制法,可以采用通常的精制法、例如再结晶或再沉淀法、提取精制、柱色谱等,并无限制。
实施例
下面通过实施例和比较例更详细地说明本发明,但只要不超出其要点,本发明并不受以下实施例的限定。本发明低聚芴单体的品质评价以及树脂组合物和透明膜的特性评价按下述方法进行。需要说明的是,特性评价方法并不限于以下的方法,本领域技术人员可适当选择。
另外,下述制造例及实施例中使用的化合物的简写符号等如下。
isb;异山梨醇(roquettefreres社制造,商品名:polysorb)
dpc;碳酸二苯酯(三菱化学株式会社制造)
chdm;1,4-环己烷二甲醇(顺式、反式混合物,sk化学社制造)
bhepf:9,9-双[4-(2-羟基乙氧基)苯基]-芴(osakagaschemicals株式会社制造)
bcf:9,9-双[4-羟基-3-甲基苯基]-芴(osakagaschemicals株式会社制造)
def:芴-9,9-二乙醇(采用日本特开2010-261008号公报所记载的方法合成出。)
chda:1,4-环己烷二羧酸(顺式、反式混合物,eastman化学社制造)
spg:螺环二醇(三菱瓦斯化学株式会社制造)
bpa:2,2-双[4-羟基苯基]丙烷(三菱化学株式会社制造)
tcddm:三环癸烷二甲醇(oxea社制造)
peg#1000:聚乙二醇数均分子量:1000(三洋化成株式会社制造)
thf:四氢呋喃(不含稳定剂,wako社制造)
(1)低聚芴单体中的铝、钠含量
如下测定低聚芴单体中的铝、钠含量。对分析试样进行湿式分解处理后,利用icp-aes(horibajobinyvon社制造ultima2c)进行分析。此外,关于钠含量,根据分析试样的不同,还合用了利用原子吸光法(varian制spectraa-220p)进行的分析。
(2)低聚芴单体中的氯含量
如下测定低聚芴单体中的氯含量。将分析试样使用燃烧装置(三菱化学社制造aqf-2100m)进行燃烧、吸收,利用离子色谱(日本dionex株式会社制造dx-500)进行分析。
(3)低聚芴单体的热分解温度(tg/dta)
低聚芴单体的玻璃化转变温度使用示差热重同时分析装置(siinanotechnology社制造tg-dta6300)进行测定。将约4mg分析试样放入该社制造的铝盘中并进行密封,在200ml/分钟的氮气流下,以升温速度10℃/分钟由室温(20~30℃)升温至600℃。根据所得到的tg数据(热重数据),将试样重量减少5wt%的温度作为5wt%失重温度。对于含有溶剂的试样的试样重量,在由1h-nmr估算的溶剂重量减少后,将不发生重量变化的重量作为试样的初期重量,将该初期重量减少5wt%的温度作为5wt%失重温度。另外,根据所得到的tg数据(热重数据),在确认不到试样重量的减少、且观测到陡峭的吸热峰的该峰顶作为试样的熔点。
(4)低聚芴单体的紫外可见区域(uv-vis)的吸收极大波长
低聚芴单体的紫外可见区域(uv-vis:280~800nm)的吸收极大波长使用紫外可见吸收分光光度计(岛津制作所社制造uv-1650pc)进行测定。作为溶剂使用四氢呋喃,使用1cm见方的石英池,在温度23±5℃进行测定。关于浓度,按照以芴环计的浓度为10μm进行精确地制备(例如,若为化合物1,则制备为5.0μm;若为化合物6b,则制备为0.33μm)。
在280nm~800nm的范围内测定吸收光谱,将吸收的极大值作为吸收极大波长(λmax)。
(5)树脂组合物的比浓粘度
树脂组合物的比浓粘度如下测定。使用森友理化工业社制造的乌氏粘度管,作为溶剂使用二氯甲烷,在温度20.0℃±0.1℃进行测定。将浓度精确地制备为0.6g/dl。
按照式:ηrel=t/t0由溶剂的通过时间t0、溶液的通过时间t求出相对粘度ηrel,按照式:ηsp=(η-η0)/η0=ηrel-1由相对粘度ηrel求出比粘度ηsp。将比粘度ηsp除以浓度c(g/dl),按照式:ηred=ηsp/c求出比浓粘度(换算粘度)ηred。该数值越高,分子量越大。
(6)树脂组合物的玻璃化转变温度(tg)
树脂组合物的玻璃化转变温度使用差示扫描量热计(siinanotechnology社制造dsc6220)进行测定。将约10mg树脂组合物试样放入该社制造的铝盘中并进行密封,在50ml/分钟的氮气流下,以升温速度20℃/分钟由30℃升温至250℃。将温度保持3分钟后,以20℃/分钟的速度冷却至30℃。在30℃下保持3分钟,再次以20℃/分钟的速度升温至200℃。根据第2次升温中得到的dsc数据求出外推玻璃化转变起始温度,该外推玻璃化转变起始温度是将低温侧的基线向高温侧延长得到的直线与在玻璃化转变的阶梯状变化部分的曲线斜率达到最大的点所画出的切线相交的交点的温度,将该外推玻璃化转变起始温度作为玻璃化转变温度。
(7)树脂组合物的熔融粘度
在测定前,将树脂组合物试样在80℃下进行5小时以上的真空干燥。使用东洋精机株式会社制造的capillograph,使用直径1mm×长度10mm的模具,测定温度240℃、剪切速度91.2sec-1时的熔融粘度。
(8)树脂组合物中的na、k、cs、fe的含有比例
在perkinelmer社制造的微波分解容器中精确称量约0.5g树脂组合物试样,加入97%硫酸(多摩化学制造的超高纯度硫酸)2ml,使其为密闭状态,在230℃下进行10分钟微波加热。冷却至室温(30℃以下)后,加入68%硝酸(多摩化学制造的超高纯度硝酸)1.5ml,使其为密闭状态,在150℃下进行10分钟微波加热后,再次冷却至室温(30℃以下),加入68%硝酸2.5ml,再次使其为密闭状态,在230℃下进行10分钟微波加热,使内容物完全分解。微波加热器使用perkinelmer社制造的multiwave3000,通过在300w至1000w之间进行输出功率的调整来调整温度。冷却至室温(30℃以下)后,将上述得到的液体利用纯水稀释,利用thermoquest社制造的icp-ms进行定量。
(9)树脂组合物中的残存单羟基化合物
精确称量约1g树脂组合物试样,溶解在二氯甲烷5ml中,之后添加丙酮,使总量为25ml。将溶液利用0.2μm盘式过滤器进行过滤,利用液相色谱法进行苯酚的定量后,计算出含有比例。
(10)树脂组合物的光弹性系数
对于在80℃下5小时真空干燥后的树脂组合物试样约4g,使用宽8cm、长度8cm、厚度0.5mm的隔垫(spacer),通过热压在热压温度200℃~250℃、预热1分钟~3分钟、压力20mpa的条件下进行1分钟加压后,连同隔垫一起取出,利用水管冷却式压力机在压力20mpa下进行3分钟加压冷却,制作片状物。从该膜切出宽5mm、长20mm的样品。
使用将包含he-ne激光器、偏振元件、补偿板、检偏器以及光检测器的双折射测定装置与振动型粘弹性测定装置(rheology社制造的“dve-3”)组合而成的装置进行测定。(详细内容参照日本rheology学会志vol.19,p93-97(1991)。)
将切出的样品固定于粘弹性测定装置,在25℃的室温下以频率96hz测定储能模量e’。同时,使射出的激光依次通过偏振元件、试样、补偿板、检偏器,并利用光检测器(光电二极管)进行拾取,使其通过锁定放大器,针对角频率ω或2ω的波形求出相对于其振幅和应变的相位差,并求出应变光学系数o’。此时,调整偏振元件与检偏器的方向,使它们正交,并使它们分别与试样的伸长方向成π/4的角度。光弹性系数c使用储能模量e’和应变光学系数o’通过下式求出。
c=o’/e’
(11)树脂组合物的折射率各向异性和相位差的波长分散性
通过上述利用热压进行的方法进行厚度100μm~200μm的膜的成型,从该膜切出宽6cm、长6cm的试样。使用分批式双向拉伸装置(island工业社制造双向拉伸装置bix-277-al),在拉伸温度为树脂组合物试样的玻璃化转变温度+15℃、拉伸速度为1000%/分钟、拉伸倍率为2倍的条件下,对该试样进行自由端单向拉伸,得到拉伸膜。由所得到的拉伸膜切出宽4cm、长4cm的样品,通过相位差测定装置(王子计测机器社制造kobra-wpr)对于该样品在测定波长450nm的相位差(re450)和550nm的相位差(re550)进行测定。将两测定值之比(re450/re550)作为相位差的波长分散性的指标。此外,在该相位差测定中,在拉伸方向的相位差的测定值表现出正值的情况下,该树脂的折射率各向异性为正。
(12)膜的韧性(弯曲测试)
通过上述利用热压进行的方法进行厚度100μm~200μm的膜的成型,由该膜制作长40mm、宽10mm的长方形试验片。将老虎钳左右接合面的间隔打开40mm,将试验片的两端固定在接合面内。接着以2毫米/秒以下的速度使左右接合面的间隔缩小,在不使膜挤出到老虎钳的接合面之外的情况下,在该接合面内将弯曲成“く”字的膜整体压缩。将至接合面间完全密合为止试验片在弯曲部裂成2片(或3片以上的破片)的情况作为“有裂纹”,将即使接合面间完全密合试验片也未裂开而弯折的情况作为“无裂纹”。对于同一种类的膜实施5次重复试验,将其中4次以上为“有裂纹”的情况作为“×:发生脆性破坏”,将3次以下为“有裂纹”的情况作为“○:未发生脆性破坏”。
(13)折射率和阿贝值的测定
由通过上述利用热压进行的方法取得的膜中切出长40mm、宽8mm的长方形试验片,作为测定试样。利用多波长abbe折射率计(株式会社atago制dr-m4/1550),使用波长656nm(c线)、589nm(d线)、486nm(f线)的干涉滤光器,测定各波长的折射率、nc、nd、nf。测定中使用单溴萘作为界面液,在20℃下进行测定。
阿贝值νd通过下式计算。
νd=(1-nd)/(nc-nf)
阿贝值越大,表示折射率的波长依赖性越小。
(14)偏光atr分析
将由本发明的树脂组合物制作的拉伸膜在热台上加热至玻璃化转变温度(tg)以上,将拉伸膜在其拉伸方向进一步进行2倍左右的拉伸,进行空气冷却,对于冷却后的试样实施偏光atr(一次反射法、ge晶体)测定。
(测定方法)
作为测定装置,使用将atraccessory“foundationthunderdome”(下文中简称为thunderdome)设置于nicolet社(现在的thermofisherscientific社)制造的magna550所得到的装置。另外,将krs-5制造的偏振片设置于thunderdome的入射侧入口处,使红外光具有线性偏光。偏振方向固定在与膜面平行的方向。通过使试样旋转,将偏振方向与膜拉伸方向平行的状态的光谱作为平行偏振光光谱,将垂直状态的光谱作为垂直偏振光光谱,来进行评价。需要说明的是,关于测定条件,以分解能4cm-1、扫描次数64次进行实施。
关于羰基的取向,对于在约1750cm-1处观测的c=o伸缩振动与在约1250cm-1处观测的c-o伸缩振动进行观测;关于芴环的取向,对于在约740cm-1处观测的苯环ch面外变角振动进行观测。
(评价方法)
作为评价方法,首先,在将显示吸光度的平行偏振光光谱与垂直偏振光光谱之差作为差示光谱时,确认到处于下述状态:差示光谱的1750cm-1在负方向明确显现出峰,1250cm-1的吸收在正方向明确显现出峰;在确认到羰基相对于拉伸方向垂直取向、即主链在拉伸方向取向的情况下,对于羰基,设为“○”。其后,在与芴环的取向相关的差示光谱的740cm-1的吸收在正方向明确显现出的情况下、即芴环相对于主链大致垂直取向的情况下,对于芴,设为“○”,在未明确显现出的情况下,对于芴,设为“×”。
[单体的合成例]
<合成例1>9,9’-二(羟基甲基)-9,9’-联芴(化合物1)的合成
[化93]
在50ml茄形瓶中装入9,9’-联芴(0.5g、1.51mmol)、多聚甲醛(42.8mg、1.43mmol)、n,n-二甲基甲酰胺(2.5ml),进行氮气置换后加入乙醇钠(4.6g、0.068mmol),进行搅拌。1小时后,添加多聚甲醛(42.8mg、1.43mmol),进行3小时搅拌。进一步添加多聚甲醛(120mg、4.0mmol),进行2小时搅拌。向反应液中滴加1n盐酸(3ml),停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(10ml)进行喷撒清洗。使所得到的粗生成物分散至甲苯(4ml)中后,进行1小时加热回流。恢复至室温(20℃)后,进行抽滤,100℃下减压干燥直至恒量,从而得到作为白色固体的9,9’-二(羟基甲基)-9,9’-联芴(化合物1)313mg(收率:53.1%、hplc纯度:98.8%)。
1h-nmr(400mhz,dmso-d6)δ7.59(d,j=7.3hz,4h),7.21(t,j=7.3hz,4h),7.03(t,j=7.1hz,4h),6.85(br,4h),5.81(t,j=4.8hz,2h),4.07(br,4h).
