混合四模板磁性印迹聚合物的制备方法及其应用与流程
本发明属于化学合成、分析化学、超分子化学及食品安全领域,涉及磁性分子印迹聚合物的制备及应用,具体地说是一种混合四模板磁性印迹聚合物的制备方法及其应用。
背景技术:
戊唑醇、联苯三唑醇和烯唑醇等均属三唑类杀菌剂,为广谱内吸杀菌剂,主要用于防治植物病害,对病原微生物有杀死作用或抑制生长作用,因其高效、广谱、低毒以及长效性而广泛地应用于农作物等病虫害的防治,同时该类农药在农作物、土壤和水体中的残留也给生物安全和人体健康带来了负面影响。
胺菊酯、三氟氯氰菊酯和四氟甲醚菊酯等都属于拟除虫菊酯类杀虫剂,是高效、广谱、速效杀虫剂、杀螨剂,以触杀和胃毒作用为主,无内吸作用。它们能有效地防治棉花、果树、蔬菜、大豆等作物上的多种害虫,也能防治动物体上的寄生虫。
扑草净、莠去津和莠灭净等都属于三嗪类除草剂,三嗪类除草剂作为预防农田杂草生长的高效除草剂在世界范围内广泛使用,由于其用量较大、性质较稳定、残留时间较长,而且具有较高水溶性,因此在使用过程中可能对土壤、农作物、地表水和其它饮用水源造成污染,从而对人类健康和环境造成严重的危害。研究表明:该类化合物及其降解产物在水体中的残留会损害免疫系统而危害人体动植物和水生生物的健康。另外,这类化合物可能引起人类癌症及先天性缺陷,同时能够干扰荷尔蒙的正常功能,世界多国已将其列入内分泌干扰剂化合物名单。
克百威和灭多威等属于氨基甲酸酯类农药,是一种高效、广谱杀虫剂,其杀虫效果显著、残留期短、代谢迅速,因此作为农作物防治虫害的常用农药。但是该类农药具有一定的毒性,对人体的急性毒作用表现在可以抑制体内乙酰胆碱酯酶,使他失去分解乙酰胆碱的功能,造成组织内乙酰胆碱的蓄积而中毒,影响正常的神经冲动传递活动,严重危害人体健康,在当前中国已经引发了严重的“食品安全危机”。
在我国,三唑类杀菌剂、菊酯类除虫剂、三嗪类除草剂和氨基甲酸酯类农药这四类农药的产量和使用量都很大,对生态环境和食品安全造成极大的危害。为确保我国的生态环境及食品安全,建立针对此四类农药的快速、高效、准确的检测方法是环境及食品安全分析工作者的迫切任务。而由于这些物质在自然环境中的残留浓度很低,并且存在多种干扰物质,常规的监测分析方法难以对其进行有效的测定。另外,复杂基体所带来的基质效应也对此四类农药的分析提出了挑战,如何在样品前处理过程中尽量减少基质效应对分析效果的影响是研究者面临的重大挑战。因此,探索一种识别性强、干扰小的富集方法具有十分重要的意义。而分子印迹技术和磁性材料的结合,不仅克服了传统固相萃取的缺点,还解决了分子印迹固相萃取过程中装柱繁琐的过程,从而具有对目标物特异性选择的特点,很好地分离富集复杂样品中的痕量分析物,还降低检测限,提高分析的精密度和准确性,为痕量组分的富集和分析提供极大的方便。
分子印迹技术(Molecular imprinted technology, MIT) 就是制备分子印迹聚合物的技术,也叫模板技术,是在模拟生物体内抗原与抗体之间相互作用的基础上发展起来的一种新型技术。通过模板分子、功能单体和交联剂的相互作用从而合成的具有三维空间结构的“受体”,其对模板分子有特异“记忆”功能,体现了高度的选择性、特殊的亲和性及卓越的分子识别能力。分子印迹聚合物(Molecular Imprinted Polymers, MIPs)的制备是将模板分子与功能单体、交联剂和引发剂等在特定的分散体系中进行共聚合,制得高交联刚性聚合物。然后通过物理或化学的方法除去MIPs中的模板分子,得到具有确定空间构型的空穴和功能基团在空穴内精确排布的聚合物。因此,MIPs具有从复杂样品中选择性提取目标分子或与其结构相近的某一类化合物的能力,适合作为固相萃取填料、固相微萃取涂层以及分子印迹薄膜来分离富集复杂样品中的痕量分析物,克服样品体系复杂、预处理繁琐等不利因素,达到样品分离净化的目的。