uv-vis(thf):λmax=269,290,302nm.
5wt%失重温度(氮气气氛下):241℃
<合成例2>
[化94]
<合成例2a>双(芴-9-基)甲烷(化合物2a)的合成
在1l四口烧瓶中转入芴(120g、722mmol)、n,n-二甲基甲酰胺(480ml),进行氮气置换后,冷却至5℃以下。加入乙醇钠(24.6g、361mmol),分多次、每次少量添加多聚甲醛(8.7g、289mmol)以使温度不超过10℃,进行搅拌。2小时后,滴加1n盐酸(440ml),停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(240ml)进行喷撒清洗。其后,使所得到的粗生成物分散在去离子水(240ml)中,进行1小时搅拌。对该悬浮液进行抽滤后,利用去离子水(120ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(480ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的双(芴-9-基)甲烷(化合物2a)84.0g(收率:84.5%、hplc纯度:94.0%)。
1h-nmr(400mhz,cdcl3)δ7.83(d,j=7.6hz,4h),7.56(dd,j1=7.6hz,j2=0.8hz,4h),7.41(t,j=7.3hz,4h),7.29(dt,j1=7.3hz,j2=1.3hz,4h),4.42(t,j=7.6hz,2h),2.24(d,j=7.6hz,2h).
<合成例2b>双(9-羟基甲基芴-9-基)甲烷(化合物2b)的合成
在500ml四口烧瓶中装入合成例2a中得到的双(芴-9-基)甲烷(100g、290mmol)、n,n-二甲基甲酰胺(400ml),进行氮气置换后,加入多聚甲醛(18.3g、610mmol)。冷却至5℃以下后,加入乙醇钠(0.698g、13mmol),进行搅拌使温度不超过10℃。1.5小时后,添加1n盐酸(32ml)使温度不超过25℃,停止反应。进一步加入水(300ml),进行搅拌,对所得到的悬浮溶液进行抽滤,利用去离子水(100ml)进行喷撒清洗。使所得到的粗生成物分散在四氢呋喃(400ml)中后,进行1小时的加热回流。恢复至室温(20℃),在抽滤后80℃下减压干燥直至恒量,得到白色固体108g(收率:91%、hplc纯度:99.1%)。所得到的白色固体中的钠含量为620ppm。其后使白色固体分散在甲苯(800ml)和水(200ml)混合液中,进行1小时加热回流,过滤、干燥后,对所得到的固体中的钠含量进行测定,结果为390ppm。进一步使所得到的白色固体分散在n,n-二甲基甲酰胺(500ml)中,加热制成均匀的溶液后,冷却至40℃以下,缓慢地滴加到0.03n的盐酸(1500ml)中。对所得到的悬浮溶液进行抽滤,使其分散在去离子水(200ml)中,进行1小时搅拌。对该悬浮液进行抽滤后,利用去离子水(100ml)进行喷撒清洗。使所得到的生成物分散在甲苯(800ml)中后,在加热回流下进行共沸脱水。恢复至室温(20℃),抽滤后,100℃下减压干燥直至恒量,从而得到作为白色固体的双(9-羟基甲基芴-9-基)甲烷(化合物2b)104g(收率87%、hplc纯度:99.8%)。固体中的钠、氯的含量分别小于10ppm。
1h-nmr(400mhz,dmso-d6)δ7.12(d,j=7.3hz,4h),7.01-6.93(m,8h),6.77(dt,j1=7.3hz,j2=1.0hz,4h),4.97(t,j=4.6hz,2h),3.31(s,2h),3.23(d,j=4.3hz,4h).
uv-vis(thf):λmax=263,292,304nm.
5wt%失重温度(氮气气氛下):289℃
m.p.:226℃
<合成例3>
[化95]
<合成例3a>1,2-双(芴-9-基)乙烷(化合物3a)的合成
在100ml四口烧瓶中装入芴(2.0g、12mmol)、四氢呋喃(35ml),进行氮气置换后,在乙醇-干冰浴中冷却至-50℃以下。分多次、每次少量添加1.6mol/l正丁基锂(7.8ml、12.5mmol)使温度不超过-40℃,进行搅拌。其后,升温至10℃,进行1小时搅拌后,加入1,2-二溴乙烷(0.55ml、6.4ml),进一步进行2小时搅拌。其后,滴加1n盐酸(0.5ml),对生成的悬浮溶液进行抽滤,水洗后,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双(芴-9-基)乙烷(化合物3a)0.63g(收率:29.2%、hplc纯度:98.0%)。另外,减压蒸馏除去滤液的溶剂,加入乙醇(25ml),进行30分钟进行搅拌。对悬浮液进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双(芴-9-基)乙烷(化合物3a)0.44g(收率:20.5%、hplc纯度:84.0%)。若将所得到的白色固体合并,则为1.07g(收率:49.7%)。
1h-nmr(400mhz,cdcl3)δ7.75(d,j=7.6hz,4h),7.37(dt,j1=7.6hz,j2=0.5hz,4h),7.27-7.34(m,8h),3.85(s,2h),1.74(t,j=2.3hz,4h).
<合成例3b>1,2-双(9-羟基甲基芴-9-基)乙烷(化合物3b)的合成
在1l四口烧瓶中装入合成例3a中得到的1,2-双(芴-9-基)乙烷(化合物3a、100g、278.9mmol)、多聚甲醛(17.6g、585.8mmol)、n,n-二甲基甲酰胺(400ml),进行氮气置换后,冷却至10℃以下。加入乙醇钠(1.80g、27.9mmol),升温至室温(20℃),进行1小时搅拌。利用hplc确认到原料消失后,将反应液滴加到0.1n盐酸(440ml)中,停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(100ml)进行喷撒清洗。其后,使所得到的粗生成物分散在n,n-二甲基甲酰胺(300ml)中,进行1小时搅拌。将该悬浮液滴加在0.005n盐酸(1000ml)中,搅拌30分钟后,进行抽滤。使所得到的粗生成物分散在去离子水(500ml)中,进行1小时搅拌。对该悬浮液进行抽滤后,利用去离子水(200ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(500ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,100℃下减压干燥直至恒量,从而得到作为白色固体的1,2-双(9-羟基甲基芴-9-基)乙烷(化合物3b)112.4g(收率:96.3%、hplc纯度:99.1%)。固体中的钠含量小于1ppm。
1h-nmr(400mhz,dmso-d6)δ7.91(d,j=7.3hz,4h),7.44(dt,j1=7.6hz,j2=1.0hz,4h),7.35(dt,j1=7.6hz,j2=1.0hz,4h),7.18(d,j=7.3hz,4h),4.79(t,j=5.3hz,2h),3.18(d,j=5.3hz,2h),1.40(s,4h).
uv-vis(thf):λmax=264,291,302nm.
5wt%失重温度(氮气气氛下):301℃
m.p.:278℃
<合成例4>
[化96]
<合成例4a>双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)的合成
在1l三口烧瓶中装入合成例2a中得到的双(芴-9-基)甲烷(化合物2a、80g、232.3mmol)、n-苄基-n,n,n-三乙基氯化铵(10.6g、46.5mmol)、二氯甲烷(400ml),进行氮气置换后,利用水浴将温度控制在15℃~20℃,加入50%氢氧化钠水溶液(64ml),结果溶液的颜色变为淡红色。其后利用5分钟滴加丙烯酸乙酯(50.5ml、465mmol)。在1小时后进一步加入丙烯酸乙酯(25.3ml、232mmol),一边通过hplc追踪反应的进行一边进行9小时搅拌。在通过hplc确认到单加成体为5%以下后,利用冰浴进行冷却,控制着温度滴加3n盐酸(293ml),进行骤冷。利用水进行清洗直至有机层的液性呈中性后,利用无水硫酸镁进行干燥、过滤,减压蒸馏除去溶剂。使所得到的粗生成物分散在甲醇(400ml)中,进行30分钟加热回流,从而进行热悬浮洗涤。其后恢复至室温(20℃),抽滤后,80℃下减压干燥至恒量,从而得到作为白色固体的双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)96.1g(收率:75.9%、hplc纯度:96.0%)。
1h-nmr(400mhz,cdcl3)δ7.03(d,j=7.6hz,4h),6.97(dt,j1=7.6hz,j2=1.5hz,4h),6.82(dt,j1=7.6hz,j2=1.3hz,4h),6.77(d,j=7.6hz,4h),3.88(q,j=7.1hz,4h),3.12(s,2h),2.23(m,4h),1.13(m,4h),1.02(t,j=7.1hz,6h).
5wt%失重温度(氮气气氛下):295℃
m.p.:141℃
<合成例4b>双[9-(3-羟基丙基)-芴-9-基]甲烷(化合物4b)的合成
在500ml四口烧瓶中装入合成例4a中得到的双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a、50g、91.8mmol)、甲苯(250ml),进行氮气置换后,利用冰浴冷却至5℃以下,在保持在10℃以下的同时滴加双(2-甲氧基乙氧基)氢化铝钠的65wt%甲苯溶液(82.7ml、275mmol),进行1小时搅拌。通过hplc确认到原料消失后,滴加乙酸乙酯(9.9ml),进行30分钟搅拌后,进一步滴加3.1n氢氧化钠水溶液,进行2小时搅拌。对所得到的悬浮溶液进行抽滤,利用去离子水(100ml)进行喷撒清洗。其后,使所得到的粗生成物分散在去离子水(150ml)中,进行30分钟搅拌。抽滤后进行喷撒清洗,直至液性呈中性,利用甲苯(50ml)进行喷撒清洗。使所得到的粗生成物分散在四氢呋喃(150ml)中,通过加热进行溶解。四氢呋喃溶液恢复至室温(20℃)后,通过短硅胶过滤柱(50g),利用四氢呋喃(350ml)洗脱,利用蒸发器对所得到的溶液进行减压蒸馏除去溶剂。使所得到的粗生成物分散在甲苯(250ml)中,进行30分钟加热回流,从而进行热悬浮洗涤。恢复至室温(20℃)后进行抽滤,之后80℃下减压干燥至恒量,从而得到作为白色固体的双[9-(3-羟基丙基)-芴-9-基]甲烷(化合物4b)35.5g(收率:83.9%、hplc纯度:99.8%)。固体中的钠含量、铝含量均小于1ppm。
1h-nmr(400mhz,cdcl3)δ7.05(d,j=7.6hz,4h),6.97(dt,j1=7.6hz,j2=1.5hz,4h),6.81(dt,j1=7.6hz,j2=1.3hz,4h),6.77(d,j=7.6hz,4h),3.19(q,j=6.3hz,4h),3.08(s,2h),1.94(m,4h),0.77(t,j=5.8hz,2h),0.47(m,4h).
5wt%失重温度(氮气气氛下):301℃
m.p.:214℃
<合成例5>双{9-[2-(2-羟基乙氧基)羰基乙基]芴-9-基}甲烷(化合物5)的合成
[化97]
在1l四口烧瓶中投入合成例4a中得到的双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a、50g、91.8mmol)、乙二醇(400ml)、二乙二醇二甲基醚(400ml),在氮气下加热使其溶解,制成均匀的溶液。进一步添加固体的乙醇钠(0.94g、13.8mmol)后,将体系内减压至100mmhg~150mmhg除去所产生的乙醇,同时使内温为90℃~95℃进行10小时搅拌。通过hplc确认到原料消失后,将反应液冷却至室温(20℃),添加1n盐酸(14ml)进行中和,之后移动容器,进一步缓慢地添加水1.2l。滤除析出的固体,在漏斗上进行喷撒水洗后,使所得到的固体溶解在乙酸乙酯中,利用5%碳酸钾水溶液进行清洗。进一步用水进行清洗直至有机层的液性成中性,之后利用无水硫酸镁干燥,进行过滤后减压蒸馏除去溶剂。将所得到的粗生成物由甲苯(125ml)中的再结晶进行精制,80℃下减压干燥直至恒量,从而得到作为白色固体的双{9-[2-(2-羟基乙氧基)羰基乙基]芴-9-基}甲烷(化合物5)46.5g(收率86.2%、hplc纯度98.1%)。
1h-nmr(400mhz,cdcl3)δ7.05-7.02(m,4h),6.97(dt,j1=7.2hz,j2=1.2hz,4h),6.81(dt,j1=7.3hz,j2=1.3hz,4h),6.77-6.75(m,4h),3.89-3.85(m,4h),3.57-3.51(m,4h),3.12(s,2h),2.28-2.21(m,4h),1.71-1.78(m,2h),1.21-1.14(m,4h).
5wt%失重温度(氮气气氛下):306℃
m.p.:143℃
<合成例6>
[化98]
<合成例6a>9,9-双[(芴-9-基)-甲基]芴(化合物6a)的合成
在1l四口烧瓶中装入芴(100g、602mmol)、n,n-二甲基甲酰胺(400ml),进行氮气置换后,冷却至5℃以下。加入乙醇钠(20.5g、301mmol),分多次、每次少量添加多聚甲醛(11.4g、379mmol)以使温度不超过10℃,进行搅拌。1.5小时后,滴加1n盐酸(330ml),停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(200ml)进行喷撒清洗。其后使所得到的粗生成物分散在去离子水(300ml)中,进行1小时搅拌。对该悬浮液进行抽滤后,利用去离子水(120ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(400ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的9,9-双[(芴-9-基)-甲基]芴(化合物6a)71.6g(收率:73.2%、hplc纯度:98.0%)。
1h-nmr(400mhz,cdcl3)δ7.87(dd,j1=5.6hz,j2=1.8hz2h),7.73(dd,j1=6.3hz,j2=1.8hz,2h),7.56(d,j=7.6hz,4h),7.47-7.54(m,4h),7.20(t,j=7.3hz,4h),7.04(dt,j1=7.6hz,j2=1.0,4h),6.63(dd,j1=7.6hz,j2=0.8,4h),3.24(t,j=4.9hz,2h),2.80(d,j=4.9hz,4h).
<合成例6b>9,9-双[(9-羟基甲基芴-9-基)-甲基]芴(化合物6b)的合成
在500ml四口烧瓶中装入合成例6a中得到的9,9-双[(芴-9-基)-甲基]芴(化合物6a、54g、104mmol)、n,n-二甲基甲酰胺(370ml),进行氮气置换后,加入多聚甲醛(7.8g、260mmol)。在室温(20℃)下加入甲醇钠(0.698g、13mmol),在不超过20℃的条件下进行搅拌。2.5小时后,在对水750ml与1n盐酸(25ml)的混合液进行搅拌后,将反应液滴入,停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(200ml)进行喷撒清洗。其后,使所得到的粗生成物分散在去离子水(500ml)中,进行1小时搅拌。对该悬浮液进行抽滤后,利用去离子水(100ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(400ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后进行抽滤。使粗生成物分散在乙醇(200ml)中,加热回流30分钟后进行抽滤,之后得到白色固体49.5g(收率:82%、lc纯度:83%)。加入thf(150ml)进行加热回流后,加入水(100ml)与乙醇(50ml)使结晶析出,抽滤后80℃下减压干燥直至恒量,从而得到白色固体36g(收率:59%、hplc纯度:92%)。进一步使其分散在乙醇(200ml)中进行加热回流,之后进行过滤,反复进行该操作,从而得到hplc纯度99%的白色固体9,9-双[(9-羟基甲基芴-9-基)-甲基]芴(化合物6b)。固体中的钠含量小于1ppm。
1h-nmr(400mhz,cdcl3)δ7.02(dt,j1=7.6hz,j2=0.8hz4h),6.95(td,j1=7.6hz,j2=0.8hz,4h),6.74(td,j1=7.2hz,j2=0.8hz,4h),6.64-6.67(m,6h),6.42(dt,j1=8.0hz,j2=0.8、2h),6.33-6.38(m,4h),3.36(d,j=6.8hz,4h),3.06(s,4h).
uv-vis(thf):λmax=266,305nm.
5wt%失重温度tg:304℃
m.p.:259℃
<合成例7>
[化99]
<合成例7a>1,2-双[9-(2-乙氧基羰基乙基)芴-9-基]乙烷(化合物7a)的合成
在1l四口烧瓶中装入合成例3a中得到的1,2-双(芴-9-基)乙烷(化合物3a、85g、237mmol)、四氢呋喃(725ml)、n,n-二甲基甲酰胺(85ml),进行氮气置换后加入乙醇钠(3.23g、47.5mmol),升温至室温(20℃),进行30分钟搅拌。利用2.5小时滴加丙烯酸乙酯(59.3ml、545mmol)。通过hplc确认到原料消失后,向反应液中滴加0.1n盐酸(55ml),停止反应。减压蒸馏除去四氢呋喃后,加入甲苯(425ml),利用纯净水进行清洗直至有机层的液性呈中性,之后利用无水硫酸镁干燥,进行过滤,减压蒸馏除去溶剂。使所得到的粗生成物分散在甲醇(400ml)中,加热回流1小时来进行热悬浮洗涤。其后恢复至室温(20℃),抽滤后,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双[9-(2-乙氧基羰基乙基)芴-9-基]乙烷(化合物7a)101g(收率:76.1%、hplc纯度:98.6%)。
1h-nmr(400mhz,cdcl3)δ7.72(d,j=7.6hz,4h),7.36(t,j=7.6hz,4h),7.27(t,j=7.3hz,4h),6.97(d,j=7.3hz,4h),3.80(q,j=7.1hz,4h),1.93(t,j=8.6hz,4h),1.33(t,j=8.6hz,4h),1.23(s,4h),1.01(t,j=7.1hz,6h).
5wt%失重温度(氮气气氛下):306℃
m.p.:150℃
<合成例7b>1,2-双[9-(3-羟基丙基)-芴-9-基]乙烷(化合物7b)的合成
在1000ml四口烧瓶中装入合成例7a中得到的1,2-双[9-(2-乙氧基羰基乙基)芴-9-基]乙烷(化合物7a、100g、179mmol)、四氢呋喃(500ml),进行氮气置换后,利用冰浴冷却至5℃以下,一边保持在15℃以下一边滴加双(2-甲氧基乙氧基)氢化铝钠的65wt%甲苯溶液(161ml、537mmol),进行1小时搅拌。通过hplc确认到原料消失后,滴加乙酸乙酯(32ml),进行45分钟搅拌后,进一步滴加3.1n氢氧化钠水溶液(257ml),进行1小时搅拌。减压蒸馏除去四氢呋喃后,对所得到的悬浮溶液进行抽滤,利用去离子水(100ml)进行喷撒清洗。其后,使所得到的粗生成物溶解在乙酸乙酯(700ml)中,利用去离子水(100ml)清洗3次。将有机层利用硫酸镁干燥后,通过短硅胶过滤柱(50g),利用四氢呋喃(800ml)洗脱,利用蒸发器对所得到的溶液进行减压蒸馏除去溶剂。使所得到的粗生成物分散在甲苯(400ml)中,加热回流30分钟,由此来进行热悬浮洗涤。恢复至室温(20℃)后进行抽滤,之后100℃下减压干燥直至恒量,从而得到作为白色固体的1,2-双[9-(3-羟基丙基)-芴-9-基]乙烷(化合物7b)75.6g(收率:89.0%、hplc纯度:98.7%)。固体中的钠含量为2ppm、铝含量小于2ppm。
1h-nmr(400mhz,dmso-d6)δ7.81(d,j=7.3hz,4h),7.35(t,j=7.3hz,4h),7.29(t,j=7.3hz,4h),7.02(d,j=7.3hz,4h),4.02(t,j=5.0hz,2h),2.93(m,4h),1.59(m,4h),1.19(s,4h),0.45(m,4h).
5wt%失重温度(氮气气氛下):312℃
m.p.:253℃
<合成例8>α,α’-双-(9-羟基甲基芴-9-基)-1,4-二甲苯(化合物8)的合成
[化100]
在1l四口茄形瓶中装入α,α’-双-(芴-9-基)-1,4-二甲苯((130g、0.3mol)、多聚甲醛(18.9g、0.63mol)、n,n-二甲基甲酰胺(520ml),进行氮气置换后加入乙醇钠(2.04g、0.03mol),在室温(20℃)下进行1小时搅拌。在1l烧杯中装入去离子水520ml与1n盐酸(45ml),进行搅拌后加入反应液,使反应骤冷。对所得到的结晶进行抽滤,利用去离子水(100ml)进行喷撒清洗。使所得到的粗生成物分散在去离子水(500ml)中后,进行抽滤,利用去离子水(100ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(500ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,70℃下减压干燥至恒量,从而得到白色固体(化合物8)130g(收率:87%、hplc纯度:97.6%)。
1h-nmr(400mhz,cdcl3)δ7.62(d,j=7.6hz,4h),7.33(t,j=8.0hz,4h),7.25(t,j=6.0hz,4h),7.19(br,4h),6.45(s,4h),3.80(d,j=6.4hz,4h),3.12(s,4h),1.42(t,j=6.4hz,2h).