磁性材料一直是科学研究的重要领域,超顺磁性和饱和磁化强度是磁性材料特有的磁特性,磁性聚合物微球作为一种新型的功能材料,在生物医学、工业应用等方面具有很大的应用前景。
近年来,磁性微球( magnetic nanoparticles,MNPs) 被广泛应用于免疫分析、固定化酶、靶向给药、细胞分离、磁共振造影、亲和色谱和环境检测等领域,在化学、生物、医学应用等领域发挥了越来越重要的作用。MNPs 是一种由磁性材料和非磁性聚合物材料复合而成的新型功能微球,通常由无机磁性材料和有机聚合物材料构成,其中磁性成分主要是铁、钴、镍,或者它们的氧化物以及合金等。由于镍、钴等存在毒性,在生物、医药等领域的应用受到严格限制,因此目前以低毒、稳定、价廉易得的四氧化三铁( Fe3O4) 作为磁性成分的研究最多。MNPs 具有超顺磁性,可以很方便地在外加磁场作用下进行导向或分离。另外,MNPs 还具有分散性好,不易凝聚,与生物分子结合迅速等特点。
大多数MIPs的制备都采用单一模板分子,这样合成的聚合物不能同时对多类多种分析物具有较高的亲和力。而在实际检测中往往不是单一一种或一类分析物,所以采用单一模板合成的 MIPs 对其它种类的化合物的吸附不能显示高的吸附量和高的选择性。与单一类型印迹分子相比,两种及多种不同印迹分子的组合能够为被分析物的重新结合和释放创造更合适的印迹空穴。混合模板制备的 MIPs 可缩短检测时间降低检测成本,通过一次聚合可同时测定多类多种物质,因此有望得到更好的应用。本发明通过磁性材料制备的混合四模板磁性印迹聚合物(MMIPs)不仅可以同时分离测定多类多种农药残留,而且可以利用聚合物的磁性快速实现磁萃取和分离,使得样品前处理过程更加方便、快速。
技术实现要素:
本发明要解决的技术问题,是提供一种混合四模板磁性印迹聚合物的制备方法,以四种化合物作为混合四模板物质,在聚合过程中添加经改性的磁性微球,合成能同时对三唑类物质、菊酯类物质、三嗪类物质和氨基甲酸酯类物质具有特异性识别的磁性分子印迹聚合物;制备过程易于控制、便于功能设计、所制混合四模板磁性印迹聚合物稳定性好,强度高,耐酸碱,不需要研磨和装柱,对三唑类、菊酯类、三嗪类物质和氨基甲酸酯类物质的吸附效率高;
本发明的另外一个目的,是提供上述方法所制混合四模板磁性印迹聚合物的一种应用。
为解决上述技术问题,本发明所采取的技术方案是:
一种混合四模板磁性印迹聚合物的制备方法,它按照如下步骤顺序进行:
⑴磁性微球的制备及改性
①磁性微球制备
将摩尔比为1-3︰1的FeCl3·6H2O和FeCl2·4H2O在碱性条件下, 以水为溶剂,于20-90℃下通过共沉淀法制备Fe3O4,作为磁性微球;
②磁性微球改性
以氨水作为催化剂,硅酸四乙酯作为硅源,在Fe3O4表面包覆一层SiO2, 得到Fe3O4@SiO2磁性复合微球;以硅烷偶联剂KH-570为表面活性剂对Fe3O4纳米颗粒进行修饰改性,赋予Fe3O4粒子表面亲有机的反应活性点, 最终得到改性后的Fe3O4@SiO2-KH570磁性微球A ;
⑵溶解
分别各取一种三唑类化合物、菊酯类化合物、三嗪类化合物和氨基甲酸酯类化合物,作为四模板物质,溶于致孔剂中,加入功能单体,得B;
其中:所述三唑类化合物、菊酯类化合物、三嗪类化合物和氨基甲酸酯类化合物的摩尔比为1-2:1:1-2:1;