5wt%失重温度(氮气气氛下):327℃
m.p.:198℃
<合成例9>
[化101]
<合成例9a>1,2-双(芴-9-基)丁烷(化合物9a)的合成
在容量70ml的sus316制高压釜中装入芴(3.5g、21mmol)、1,4-丁二醇(4.9g、54mmol)、85%koh(1.52g、23mmol)、四乙二醇二甲基醚(4.9g),在氮气气氛下在250℃进行8小时反应。
冷却后,使内容物分散在四氢呋喃和水中,利用稀盐酸中和。由所得到的悬浮溶液中过滤析出粉末,进行水洗,得到作为白色固体的1,4-双(芴-9-基)丁烷(化合物9a)1.7g(收率:41.9%、hplc纯度:97.4%)。
1h-nmr(400mhz,cdcl3)δ7.72(d,j=7.6hz,4h),7.42(m,4h),7.25-7.36(m,8h),3.89(t,j=5.8hz,2h),1.96-1.86(m,4h),1.15-1.05(m,4h).
<合成例9b>1,2-双(9-羟基甲基芴-9-基)丁烷(化合物9b)的合成
在500ml四口烧瓶中装入合成例9a中得到的1,2-双(芴-9-基)丁烷(化合物9a、37.0g、95.7mmol)、多聚甲醛(6.03g、201mmol)、n,n-二甲基甲酰胺(148ml),进行氮气置换后,冷却至10℃以下。加入乙醇钠(0.65g、9.6mmol),升温至室温(20℃),进行1小时搅拌。通过hplc确认到原料消失后,将反应液滴加到0.1n盐酸(162ml)中,停止反应。对所得到的悬浮溶液进行抽滤,利用去离子水(37ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(185ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双(9-羟基甲基芴-9-基)丁烷(化合物9b)39.8g(收率:93.1%、hplc纯度:99.1%)。
1h-nmr(400mhz,dmso-d6)δ7.71-7.66(m,4h),7.38-7.24(m,4h),3.71(d,j=6.3hz,4h),1.89-1.81(m,4h),1.22(t,j=6.3hz,2h),0.51-0.44(m,4h).
uv-vis(thf):λmax=291,302nm.
5wt%失重温度(氮气气氛下):314℃
m.p.:212℃
<合成例10>
<合成例10a>1,1-双(芴-9-基)乙烷(化合物10a)的合成
[化102]
在500ml四口烧瓶中装入芴(100g、0.6mol)、n,n-二甲基甲酰胺(150ml),进行氮气置换后,将内温冷却至5℃以下。加入tritonb(5.3ml、0.012mol)之后,利用1小时的时间滴加乙醛(15.6g、0.319mol)的n,n-二甲基甲酰胺(40ml)溶液。通过hplc确认到原料消失后,滴加1n盐酸(18.1ml),停止反应。进一步加入甲醇(300ml),对所得到的悬浮溶液进行抽滤,利用甲醇(100ml)进行喷撒清洗。使所得到的粗生成物分散在甲醇(300ml)中后,进行30分钟加热回流。恢复至室温(20℃),对所得到的悬浮溶液进行抽滤,利用甲醇(80ml)进行喷撒清洗。减压干燥至在50℃为恒量,从而得到作为白色固体的1,1-双(芴-9-基)乙烷(化合物10a)95g(收率:88%、hplc纯度:95%)。
1h-nmr(400mhz,cdcl3)δ.7.75(dd,j1=16.4hz,j2=7.4hz,4h),7.55(d,j=7.6hz,2h),7.40(t,j=7.6hz,2h),7.31(q,j=7.6hz,4h),7.17(t,j=7.6hz,2h),6.98(d,j=7.6hz,2h)3.98(d,j=4.4hz,2h),3.0-3.1(m,1h),0.83(d,j=6.8hz,3h).
<合成例10b>1-双(9-羟基甲基芴-9-基)乙烷(化合物10b)的合成
在1l四口烧瓶中装入合成例10a中得到的1,1-双(芴-9-基)乙烷(化合物10a、100g、0.28mol)、多聚甲醛(45g、1.4mol)、n,n-二甲基甲酰胺(400ml),进行氮气置换后,冷却至10℃以下。加入乙醇钠(4g、0.056mol),慢慢地升温至室温(20℃),进行1小时搅拌。通过hplc确认到原料消失后,滴加甲醇(50ml)与1n盐酸(112ml),停止反应。加入甲苯(400ml)进行分液,利用甲苯(200ml)提取水层。将甲苯层合并,利用蒸发器蒸馏除去甲苯,得到226g的反应混合物。将甲醇(160ml)与1n盐酸(120ml)加入甲苯(400ml)加热回流1小时后,冷却至室温(20℃),之后对所得到的悬浮溶液进行抽滤,利用去离子水(100ml)和甲苯(150ml)进行喷撒清洗。使所得到的粗生成物分散在甲苯(380ml)中后,使用dean-stark装置,在加热回流条件下进行脱水。恢复至室温(20℃)后,进行抽滤,60℃下减压干燥至恒量,从而得到作为白色固体的双(9-羟基甲基芴-9-基)甲烷(化合物10b)69g(收率59%、hplc纯度:99%)。固体中的钠、氯的含量分别小于10ppm。
1h-nmr(400mhz,chcl3-d3)δ7.37(dd,j1=15.0hz,j2=8.0hz,4h),7.18-7.23(m,4h),7.13(dt,j1=8.0hz,j2=1.2hz,2h),7.02(dt,j1=7.3hz,j2=1.2hz,2h),6.87-6.96(m,4h),3.85(dd,j1=12hz,j2=6.4hz,2h),3.69(q,j=7.2hz,1h),3.18(dd,j1=11.2hz,j2=6.4hz,2h),1.49(d、j=7.2hz,2h),1.06(t,j=6.8hz,2h).
5wt%失重温度(氮气气氛下):283℃
m.p.:180℃
<合成例11>1,2-双[9-(2-甲氧基羰基丙基)芴-9-基]乙烷(化合物11)的合成
[化103]
在1l四口烧瓶中装入合成例3a的方法中得到的1,2-双(芴-9-基)乙烷(化合物3a、45g、0.126mol)、n,n-二甲基甲酰胺(360ml),进行氮气置换后,利用冰水冷却,加入苄基三甲基氢氧化铵(40%甲醇溶液)(2.86ml、6.3mmol),在冰水冷却下利用40分钟时间滴加甲基丙烯酸甲酯(28.1ml、0.264mol),慢慢地升温至室温(20℃)。通过hplc确认到原料消失后,向反应液中滴加1n盐酸(13.5ml)与水(225ml),停止反应。对析出的结晶进行过滤,使所得到的粗生成物分散在甲醇(350ml)中,加热回流1小时来进行热悬浮洗涤。其后恢复至室温(20℃),抽滤后,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双[9-(2-甲氧基羰基丙基)芴-9-基]乙烷(化合物11)65g(收率:93%、hplc纯度:98%)。
1h-nmr(400mhz,cdcl3)δ7.72(dd,j1=10hz,j2=8hz,4h),7.37(t,j=7.2hz,2h),7.33(t,j=6hz,2h),7.21-7.30(m,4h),6.95(t,j=9.2hz,2h),6.91(t,j=7hz,2h),3.04(s,6h),2.23(dd,j1=12hz,j2=9hz,2h),1.56(d,j=14hz,2h),1.36-1.46(m,2h),1.05-1.16(m,4h),0.58(d,j=6.8hz,6h).
5wt%失重温度(氮气气氛下):292℃
m.p.:147℃
<合成例12>
[化104]
<合成例12a>双[9-(2-甲氧基羰基乙基)芴-9-基]甲烷(化合物12)的合成
在300ml三口烧瓶中装入合成例2a中得到的双(芴-9-基)甲烷(化合物2a、10g、29.0mmol)、n-苄基-n,n,n-三乙基氯化铵(1.32g、5.8mmol)、四氢呋喃(50ml),进行氮气置换后,利用水浴控制在15℃~20℃,加入50%氢氧化钠水溶液(8ml)后,溶液的颜色变为淡红色。其后利用3小时的时间滴加丙烯酸甲酯(7.81ml、87.1mmol)。一边通过hplc追踪反应的进行一边进行3小时搅拌。在通过hplc确认到单加成体为10%以下后,利用冰浴进行冷却,温度均衡地滴加3n盐酸(21ml),进行骤冷。废弃水层后,添加甲苯(20ml),利用去离子水清洗有机层。减压蒸馏除去溶剂,在固体开始析出的时刻解除减压,添加甲醇(40ml),进行30分钟搅拌。其后进行抽滤,100℃下减压干燥直至恒量,从而得到作为白色固体的双[9-(2-甲氧基羰基乙基)芴-9-基]甲烷(化合物12a)7.05g(收率:47%、hplc纯度:80%)。
1h-nmr(400mhz,cdcl3)δ7.03(d,j=7.3hz,4h),6.97(dt,j1=6.8,j2=1.3hz,4h),6.75-6.83(m,8h),3.38(s,6h),3.12(s,2h),2.24(t,j=8.1hz,4h),1.14(t,j=8.1hz,4h).
<合成例12b-1>双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷(化合物12b)的合成
在300ml四口烧瓶中装入合成例12a的方法中得到的双[9-(2-甲氧基羰基乙基)芴-9-基]甲烷(化合物12a、6.0g、11.61mmol)、碳酸二苯酯(12.1g、56.6mmol)、邻钛酸四异丙酯(0.49ml、1.66mmol),升温至145℃,进行3小时搅拌。通过hplc确认到反应终止后,加入甲苯(15ml),加热回流1小时。冷却至50℃后,加入甲醇(18ml)。冷却至室温(20℃)后,进行抽滤。使所得到的白色固体分散在甲苯(12ml)中,加热回流1小时。冷却至50℃后,加入甲醇(18ml)。冷却至室温(20℃)后,进行抽滤,100℃下减压干燥直至恒量,从而得到作为白色固体的双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷(化合物12b)5.39g(收率:64%、hplc纯度:98.1%)。
1h-nmr(400mhz,cdcl3)δ7.23-7.28(m,4h),7.07-7.16(m,6h),7.03(dt,j1=6.9hz,j2=2.0,4h),6.78-6.90(m,12h),3.20(s,2h),2.37(t,j=8.3hz,4h),1.40(t,j=8.3hz,4h).
5wt%失重温度(氮气气氛下):336℃
m.p.:176℃
<合成例12b-2>双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷(化合物12b)的合成
[化105]
在1l四口烧瓶中装入合成例4a的方法中得到的双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a、50.0g、91.80mmol)、碳酸二苯酯(98.3g、459mmol)、邻钛酸四异丙酯(1.3ml、4.59mmol),将真空度调整为3kpa,在145℃~150℃的温度范围一边蒸馏除去副产物一边进行6小时搅拌。冷却到90℃,通过hplc确认反应终止后,加入甲苯(100ml),冷却至50℃。向其中加入甲醇(250ml),冷却至5℃后,进行抽滤。使所得到的白色固体分散在甲苯(100ml)中,加热回流30分钟。冷却至50℃后,加入甲醇(200ml)。冷却至室温(20℃)后,进行抽滤,100℃下减压干燥直至恒量,从而得到作为白色固体的双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷(化合物12b)50g(收率:85%、hplc纯度:98.1%)。
1h-nmr(400mhz,cdcl3)δ7.23-7.28(m,4h),7.07-7.16(m,6h),7.03(dt,j1=6.9hz,j2=2.0,4h),6.78-6.90(m,12h),3.20(s,2h),2.37(t,j=8.3hz,4h),1.40(t,j=8.3hz,4h).
5wt%失重温度(氮气气氛下):336℃
m.p.:176℃
<合成例13>1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷(化合物13)的合成
[化106]
在1l四口烧瓶中装入合成例7a的方法中得到的1,2-双[9-(2-乙氧基羰基乙基)芴-9-基]乙烷(化合物7a、100.0g、179mmol)、碳酸二苯酯(115g、537mmol)、邻钛酸四异丙酯(2.62ml、8.95mmol),进行氮气置换后,升温至135℃,进行24小时搅拌。在途中,在经过12小时的时刻、经过20小时的时刻,分次追加碳酸二苯酯(38.3g、179mmol)。通过hplc确认到反应终止后,加入甲苯(400ml),加热回流1小时。冷却至室温(20℃)后,进行抽滤。使所得到的白色固体分散在甲苯(300ml)中,加热回流1小时。冷却至室温(20℃)后,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷(化合物13)82g(收率:70.0%、hplc纯度:98.0%)。
1h-nmr(400mhz,cdcl3)δ7.76(d,j=7.6,4h),7,41(dt,j1=7.3,j2=1.0,4h),7.32(dt,j1=7.3,j2=1.0,4h),7.22(t,j=8.3,4h),7.11(t,j=7.6,2h),7.03(d,j=7.6,4h),6.78(d,j=8.6,4h),2.06(t,j=8.1,4h),1.60(t,j=8.1,4h),1.29(s,4h)
5wt%失重温度(氮气气氛下):337℃
m.p.:232℃
<合成例14>
[化107]
<合成例14a>1,2-双(9-羟基芴-9-基)乙烷(化合物14a)的合成
在1l四口烧瓶中装入合成例3a的方法中得到的1,2-双(芴-9-基)乙烷(化合物3a、20g、59mmol)、n,n-二甲基甲酰胺(200ml),加入亚磷酸三丁酯(37.9ml、140mmol),进行氮气置换后,加入苄基三甲基氢氧化铵(40%甲醇溶液)(25ml),使空气(100ml/分钟)与氮气(300ml/分钟)的混合气体在反应体系中流通。搅拌3小时后,加入苄基三甲基氢氧化铵(40%甲醇溶液)(10ml),搅拌5小时。进一步加入苄基三甲基氢氧化铵(40%meoh溶液)(10ml),进而搅拌1小时。加入1n盐酸(200ml),停止反应,加入乙酸乙酯(400ml),进行分液操作。进一步利用饱和食盐水(100ml)对有机层进行3次清洗。利用硫酸镁使有机层干燥后,进行过滤,减压蒸馏除去有机溶剂。在所得到的悬浮溶液中添加甲苯(100ml)、己烷(200ml),搅拌30分钟后,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的1,2-双(9-羟基芴-9-基)乙烷(化合物14a)13.9g(收率:63.8%、hplc纯度:92.5%)。
1h-nmr(400mhz,cdcl3)δ7.73(d,j=7.3hz,4h),7.35(dt,j1=7.6hz,j2=1.0,6h),7.26(dt,j1=7.6hz,j2=1.0,4h),7.11(d,j=7.3hz,4h),5.35(s,2h),1.40(s,4h).
<合成例14b>双{[4-(2-羟基乙氧基)苯基]芴-9-基}乙烷的合成
在300ml四口烧瓶中装入合成例14a的方法中得到的1,2-双(芴-9-基)乙烷(化合物14a、17g、45mmol)、苯氧基乙醇(37g、267mmol),进行氮气置换后,冷却至10℃以下。加入三氟化硼-二乙醚络合物(5.6ml、45mmol),在室温(20℃)下搅拌3小时后,进一步加入三氟化硼-二乙醚络合物(5.6ml、45mmol)、氯仿(35ml),在40℃下搅拌4小时、在60℃下搅拌2小时。进一步加入三氟化硼-二乙醚络合物(5.6ml、45mmol),过热回流2小时。冷却至室温(20℃)后,使用饱和碳酸氢钠水溶液进行中和,之后通过抽滤除去不溶物。添加乙酸乙酯(120ml),将有机层利用饱和食盐水清洗2次、利用去离子水清洗1次,利用硫酸镁干燥后,进行过滤,减压蒸馏除去有机溶剂。再次溶解于乙酸乙酯(150ml)中,加入活性炭(日本norit株式会社、sxplus、ph=7、2.5g),进行1小时搅拌后,进行硅藻土过滤,减压蒸馏除去有机溶剂。加入甲醇(100ml),搅拌1小时,进行抽滤,80℃下减压干燥至恒量,从而得到作为白色固体的双{[4-(2-羟基乙氧基)苯基]芴-9-基}乙烷(化合物14b)15.8g(收率:56.1%、hplc纯度:86%)。
1h-nmr(400mhz,cdcl3)δ7.77(d,j=7.3hz,4h),7.36(dt,j1=7.6hz,j2=1.0,4h),7.22(dt,j1=hz,j2=1.0,4h),6.92(d,j=7.6hz,4h),6.73(d,j=9.1hz,4h),6.59(d,j=9.1hz,4h),3.91-3.93(m,4h),3.83-3.87(m,4h),1.92(t,j=6.3hz,2h)1.82(s,4h).
[聚合物的合成例]
<实验例1>
将isb59.43质量份(0.407摩尔)、双[9-(3-羟基丙基)-芴-9-基]甲烷(化合物4b)28.40质量份(0.062摩尔)、dpc101.32质量份(0.473摩尔)和作为催化剂的乙酸钙1水合物8.25×10-4质量份(4.68×10-6摩尔)投入到反应容器中,在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚碳酸酯的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例2>
除了使用isb46.69质量份(0.320摩尔)、双[9-(3-羟基丙基)-芴-9-基]甲烷(化合物4b)42.60质量份(0.092摩尔)、dpc89.14质量份(0.416摩尔)和作为催化剂的乙酸钙1水合物7.26×10-4质量份(4.12×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例3>
除了使用isb38.20质量份(0.261摩尔)、双[9-(3-羟基丙基)-芴-9-基]甲烷(化合物4b)37.86质量份(0.082摩尔)、chdm12.71质量份(0.088摩尔)、dpc93.41质量份(0.436摩尔)和作为催化剂的乙酸钙1水合物7.61×10-4质量份(4.32×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例4>
除了使用isb50.94质量份(0.349摩尔)、双(9-羟基甲基芴-9-基)甲烷(化合物2b)18.79质量份(0.046摩尔)、chdm16.95质量份(0.118摩尔)、dpc110.89质量份(0.436摩尔)和作为催化剂的乙酸钙1水合物7.61×10-4质量份(9.03×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例5>
除了使用isb42.45质量份(0.290摩尔)、9,9-双[(9-羟基甲基芴-9-基)-甲基]芴(化合物6b)28.72质量份(0.049摩尔)、chdm16.95质量份(0.118摩尔)、dpc98.93质量份(0.462摩尔)和作为催化剂的乙酸钙1水合物8.06×10-4质量份(4.57×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例6>
除了使用isb48.39质量份(0.331摩尔)、1,2-双(9-羟基甲基芴-9-基)乙烷(化合物3b)16.95质量份(0.040摩尔)、chdm21.18质量份(0.147摩尔)、dpc112.18质量份(0.524摩尔)和作为催化剂的乙酸钙1水合物9.14×10-4质量份(5.19×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例7>
除了使用isb29.71质量份(0.203摩尔)、1,2-双[9-(3-羟基丙基)-芴-9-基]乙烷(化合物7b)26.55质量份(0.056摩尔)、chdm5.93质量份(0.041摩尔)、dpc64.99质量份(0.303摩尔)和作为催化剂的乙酸钙1水合物2.65×10-3质量份(1.50×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例8>
除了使用1,2-双[9-(3-羟基丙基)-芴-9-基]乙烷(化合物7b)33.18质量份(0.070摩尔)、isb23.77质量份(0.163摩尔)、chdm5.93质量份(0.041摩尔)、dpc59.22质量份(0.276摩尔)和作为催化剂的乙酸钙1水合物2.41×10-3质量份(1.37×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例9>
除了使用1,2-双(9-羟基甲基芴-9-基)丁烷(化合物9b)22.49质量份(0.050摩尔)、isb30.31质量份(0.207摩尔)、chdm8.90质量份(0.062摩尔)、dpc69.12质量份(0.323摩尔)和作为催化剂的乙酸钙1水合物2.81×10-3质量份(1.60×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例10>
除了使用1,2-双(9-羟基甲基芴-9-基)乙烷(化合物3b)32.95质量份(0.079摩尔)、isb20.80质量份(0.142摩尔)、chdm8.90质量份(0.062摩尔)、dpc61.18质量份(0.286摩尔)和作为催化剂的乙酸钙1水合物4.98×10-3质量份(2.83×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例11>
除了使用α,α’-双(9-羟基甲基芴-9-基)-1,4-二甲苯(化合物8)26.60质量份(0.054摩尔)、isb23.77质量份(0.163摩尔)、chdm11.86质量份(0.082摩尔)、dpc64.63质量份(0.302摩尔)和作为催化剂的乙酸钙1水合物5.26×10-3质量份(2.99×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<实验例12>
将双(9-羟基甲基芴-9-基)甲烷(化合物2b)23.70质量份(0.059摩尔)、chdm22.53质量份(0.156摩尔)、chda33.62质量份(0.195摩尔)和作为催化剂的钛酸四正丁酯6.65×10-3质量份(1.95×10-5摩尔)投入到反应容器中,在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,在常压下进行180分钟的反应。将所产生的水抽出到反应容器外。
接下来,作为反应第2步的工序,利用30分钟将加热槽的温度升温至240℃,同时利用30分钟将压力减压至13.3kpa以下。进一步利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表2。
<实验例13>
除了使用双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)34.12质量份(0.063摩尔)、chdm25.81质量份(0.179摩尔)、chda20.03质量份(0.116摩尔)和作为催化剂的钛酸四正丁酯6.09×10-3质量份(1.79×10-5摩尔)以外,与实验例12同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表2。
<实验例14>