所述功能单体,与所述四模板物质总量间的摩尔比为2-10︰1
所述四模板物质与致孔剂的质量体积比为1 mmol︰50-150 mL ;
⑶预聚合
将B于频率200Hz下超声30-60min,在4℃冰箱中放置8-14h,得C;
⑷聚合
向C中加入A、交联剂乙二醇二甲基丙烯酸酯和引发剂偶氮二异丁腈,于频率200Hz下超声10-30min,再通入高纯氮气至氮气充满容器,将容器密封后得D;
将D置于50-75℃的恒温水浴振荡器中,于150 rpm下振荡反应18-24h,得E;
⑸模板洗脱
将E磁分离得到聚合物产物F,用甲醇-乙酸混合溶液脱除混合四模板物质,然后用甲醇浸泡除去过量乙酸,得G;
⑹干燥
将G于60℃下烘干至恒重,得H。
作为本发明的限定:
(一)
所述三唑类化合物为联苯三唑醇;
所述菊酯类化合物为三氟氯氰菊酯;
所述三嗪类化合物为莠去津;
所述氨基甲酸酯类化合物为克百威;
所述功能单体为α-甲基丙烯酸或丙烯酰胺;
所述致孔剂为乙腈;
(二)所述交联剂的摩尔量,与所述三唑类化合物、菊酯类化合物、三嗪类化合物和氨基甲酸酯类化合物的总量间的摩尔比为15-40︰1;
当交联剂用量较低时,制备出来的聚合物交联度太低,没有一定的刚性,难以形成稳定的识别空腔,导致的吸附容量降低;相反,交联度过高,进入孔穴的通道就会过于坚硬,无一定的弹性,造成模板分子很难进入孔穴,同时也不利于模板分子的洗脱;
(三)所述引发剂的摩尔量,与所述三唑类化合物、菊酯类化合物、三嗪类化合物和氨基甲酸酯类化合物的总量间的摩尔比为0.4-1.2︰1;
引发剂的用量过大,则浪费试剂,引发剂的用量过小,则不能引发聚合反应。
(四)所述步骤(4)中A的质量,与步骤(2)中所述三唑类化合物、菊酯类化合物、三嗪类化合物和氨基甲酸酯类化合物的总摩尔数之比为0.1-2g︰1mmol。
(五)所述甲醇-乙酸混合溶液中,甲醇与乙酸的体积比为9︰1。
本发明还提供了混合四模板磁性印迹聚合物的一种应用,它用于特异性吸附三唑类物质、菊酯类物质、三嗪类物质和氨基甲酸酯类物质。
作为本发明混合四模板磁性印迹聚合物应用的限定,它用于对食品、饲料或其它样品中三唑类、菊酯类农药、三嗪类农药和氨基甲酸酯类农药残留分析时,通过吸附作用实现样品的净化和残留农药的富集。
本发明的制备方法作为一个整体,过程易于控制、便于功能设计等优点,制备的混合四模板磁性印迹聚合物微球具有对三唑类、菊酯类物质、三嗪类物质和氨基甲酸酯类物质特异性识别位点,选择性强,稳定性好、吸附量较大,操作简便。
由于采用了上述的技术方案,本发明与现有技术相比,所取得的技术进步在于:
本发明提供的混合四模板磁性印迹聚合物具备以下优点:
①稳定性好,强度高,耐酸碱,聚合物上具有多种特异性的空间位点,可实现特异性吸附;
②使用时不需要研磨、过筛和装柱等繁琐的过程;
③可同时对三唑类、菊酯类、三嗪类和氨基甲酸脂类物质进行特异性吸附,且吸附效率高,吸附量较大。
本发明适用于制备混合四模板磁性印迹聚合物,所制产品可进一步应用于食品、饲料及其它样品中三唑类、菊酯类、三嗪类和氨基甲酸酯类农药残留分析时样品的富集和净化。
附图说明
图1为联苯三唑醇化学结构式;
图2为三氟氯氰菊酯化学结构式;
图3为莠去津化学结构式;
图4为克百威化学结构式;
图5为实施例1中混合四模板磁性分子印迹聚合物的印迹原理示意图;
图6为不同功能单体体系的紫外吸收光谱图(a-混合四模板物质-MAA体系,b-混合四模板物质-AM体系;其中:图a和图b中C为混合四模板物质吸光值;E1为混合物体系实际测定吸光值;E2为混合物体系理论吸光值);