将双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)26.49质量份(0.049摩尔)、chdm10.37质量份(0.072摩尔)和作为催化剂的钛酸四正丁酯14.65×10-3质量份(4.30×10-5摩尔)投入到反应容器中,在氮气气氛下,在220℃在常压下反应120分钟。接下来利用30分钟将压力减压至13.3kpa,在13.3kpa保持30分钟,将所产生的乙醇抽出到反应容器外。其后,将反应液暂且冷却到室温(20℃),向该反应容器中投入isb31.43质量份(0.215摩尔)、dpc51.66质量份(0.241摩尔),在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例15>
将双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)16.76质量份(0.031摩尔)、chdm7.16质量份(0.050摩尔)和作为催化剂的钛酸四正丁酯16.89×10-3质量份(4.96×10-5摩尔)投入到反应容器中,在氮气气氛下,在220℃于常压下反应120分钟。接下来,利用30分钟将压力减压至13.3kpa,在13.3kpa保持30分钟,将所产生的乙醇抽出到反应容器外。其后,将反应液暂且冷却到室温(20℃),向该反应容器中投入isb41.11质量份(0.281摩尔)、dpc65.01质量份(0.303摩尔),在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例16>
将双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)20.90质量份(0.037摩尔)、chdm10.79质量份(0.075摩尔)和作为催化剂的钛酸四正丁酯15.91×10-3质量份(4.68×10-5摩尔)投入到反应容器中,在氮气气氛下,在220℃于常压下反应120分钟。接下来,利用30分钟将压力减压至13.3kpa,在13.3kpa保持30分钟,将所产生的乙醇抽出到反应容器外。其后,将反应液暂且冷却到室温(20℃),向该反应容器中投入isb34.62质量份(0.237摩尔)、dpc59.44质量份(0.277摩尔),在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例17>
将双(9-羟基甲基芴-9-基)甲烷(化合物10b)20.31质量份(0.049摩尔)、chdm22.45质量份(0.156摩尔)、chda34.81质量份(0.202摩尔)和作为催化剂的钛酸四正丁酯51.61×10-3质量份(1.52×10-4摩尔)投入到反应容器中,在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,在常压下反应150分钟。其后,利用30分钟升温至240℃,同时将压力减压至13.3kpa。进一步利用15分钟减压至0.10kpa以下,将所产生的水抽出到体系外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表2。
<实验例18>
将双[9-(2-乙氧基羰基乙基)芴-9-基]甲烷(化合物4a)22.65质量份(0.042摩尔)、chdm10.77质量份(0.075摩尔)和作为催化剂的钛酸四正丁酯15.54×10-3质量份(4.57×10-5摩尔)投入到反应容器中,在氮气气氛下,在220℃于常压下反应120分钟。接下来,利用30分钟将压力减压至13.3kpa,在13.3kpa保持30分钟,将所产生的乙醇抽出到反应容器外。其后,将反应液暂且冷却至室温,向该反应容器中投入isb33.58质量份(0.230摩尔)、dpc56.96质量份(0.266摩尔),在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚酯碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例19>
将1,2-双[9-(2-乙氧基羰基乙基)芴-9-基]乙烷(化合物7a)28.37质量份(0.051摩尔)、chdm11.01质量份(0.076摩尔)和作为催化剂的钛酸四正丁酯14.17×10-3质量份(4.16×10-5摩尔)投入到反应容器中,在氮气气氛下,在220℃于常压下反应120分钟。接下来,利用30分钟将压力减压至13.3kpa,在13.3kpa保持30分钟,将所产生的乙醇抽出到反应容器外。其后,将反应液暂且冷却至室温,向该反应容器中投入isb29.40质量份(0.201摩尔)、dpc49.17质量份(0.230摩尔),在氮气气氛下,使加热槽温度为150℃并一边根据需要进行搅拌一边进行原料的溶解(约10分钟)。溶解后,作为反应第1步的工序,利用30分钟的时间升温至220℃,常压下进行60分钟反应。接下来利用90分钟的时间将压力由常压减压至13.3kpa,在13.3kpa保持30分钟,将产生的苯酚抽出到反应容器外。
接下来,作为反应第2步的工序,利用15分钟将加热槽的温度升温至240℃,同时利用15分钟将压力减压至0.10kpa以下,将产生的苯酚抽出到反应容器外。达到规定的扭矩后,终止反应,将生成的聚合物挤出到水中,得到聚酯碳酸酯树脂的颗粒。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例20>
除了使用1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷(化合物13)27.36质量份(0.042摩尔)、chdm12.05质量份(0.084摩尔)、isb31.72质量份(0.217摩尔)、dpc55.45质量份(0.259摩尔)和作为催化剂的乙酸钙1水合物2.65×10-3质量份(1.50×10-5摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例21>
除了使用1,2-双[9-(2-苯氧基羰基乙基)芴-9-基]乙烷(化合物13)33.25质量份(0.051摩尔)、chdm10.99质量份(0.076摩尔)、isb29.42质量份(0.201摩尔)、dpc48.57质量份(0.227摩尔)和作为催化剂的乙酸钙1水合物4.89×10-4质量份(2.78×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<实验例22>
除了使用双[9-(2-苯氧基羰基乙基)芴-9-基]甲烷(化合物12b)26.63质量份(0.042摩尔)、chdm10.78质量份(0.075摩尔)、isb33.58质量份(0.230摩尔)、dpc56.33质量份(0.263摩尔)和作为催化剂的乙酸钙1水合物5.36×10-4质量份(3.04×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表3。
<比较实验例1>
使用9,9-双(4-羟基-3-甲基苯基)芴(bcf)61.79质量份(0.163摩尔)、2,2-双(4-羟基苯基)丙烷(bpa)30.49质量份(0.134摩尔)、dpc67.40质量份(0.315摩尔)和作为催化剂的乙酸钙1水合物2.61×10-3质量份(1.48×10-5摩尔)。并使最终聚合温度为280℃,除此以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<比较实验例2>
使用9,9-双(4-羟基-3-甲基苯基)芴(bcf)32.20质量份(0.085摩尔)、螺环二醇(spg)60.43质量份(0.199摩尔)、dpc63.18质量份(0.295摩尔)和作为催化剂的乙酸钙1水合物2.50×10-3质量份(1.42×10-5摩尔)。并使最终聚合温度为280℃,除此以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<比较实验例3>
使用9,9-双(4-羟基-3-甲基苯基)芴(bcf)34.63质量份(0.091摩尔)、isb53.48质量份(0.366摩尔)、dpc100.93质量份(0.471摩尔)和作为催化剂的乙酸钙1水合物4.03×10-3质量份(2.29×10-5摩尔)。并使最终聚合温度为250℃,除此以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<比较实验例4>
使用9,9-双(4-(2-羟基乙氧基)苯基)芴(bhepf)80.49质量份(0.184摩尔)、2,2-双(4-羟基苯基)丙烷(bpa)13.23质量份(0.058摩尔)、dpc53.29质量份(0.249摩尔)和作为催化剂的乙酸钙1水合物1.28×10-3质量份(7.25×10-6摩尔)。并使最终聚合温度为260℃,除此以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<比较实验例5>
除了使用9,9-双(4-(2-羟基乙氧基)苯基)芴(bhepf)62.40质量份(0.142摩尔)、isb28.78质量份(0.197摩尔)、dpc73.40质量份(0.343摩尔)和作为催化剂的乙酸镁四水合物7.28×10-4质量份(3.39×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1。
<比较实验例6>
除了使用def27.22质量份(0.107摩尔)、isb59.43质量份(0.407摩尔)、dpc111.69质量份(0.521摩尔)和作为催化剂的乙酸钙1水合物4.53×10-4质量份(2.57×10-6摩尔)以外,与实验例1同样地进行。
所得到的树脂组合物的玻璃化转变温度等以及膜成型并进行拉伸时的拉伸膜的折射率各向异性、相位差比(re450/re550)、膜的韧性等的测定结果列于表1~3。
[表2]
由表1~3可知,本发明的树脂组合物具有正折射率各向异性,膜的韧性等物性也良好,可以说在相位差膜或其它光学膜、透镜等用途中也是有用的。
比较实验例1~3中,膜的韧性不合适;与此相对,实验例中示出的本发明的全部树脂组合物显示出良好的韧性。此外,实验例中示出的本发明的全部树脂组合物的玻璃化转变温度位于优选的90℃以上170℃以下的范围,结果显示出,从熔融加工性与耐热性这两方面考虑取得了平衡。
<针对显示出逆波长分散性的相位差膜的研究>
实验例1~3、实验例7以及8、比较实验例1~5中得到的拉伸膜的光学特性的测定结果汇总于表4。
在假设为显示出逆波长分散性的相位差膜用途的情况下,若将相位差比(re450/re550)为同等程度的实验例1与比较实验例3进行比较,则使用化合物4b作为含芴环单体的实验例1的相位差比(re450/re550)为0.99,使用bcf的比较实验例3的相位差比(re450/re550)为1.00,二者显示出了同等程度的相位差比;但实验例1中含芴环单体的比例较少,为13.2mol%,与此相对,在比较实验例3中含芴环单体的比例较多,为20.0mol%,可以说使用化合物4b的实验例1表现逆波长分散性的效果更高。并且,使用bcf的比较实验例3在膜的韧性方面也有问题。
若将使用化合物4b作为含芴环单体的实验例2与使用bhepf的比较实验例5进行比较,则尽管实验例2中含芴环单体的比例(摩尔分数)较低,但相位差比(re450/re550)为0.80;另一方面,含芴环单体的比例(摩尔分数)高于实验例2的比较实验例5的相位差比(re450/re550)为0.90,因而可以说化合物4b在与bhepf相比为少量的情况下即能够表现出强逆波长分散性。
此外,若将使用化合物4b作为含芴环单体、但其含有比例(摩尔分数)不同的实验例1、实验例2以及实验例3进行比较,则实验例3的相位差比(re450/re550)为0.93、实验例2的相位差比(re450/re550)为0.80,因而可以说,通过改变含芴环单体的含有比例,可以在优选的范围内调整相位差比。
若将含芴环单体的比例(摩尔分数)为同等程度的实验例7与比较实验例3进行比较,则使用化合物7b作为含芴单体的实验例7的相位差比(re450/re550)为0.97,显示出了逆波长分散性;与此相对,使用bcf的比较实验例3的相位差比为1.00,逆波长分散性的表现极弱。由此可以说,使用化合物7b的实验例7表现逆波长分散性的效果高。进一步地,若将使用同一化合物7b作为含芴环单体、其含有比例不同的实验例7和实验例8进行比较,则实验例8的相位差比(re450/re550)为0.54,因此可以说,通过改变含芴环单体的含有比例,可以在优选的范围内调整相位差比。
比较实验例1和比较实验例2均为使用bcf作为含芴环单体的聚碳酸酯,尽管比较实验例1中的bcf含有比例(摩尔分数)大幅高于比较实验例2,但比较实验例1的相位差比(re450/re550)为0.98,显示出了大于比较实验例2的相位差比(re450/re550)的值(比较例2中,该相位差比为0.92),逆波长分散性的表现效率差。进一步地,如由表1所知,比较实验例1的光弹性系数为42×10-12pa-1,与比较实验例2的17×10-12pa-1相比,比较实验例1显示出了大幅增大的光弹性系数值。据认为,其原因在于在共聚单体中使用了芳香性的bpa。基于此,在共聚单体中优选使用非芳香性的单体,据推测,在本发明的树脂组合物的情况下也可以说是同样的情况。同样地,在将使用bhepf作为含芴环单体的比较实验例4与比较实验例5进行比较的情况下,对于在共聚单体中使用芳香性bpa的比较实验例4来说,为了表现出同等程度的相位差比,需要提高bhepf的比例,光弹性系数也增大。
下面,实验例1、3、7、8、13~16、18~22中得到的拉伸膜的光学特性的测定结果汇总于表5。
若将含芴环单体的比例(摩尔分数)为同等程度、并且使用了除两末端以外均具有相同结构的化合物4a与4b的实验例1与实验例13进行比较,则使用了两末端为羟基丙基的化合物4b的实验例1的聚碳酸酯的相位差比(re450/re550)为0.99,与此相对,使用了两末端为乙氧基羰基乙基的化合物4a的实验例13的聚酯的相位差比(re450/re550)为0.36,可知实验例13中表现出了强逆波长分散性。基于此,可认为实验例13中使用的化合物4a表现逆波长分散性的效果高于实验例1中使用的化合物4b,通过减少化合物4a的含有比例,能够在优选的范围内调整相位差比。
此外,同样地,若将使用了除两末端以外均具有相同结构的化合物7a与7b的实验例8与实验例16进行比较,则可知,使用了两末端为羟基丙基的化合物7b的实验例8的聚碳酸酯的相位差比(re450/re550)为0.97、使用了两末端为乙氧基羰基乙基的化合物7b的实验例16的聚酯碳酸酯的相位差比为0.96,二者为同等的;与此相对,实验例16中,含芴环单体的比例(摩尔分数)少。基于此,可以说,实验例16中使用的化合物7a表现逆波长分散性的效果高于实验例8中使用的化合物7b。进一步地,在增加了化合物7a的含有比例的实验例19中,将相位差比(re450/re550)调整至属于特别优选范围的0.83。
对于使用了与实验例13相同的化合物4a的实验例14的聚酯碳酸酯来说,也可以说是同样的,若与使用了化合物4b的实验例3的聚碳酸酯相比,可以说表现逆波长分散性的效果高。在减少了化合物4a的含有比例的实验例15中,逆波长分散性的表现弱,因而在使用化合物4a的情况下,将含芴环单体的比例(摩尔分数)设定在实验例14与实验例15之间的实验例18在更优选的范围内具有相位差比(re450/re550)。
若将含芴环单体的比例(摩尔分数)相同的、使用了化合物7a的实验例19与使用了化合物13的实验例21的聚酯碳酸酯进行比较,则据信尽管聚酯碳酸酯的末端结构不同,但为大致等同的树脂组合物,因而实验例19的相位差比(re450/re550)为0.83,实验例21采取与0.77相近的值。但是,实验例19是通过酯化与聚合的两步制造的,与此相对,实施例20能够通过一步进行聚酯碳酸酯的制造,与使用二乙基酯的化合物7a相比,据信使用反应性优异的二苯基酯的化合物13能够缩短制造工序。在化合物13的含有比例降低的实验例20中,逆波长分散性的表现稍弱,因而,在使用化合物13的情况下,据信例如通过将含芴环单体的比例(摩尔分数)设定在实验例20与实验例21之间,可在更优选的范围内调整相位差比(re450/re550)。
对于除了两末端以外具有相同结构的二乙基酯的化合物4a与二苯基酯的化合物12b来说,也可以说是同样的,使用了化合物12b的实验例22所具有的含芴环单体的比例(摩尔分数)为使用了化合物4a的实验例14与实验例15之间,实验例22的相位差比(re450/re550)也在之间存在,因而可认为逆波长分散性的表现大致同等。但是,在实验例14、实验例15中,是通过酯化与聚合的两阶段制造的,与此相对,实施例20能够以一步进行聚酯碳酸酯的制造,与二乙基酯的化合物4a相比,据信使用反应性优异的二苯基酯的化合物12b能够缩短制造工序。
根据上述内容,在假定为显示出逆波长分散性的相位差膜用途的情况下,含有具有本发明低聚芴重复单元的聚合物的树脂组合物显示出了在低聚芴单体的较少的质量和/或摩尔分数下即能够达成各种物性、正固有双折射、相位差比的情况。
<针对显示出平坦分散性的相位差膜、宽带域零双折射材料进行的研究>
实验例4~6、9~12和17中得到的拉伸膜的光学特性的测定结果汇总于表6。
在使用在侧链具有羟基甲基的低聚芴二醇作为含芴环单体的实验例4~6和实验例9~12和17的树脂组合物中,全部显示出了相位差比(re450/re550)为1.0以上1.06以下的平坦分散性,膜的韧性等物性也良好,因而在显示出平坦分散性的相位差膜用途及其它光学膜中是有用的。
一般来说,如使用了化合物4b的实验例1~3及使用了化合物7b的实验例7和8那样,使用了含芴环单体的树脂组合物根据含芴环单体比例的不同,具有相位差比(re450/re550)大幅变化的倾向。与此相对,实验例6和实验例10为使用了在侧链具有羟基甲基的含芴环单体——化合物3b、isb、chdm的聚碳酸酯,但其具有下述特异性特征:即使改变化合物3b的比例,相位差比(re450/re550)仍为1.04,存在相位差比几乎不会发生变化的含有比例的范围。这并不限于实验例6和实验例10,为使用了在侧链具有羟基甲基的低聚芴二醇的实验例4、实验例5、实验例9、实验例11以及实验例12中的共同特征。因此,使用了具有羟基甲基的低聚芴二醇的本发明的树脂组合物能够在保持平坦分散性的同时对双折射进行控制,由此,通过以适当的摩尔分数使用低聚芴二醇,可期待其在宽带域的零双折射材料中的应用。
此外,在使用了同样具有羟基甲基的低聚芴二醇的实验例4与实验例12、实验例6与实验例10中,具有羟基甲基的低聚芴二醇的比例高的树脂组合物具有折射率高、阿贝值低的倾向。据信这是使用了具有羟基甲基的低聚芴二醇的本发明的树脂组合物中的共同特征,例如在增加具有羟基甲基的低聚芴二醇的比例的情况下,据信可在宽带域显示出零双折射,折射率与阿贝值可调整在更优选的范围,因而可期待其在摄像系统的光学透镜中的应用。
根据上述内容,使用了本发明的在侧链具有羟基甲基的低聚芴二醇的树脂组合物具有能够以任意的比例达成平坦分散性、也容易进行双折射的调节的倾向。
<针对宽带域低双折射材料的研究>
在使用了同样含芴环单体的实验例4与实验例12、实验例6与实验例10中,尽管含芴环单体的比例(摩尔分数)不同,但关于其相位差来,re450/re550与re630/re550这两者几乎未发生变化,可认为在任意的比例下显示出平坦分散。因此,在含芴环单体的比例(摩尔分数)高的区域,推测在宽带域显示出零双折射。此外,在使用了化合物3b的实验例6与实验例10中,化合物3b的比例高的实验例10的折射率高、阿贝值低。
并且,在使用了在侧链具有羟基甲基的低聚芴二醇的实验例4~6和实验例9~12及实验例17的树脂组合物中,全部显示出了相位差比(re450/re550)为1.0以上1.06以下的平坦分散性,膜的韧性等物性也良好,因而在显示出平坦分散性的相位差膜用途或其它光学膜中是有用的。
如上述实验例中所示,在使用了含芴环单体的树脂组合物中,根据含芴环单体的结构的不同,在相同摩尔分数下,具有显示出强逆波长分散性的物质、显示出中等程度的逆波长分散性的物质、显示出平坦分散性的物质,因而可根据目的适当选择含芴环单体、或适当选择摩尔分数等树脂组成。
<芴比例>
表7汇总示出了利用合成例合成的来源于含芴环单体的重复单元的化学结构式与芴比例。需要说明的是,芴比例由下式计算出。其中,下式中的芴骨架的分子量以碳原子13个(不包括氢原子)进行计算。
芴比例(%)=芴骨架的分子量/重复单元的分子量x100
另外,在表8中示出了本发明树脂组合物的代表性合成例与比较实验例6的投料时的单体组成以及树脂组合物中来源于各单体的重复单元的组成的分析值。需要说明的是,树脂组合物中的重复单元的组成由1h-nmr光谱进行计算。
[表8]
如上所述,据信通过提高重复单元中的芴所占的比例,能够有效地表现出所期望的光学特性,由于合成例中合成的单体的芴比例均为50%以上,因而可认为使用了它们的本发明的树脂组合物适于有效表现出所期望的光学特性。与此相对,对于比较实验例中使用的bcf、bhepf来说,来源于其的重复单元中的芴比率小于50%,对于表现出所期望的光学特性是无效的,为了表现出所期望的光学特性,认为需要提高具有芴的单体的使用比例。另一方面,对于比较实验例6中使用的def,来源于其的重复单元中的芴比例高,认为具有少量既能够表现出所期望的光学特性的可能性。但是,如表8所示,本发明的树脂组合物中,投料时的单体组成与树脂组合物中的重复单元的组成的摩尔比的偏差为0.3%以内,与此相对,在使用了def的比较实验例6的聚碳酸酯树脂组合物中,偏差为1.0%。关于这一点,据认为,在聚合时def会变化为低沸点的分解物,投料的def的摩尔比与聚合后的树脂组合物中的def来源的重复单元的摩尔比具有较大偏差。因此,难以调整为所期望的光学特性,具有缺乏光学特性的再现性的缺点。此外,使用def制造的树脂组合物还具有tg倾向于降低的问题。
<偏光atr分析与构型的能量和角度的计算>
为了研究低聚芴的化学结构与光学特性的关系,如下对来源于低聚芴单体的重复单元的特定构象(构型)的能量进行计算并对该构象中芴环与主链所成的角度进行计算。
关于软件,在am1法中使用美国wavefunction社制造的pcspartanpro1.0.5(windows(注册商标)32位版本),在b3lyp/6-31g*法中使用美国gaussian社制造的gaussian03rev-b.05(windows(注册商标)32位版本)。需要说明的是,聚焦判定值等与计算精度相关的输入值全部使用该软件的默认值。
此处,关于来源于二醇单体的重复单元,假设为通过碳酸酯键进行聚合的树脂,对于将两羟基碳酸甲酯化而成的结构进行计算;关于来源于二酯单体的重复单元,假设为通过酯交换反应进行聚合的树脂,对于将两酯基甲基酯化而成的结构进行计算。
首先,对于来源于化合物3b、化合物7b、化合物4b的结构中的单侧的侧链,如下所示,将下述键接a与键接b所成的二面角为180°(即侧链为反式构象)以及为60°和-60°(即侧链为两种的歪扭构象)作为初期结构,通过am1法求出平衡结构与其能量(生成热),该键接a是芴9位碳原子与在相邻的芴侧与芴键合的碳原子间的键接,该键接b是与芴键合的侧链的原子同与该原子结合的主链的原子间的键接。此处,将各化合物中的具有与芴9位碳原子键合的碳酸甲酯或甲基酯的取代基称为侧链。设这3种构象中最稳定的构象为0(kj/mol),将此时的能量差(kj/mol)记载于表10。需要说明的是,在两种歪扭构象为对称形的情况下,为相同值。此外,在能量差为4kj/mol以上的情况下,将为0(kj/mol)的构象作为稳定构象。进一步将am1法的结果作为初期结构,使用b3lyp/6-31g*法同样地求出能量差。
接下来,使用am1法对于各单体中存在的2个侧链的两方为反式构象、以及为两种歪扭构象的构象异构体的能量差进行计算(表11~13)。此外,对于反式构象与歪扭构象(两种歪扭构象中的稳定构象),计算出主链与芴环所成的角度并进行记载。
需要说明的是,主链与芴环所成的角度如下来确定。首先,将两末端的甲基碳原子彼此连接的直线作为主链方向,将通过芴的3位、6位、9位碳原子的平面作为芴平面。此时,与主链方向交叉的芴平面上的直线无限存在,但与主链方向的角度最小的芴平面上的直线是唯一确定的。将该角度作为主链与芴环所成的角度。
[化108]