图7不同比例的功能单体与混合四模板物质对预组装体系紫外光谱的影响(a-不同比例C与MAA混合物的紫外光谱;b-图a中混合物最大吸收波长和对应吸光值变化曲线;其中:图a中C为混合四模板物质,图a右上角标注为摩尔比,图b中1-6分别是C与MAA摩尔比为1︰0-1︰10);
图8不同致孔剂对体系紫外光谱的影响(a-混合四模板物质-MAA-乙腈体系,b-混合四模板物质-MAA-甲醇体系;其中:图a和图b中C为混合四模板物质);
图9为四氧化三铁(Fe3O4)的红外吸收光谱图;
图10为包裹着二氧化硅的四氧化三铁(Fe3O4@SiO2)的红外吸收光谱图;
图11为混合四种模板磁性分子印迹聚合物(MMIP)的红外吸收光谱图;
图12为混合四种模板磁性非分子印迹聚合物(MNIP)的红外吸收光谱图;
图13为实施例1中磁性印迹聚合物在不同放大倍数下的透射电子显微镜(TEM)图;
图14为实施例1制备的混合四模板磁性印迹聚合物对模板分子及其结构类似物竞争性吸附能图;
图15为11种农药的紫外光谱扫描图;
图16为以甲醇-水为流动相时混合标准溶液的分离色谱图;
图17为小麦提取液未经MMIP处理的高效液相色谱图;
图18为小麦提取液经MMIP处理后的洗脱液的高效液相色谱图;
图19为小麦提取液加标经MMIP处理后的洗脱液的高效液相色谱图;
图14和图19中:1-灭多威;2-克百威;3-莠灭净;4-莠去津;5-扑草净;6-联苯三唑醇;7-戊唑醇;8-烯唑醇;9-胺菊酯10-三氟氯氰菊酯;11-四氟甲醚菊酯;
本发明下面将结合说明书附图和具体实施例作进一步详细说明。
具体实施方式
下述实施例中所使用的试验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业渠道得到。
实施例1 一种混合四模板磁性印迹聚合物的制备方法
本实施例所提供的混合四模板磁性印迹聚合物的制备方法,印迹反应原理见图5,该制备方法按照如下的步骤顺序进行:
(11)磁性微球的制备及改性
①分别称取0.018mol FeCl3·6H2O和0.01mol FeCl2·4H2O,放入250 mL的三口烧瓶中,然后向其加入80 mL的超纯水,向其中通入N2,在800 rpm下连续搅拌,于80℃水浴温度下,逐滴加入10 mL浓氨水,熟化30 min,用磁铁分离产物;
使用除氧超纯水将产物洗至中性,用乙醇再洗3次,最后在50℃真空条件下干燥24 h,得到试验用Fe3O4;
②称取0.3 g Fe3O4,加入到250 mL三口瓶中,向其中分别加入4 mL超纯水和50 mL异丙醇,超声混匀20 min。在800 rpm连续机械搅拌下,加入5 mL氨水,再逐滴加入2 mL 硅酸四乙酯,室温条件下反应12 h后,磁铁分离产物,超纯水洗涤产物至中性后,再用乙醇洗3次,之后于50℃真空干燥24 h,得到Fe3O4@SiO2。
称取0.2 g Fe3O4@ SiO2,加入到250 mL三口瓶中,加入50 mL甲醇,超声混匀10 min。在550 rpm连续机械搅拌下,逐滴加入3 mL KH570,室温下反应24 h后,磁铁分离产物,乙醇和水交替洗涤产物(最后用乙醇洗)数次后,于50℃真空干燥24 h,得到试验用Fe3O4@ SiO2-KH570,记为A1;
(12)溶解
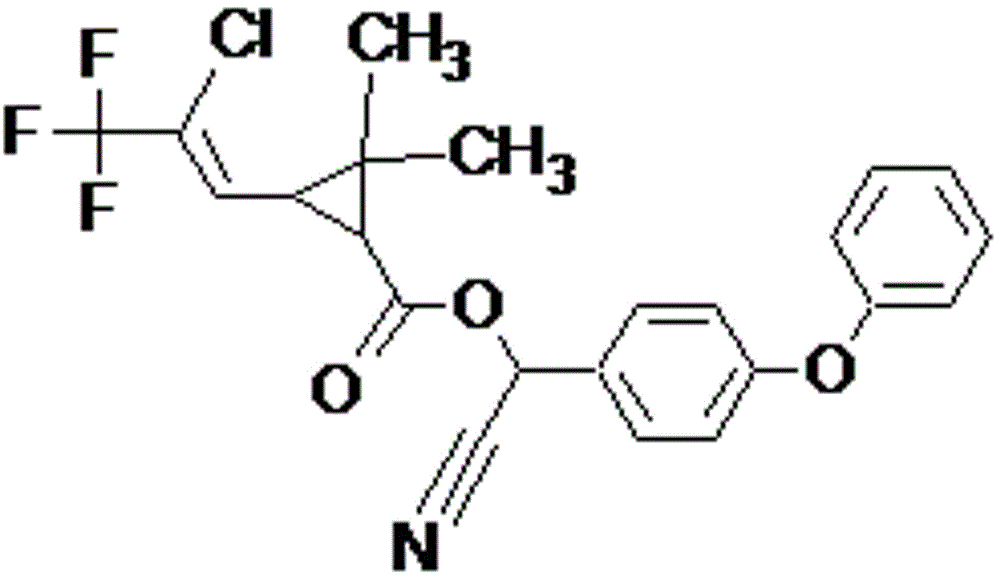