首先,在表9中示出了由含有化合物7b、化合物4b、化合物3b、化合物9b、化合物2b作为含芴环单体的树脂组合物(分别为实验例8、2、6、9和12中得到的树脂组合物)制作的膜的偏光atr分析的结果以及有无逆波长分散性。在全部膜中,在来源于羰基取向的1245cm-1的峰中,确认处于明确表现出差示光谱的状态。即,在1245cm-1的峰强度(在实验例8、2、6和9中的1170cm-1、1420cm-1间、实验例12中的1080cm-1、1280cm-1间以直线引出基线时的最强峰的强度)中,在拉伸方向与垂直方向的强度比(2色比)表示大值(拉伸方向的强度/垂直方向的强度>1.2)的情况下定义为判定○,全部膜为○。它们之中,有明确表现出与未表现出来源于芴环取向的740cm-1的吸收的差示光谱的情况。即,740cm-1的峰强度(在全部实验例中在715cm-1、830cm-1间以直线引出基线时的最强峰的强度)中,在拉伸方向与垂直方向的强度比(2色比)表示大值(拉伸方向的强度/垂直方向的强度>1.2)情况判定为○、将1.2以下的情况定义为×,此时实验例8和2为○,实验例6、9和12为×。确认该判定结果与逆波长分散性的相关性,结果在明确表现出来源于芴环取向的740cm-1的吸收的差示光谱的情况下(即2色比为判定○的情况)具有逆波长分散性(re450/re550≦1.0),在未表现出该差示光谱的情况下(2色比为判定×的情况)无逆波长分散性(re450/re550>1.0)。
[表9]
接下来,对于化合物7b、化合物4b、化合物3b,在表10中示出了利用am1法和b3lyp/6-31g*法对来源于这些化合物的重复单元的反式、歪扭的各构象的生成能量差进行计算的结果。如上所述,假设各二醇单体为可在聚碳酸酯树脂中采取的结构时,为进行简化,将单体的两末端假设为碳酸甲酯进行计算。并且求出在各化合物中所具有的2个末端中仅一个为反式构象的情况与为歪扭构象的情况下的差。此处,在能量差为4kj/mol以上的情况下,将能量(生成热)低的构象作为稳定构象。
[表10]
如表10所示,关于将am1法与b3lyp6-31g*法合用得到的结果,可知化合物7b和4b的稳定构象为反式构象,另一方面,化合物3b的稳定构象为歪扭构象。
接下来,对于下述[x]组中示出的各化合物,在表11~14中示出了通过am1法对来源于各化合物的重复单元的反式、歪扭的各构象的能量差进行计算的结果。关于二醇单体,对于将两羟基碳酸甲酯化得到的结构进行计算;关于二酯单体,对于将酯基制成甲基酯的结构进行计算。另外,此处,为了对各重复单元的聚合物中的立体结构进行推测,使各化合物中存在的2个侧链的两者进行变化。即,表11~14中的反式构象是指各单体中存在的2个侧链的两者为反式构象的结构,歪扭构象是指各单体中存在的2个侧链的两者为歪扭构象的结构。此外,歪扭构象有60°和-60°两种,针对2个侧链的两者为60°和-60°者两种情况进行计算。此处,在能量差为4kj/mol以上的情况下,能量(生成热)低的构象为稳定构象。
此外,对于反式构象与歪扭构象(两种歪扭构象中的稳定构象),将两末端甲基的碳原子彼此连结的直线确定为主链方向,将其与芴环平面的角度(主链方向和与其相交的平面上的直线所成的角度的最小值)记载为主链与芴环所成的角度。
需要说明的是,对于表13中示出的重复单元,由于在对位取代的亚苯基与芴的9位碳原子键合,它们即使二面角发生变化,侧链的位置关系也不变,因而不存在反式构象、歪扭构象。因此,对于所认为的一种结构,求出主链与芴环所成的角度。
[化109]
[x]
[表11]
[表12]
[表13]
需要说明的是,表13中的*是指下述官能团。
[化110]
[表14]
如上述实验例1~24所示,对于本发明的包含具有2价低聚芴作为重复单元的聚合物的树脂组合物,根据低聚芴单体所具有的末端基团种类的不同,其特性大不相同。关于该现象,可由偏光atr分析结果以及平衡结构的计算结果明确。
表9中,根据使用末端基团为羟基丙基的低聚芴二醇(化合物7b和化合物4b、上述式(1)中的r1=r2=亚丙基)制造的树脂组合物的偏光atr分析的结果可以说为如下情况。与其说对来源于主链中所含有的碳酸酯键的羰基的差示光谱进行了观测,不如可以说主链在拉伸方向进行取向,进一步地,根据还观测到芴环的差示光谱的情况,确认到芴环相对于聚合物的主链垂直取向。
另一方面,在末端基团为羟基甲基的低聚芴二醇(化合物3b、化合物9b、化合物2b、上述式(1)中的r1=r2=亚甲基)的情况下,尽管确认到了羰基的差示光谱,但对于芴环却并未观测到明确的差示光谱。由于观测到了羰基的差示光谱,因而主链在拉伸方向取向;另一方面,由于未观测到芴环的差示光谱,因而可以说芴环相对于拉伸方向(即主链的取向方向)与垂直方向处于大致等价的状态。
其结果,根据各重复单元的平衡结构的计算结果可推测出如下内容。
对于末端基团为羟基丙基的化合物7b和化合物4b,在使单侧末端基团旋转的表10的计算中为下述结果:在am1法中,反式构象与歪扭构象的能量(生成热)大致同等;在作为更高精度的计算方法的b3lyp/6-31g*法中,反式构象为稳定的。可以说,为了表现出高逆波长分散性,芴环采取与主链正交的构象是重要的,如下所述,在计算中反式构象稳定的化合物7b和化合物4b容易采取芴环相对于主链正交的构象,因而在偏光atr分析中确认了芴环的取向,并且可认为得到了高逆波长分散性。
另一方面,在末端基团为羟基甲基的化合物3b中,在am1法、b3lyp/6-31g*法的任意一种中,歪扭构象均为稳定的。由于在歪扭构象中芴环相对于主链取倾斜配置,因而可认为,不管是否具有芴环,均观测不到逆波长分散性。根据该结果,在化合物3b的偏光atr分析中未观测到芴环的显著差示光谱,可以说明无逆波长分散性,可认为其原因在于其具有即便使单体量变化,波长分散性也不发生变化的特异性质。
[化111
如表11所示,在树脂组合物含有具有式(1)中的r1和r2为亚甲基以外的基团的低聚芴作为重复单元的聚碳酸酯聚合物的情况下,与化合物7b和化合物4b同样地,在am1法中,反式构象与歪扭构象的能量(生成热)大致同等、或者反式构象为稳定构象。经碳酸甲酯进行末端基团改性的化合物7b的结构式以及反式构象与歪扭构象的空间填充模型示于图1(a)~(c)中。可知在末端基团中,与芴环离开2个原子的亚甲基(结构式中以及空间填充模型中被圆包围的部分)在反式构象中未见到较大的立体排斥;另一方面,在歪扭构象中,与相邻的芴环以及相邻的亚甲基的氢原子产生排斥,呈不稳定化。
如表9所示,化合物7b和化合物4b在偏光atr分析中观测到羰基和芴环的取向,显示出逆波长分散性。关于这些化合物,根据表11所示的am1法的计算结果,反式构象与歪扭构象的稳定性均为同等程度、或反式构象为稳定构象,歪扭构象不是稳定构象。即,在am1法中歪扭构象不是稳定构象的情况下,可认为芴环占据与主链正交的反式构象的重复结构的作用大,显示出逆波长分散性。进一步地,在式(1)中的r1和/或r2是碳原子数为2以上的基团的情况下,更具体地说,末端基团中,在与芴环离开2个原子的位置存在亚甲基或者空间位阻大于亚甲基的基团的情况下,由于空间位阻,歪扭构象不利,因而歪扭构象不是稳定构象。
根据表11所示的计算结果,对于化合物7b,主链与芴环所成的角度在反式构象中为89.3°、在歪扭构象中为47.8°;对于化合物4b,在反式构象中为74.5°、在歪扭构象中为66.0°。关于偏光atr分析的差示光谱,在角度为45°附近时未观测到,角度越接近90°,越观测到更大的差示光谱。由于在偏光ir分析中观测到差示光谱,因而对于化合物7b和化合物4b而言,反式构象的作用大。
在包括在偏光atr分析中观测到芴环的取向、显示出逆波长分散性的化合物7b和化合物4b的情况下,表11中的反式构象中,主链与芴环所成的角度全部为60°以上。在角度为45°附近时,在偏光atr分析中未观测到芴环的差示光谱,也未表现出逆波长分散性。另一方面,在反式构象的角度为50°以上、优选为60°以上、更优选为70°以上时表现出逆波长分散性,可以预计表11所示的重复结构均表现出逆波长分散性。
另一方面,在为含有表12所示的具有式(1)中的r1与r2为亚甲基的低聚芴作为重复单元的聚碳酸酯聚合物的树脂组合物的情况下,与化合物3b同样地,在am1法中,歪扭构象为稳定构象。经碳酸甲酯进行末端基团改性的化合物3b的结构式以及反式构象与歪扭构象的空间填充模型示于图2(a)~(c)中。可知在末端基团中,与芴环离开2个原子的氧原子(结构式中以及空间填充模型中被圆包围的部分)在歪扭构象也未发生在化合物7b中对应的亚甲基中见到的排斥,因而歪扭构象呈相对稳定化。
如表9所示,化合物3b、化合物2b和化合物9b在偏光atr分析未观测到芴环的取向,未显示出逆波长分散性。根据表12所示的am1法的计算结果,这些化合物中均是歪扭构象为稳定构象。即,在具有歪扭构象为稳定构象的重复结构的树脂组合物中,由于芴环相对于主链倾斜,因而认为未显示出逆波长分散性。进一步地,在通式(1)中的r1和r2是碳原子数为1的基团、例如是亚甲基的情况下,由于歪扭构象的空间位阻减轻,因而歪扭构象为稳定构象。
此外,在表12中,在为化合物3b、化合物2b和化合物9b的稳定构象的歪扭构象中,关于主链与芴环所成的角度,在化合物3b中为49.1°、在化合物2b中为59.9°、在化合物9b中为55.5°。在角度为45°附近时未观测到偏光ir分析的差示光谱。在这些化合物中,由于未观测到芴环的差示光谱,因而可认为歪扭构象优先。
在包括在偏光atr分析中未观测到芴环的取向、未显示出逆波长分散性的化合物3b、化合物2b和化合物9b的情况下,表12中的歪扭构象中,主链与芴环所成的角度全部小于60°。在角度为45°附近时,在偏光atr分析中未观测到芴环的差示光谱、也未表现出逆波长分散性。在歪扭构象的角度小于60°、优选为55°以下、更优选为50°以下时,逆波长分散性的表现弱,可以预计表12所示的重复结构均未表现出逆波长分散性或逆波长分散性弱。
如表13所示,对于在对位取代的亚苯基与芴的9位碳原子键合的重复单元,在已知具有逆波长分散性的bpef以及具有低聚芴基的重复单元中,角度全部为70°以上。由此可以说具有逆波长分散性。
如表14所示,在二酯单体中也可以说是同样的。式(1)中的r1与r2为亚甲基的化合物(化合物26)中,歪扭构象比反式构象稳定16.9kj/mol(am1法),可认为未得到逆波长分散性。另一方面,对于在与芴环离开2个原子的位置具有亚甲基或具有空间位阻大于亚甲基的基团的化合物,在am1法中,反式构象均为稳定构象,可认为表现出逆波长分散性。
此外,在对表11的二醇单体与表14的二酯单体进行比较的情况下,在am1法中,二酯单体的反式构象更为稳定,可认为在全部构象中的反式构象的作用更大,因而可认为表现出更强的逆波长分散性。例如,若在r3为亚乙基的化合物中进行比较,则在二醇单体中,在r1和r2为亚乙基的化合物(化合物18)或为亚丙基的化合物(化合物7b)中,反式构象分别为2.4kj/mol、0.8kj/mol,是稳定的;在r1和r2为亚乙基的二酯单体(化合物7a)中,反式构象为4.7kj/mol,是稳定的,二酯单体中的反式构象的稳定性高。此外,关于r3为亚甲基的化合物,r1和r2为亚丙基的二醇单体(化合物4b)的歪扭构象为3.3kj/mol,该二醇单体是稳定的;r1和r2为亚乙基的二酯单体(化合物4a)的反式构象为3.4kj/mol,该二酯单体是稳定的。
如上所述,在与芴环离开2个原子的位置存在亚甲基的情况下,如下图所示,在反式构象中未见到大的立体排斥,而另一方面,在歪扭构象中,由于与芴环上的氢原子的立体排斥以及与位于与芴环9位碳原子直接键合的碳原子上的氢原子的立体排斥,可认为反式构象更为稳定。该效果在该亚甲基具有取代基的情况下更为显著。
[化112]
尽管参照特定实施方式对本发明进行了详细说明,但对本领域技术人员来说,已知可在不脱离本发明的精神和范围的情况下对本发明施以各种变更、修正。本申请基于2012年10月16日提交的日本专利申请(日本特愿2012-228946)、2013年6月21日提交的日本专利申请(日本特愿2013-130882),以参考的形式将其内容引入本说明书。
- 还没有人留言评论。精彩留言会获得点赞!