分别将0.15 mmol的联苯三唑醇、0.1 mmol的三氟氯氰菊酯、0.15mmol莠去津和0.1 mmol克百威组成的混合四模板物质溶于50mL致孔剂乙腈中,再加入2.4 mmol的功能单体α-甲基丙烯酸,得B1;联苯三唑醇、三氟氯氰菊酯、莠去津与克百威的化学结构式参见图1-图4;
(13)预聚合
将B1置于频率200Hz下超声30min,放在4℃冰箱中12h,得C1;
(14)聚合
向C1中加入0.1 g Fe3O4@SiO2-KH570磁性微球A1、8 mmol交联剂乙二醇二甲基丙烯酸酯和0.25 mmol引发剂偶氮二异丁腈,在频率200Hz下超声脱气15 min,通入氮气10 min至充满容器,将容器密封后得D1;
将D1置于60℃恒温水浴振荡器中,150 rpm振荡反应24 h,得E1;
(15)模板洗脱
将E1用磁铁磁分离得到聚合物产物F1,再用体积比为9︰1的甲醇-乙酸混合溶液脱除模板物质,然后用甲醇浸泡除去过量乙酸,得G1;
⑴ 干燥
将G1于60℃真空干燥至恒重,得H1;
本实施例制备过程易于控制,便于功能设计,成本低,磁性微球稳定性好、强度高,耐酸碱,印迹聚合物微球具有特异性的空间位点,可实现特异性吸附。
实施例2-5 混合四模板磁性印迹聚合物微球的制备方法
实施例2-5分别为一种混合四模板磁性印迹聚合物微球的制备方法,它们的制备过程与实施例1相同,不同之处仅在于:相应的技术参数有所不同,具体的结果见表1。
表1 混合四模板磁性印迹聚合物微球技术参数
实施例2-5制备过程易于控制,成本低,印迹聚合物无需研磨和装柱,微球稳定性好、强度高、耐酸碱,印迹聚合物具有特异性的空间位点,可实现特异性吸附。
实施例6 功能单体的选择及模板分子与功能单体比例确定试验
本实施例对聚合过程中,功能单体的选择及模板物质与功能单体的比例确定进行了探究。本实施例采用了紫外光谱分别对混合四模板物质-MAA体系及混合四模板物质-AM体系进行了紫外光谱扫描。
分别配制一定浓度的MAA-乙腈溶液和AM-乙腈溶液以及混合四模板物质-乙腈溶液,同时配制功能单体与模板物质的混合乙腈溶液,使其摩尔比分别为1:0,1:2,1:4,1:6,1:8,1:10,在波长190-400nm之间进行紫外光谱扫描。
模板分子与功能单体若不发生作用则在同一波长下紫外吸光值是两种物质吸光值的加和,即混合物的理论吸光值;而混合物的实际吸光值为混合物溶液在同一波长下实际测定的紫外吸光值。实际测定值与理论吸光值差值越大,表明模板分子与功能单体相互作用也越大。
由图6可以看出,混合物实际测定吸光值远远小于混合物理论吸光值,这充分说明混合四模板物质与功能单体发生了相互作用,且从吸光值差值上能够看出混合四模板物质与功能单体相互作用强度较大。从最大吸收峰能看出混合物的最大吸收波长向长波段发生了移动,即发生了红移,这是因为混合四模板物质可与功能单体形成氢键,氢键的形成使吸收峰的波长向长波段方向移动。由结果可知,混合四模板物质-MAA实际测定值与理论值差值为0.207,混合四模板物质-AM实际测定值与理论值差值为0.173。从差值大小上明显表明,混合四模板物质与MAA结合力较大,充分证明了MAA作为功能单体合成的磁性分子印迹聚合物对混合四模板物质具有更好的稳定性和特异识别能力。
在制备磁性分子印迹聚合物时,模板分子与功能单体的比例不同时,混合四模板物质的特异性吸附能力也不同,功能单体过多或者过少都会降低磁性分子印迹聚合物的特异性吸附能力。因为过多的功能单体会被无规则的固定在交联剂内,而导致非特异性识别位点增加使吸附性能降低;过少形成的识别位点少。现今大部分研究者利用静态吸附试验来确定最优比例,由于其存在试验周期长、工作量大等缺点,本试验采用紫外光谱法进行测定分析。
由图7中 a图可知,混合物的最大吸收波长发生了红移,原因为混合四模板物质与功能单体形成了氢键。当吸光值变化趋近于平缓时,这就说明体系在平缓区域所对应的混合物在这个范围内趋向于稳定,这时进入平缓区域的转折点所对应的混合物比例是最优的比例。
从图7中b图可知混合四模板物质与MAA在摩尔比为1:6(图中对应点为3)之后吸光值和波长变化比较平缓。由此可以推断混合四模板物质:MAA=1:6是聚合时最优浓度比。
实施例7 致孔剂选择试验
本实施例分别对混合四模板物质-MAA-乙腈体系、混合四模板物质-MAA-甲醇体系进行了紫外光谱扫描。
从图8a可知,在乙腈中,混合四模板物质与MAA浓度比增加会使溶液最大吸收波长发生红移,从203 nm红移至213 nm,红移量为10 nm;还可明显看出,随着MAA加入量增加,溶液的吸收强度有所增大。试验结果表明,混合四模板物质与MAA之间产生了较强的分子间作用力。
从图8b可知,在甲醇溶液中,随着混合四模板物质与MAA浓度比增加,溶液最大吸收波长也发生红移,从209 nm红移至213 nm,仅红移了4 nm;还可明显看出,MAA加入量增加使溶液的吸收强度也有所增强。试验也表明混合四模板物质与MAA之间产生了分子间作用力,但弱于在乙腈溶液中的作用力。综合上述,本试验选择乙腈作为致孔剂。
实施例8 致孔剂用量选择试验
致孔剂的用量对聚合物的制备具有关键作用,若溶剂量过低会导致聚合物交联紧密成为块状,即传统的本体聚合方式;溶剂量过高则导致聚合物交联度低不能形成微球沉淀。在其它合成条件不变的情况下,通过改变溶剂用量研究了其对分子印迹聚合物粒径及机械强度的影响。
由表2可以看出,随着致孔剂用量的增加,聚合物微球的粒径逐渐减小,机械强度也在逐渐降低。当乙腈用量过少时,体系生成块状聚合物(不属于沉淀聚合产物),虽然机械强度高,但聚合物需研磨才能使用,破坏了部分价键,进而降低了聚合物的识别性能。当乙腈用量为50 mL时,粒径均匀,机械强度较强,吸附性能良好。当乙腈的用量过大时,生成的聚合物性能较差,不符合要求。
实施例9 聚合温度选择试验
由表2结果可以得到,随着反应温度的升高,引发与聚合速率均加快,但是聚合温度过高时,体系容易发生爆聚,温度在65℃时,体系在8h左右即生成大量磁性分子印迹聚合物;60℃时,体系在10h左右出现大量磁性分子印迹聚合物;55℃时,反应24h以后,才生成较多的磁性分子印迹聚合物。在50℃下聚合,反应24h之后,反应液无明显颜色变化,无聚合物生成。温度越低,生成聚合物的识别性能越好;但是温度过低,无法达到引发剂AIBN的热分解温度,聚合反应将不能发生。
为了比较印迹效果,定义印迹因子 IF, IF=QMMIP/QMNIP注:QMMIP为磁性印迹聚合物吸附模板分子的量,QMNIP为磁性非印迹聚合物吸附模板分子的量。 其中 IF 值越大表明分子印迹的效果越好。
表2 致孔剂用量、聚合温度对聚合物吸附性能的影响
实施例10 不同比例的混合四模板物质试验
由表3试验数据分析得出,混合四模板物质比例为1.5:1:1.5:1时吸附量大而且印迹效果好。原因为当四种模板物质比例合适时,才能与功能单体最大限度的发生作用,形成更多的氢键,从而使聚合物具有更多的特异性吸附位点。
表3 混合四模板不同比例对聚合物吸附性能的影响
实施例11 混合四模板磁性印迹聚合物的表征
为了更好地说明实施例1中制备的混合四模板磁性印迹聚合物的结构特征,本实施例分别对Fe3O4、Fe3O4@SiO2、MMIP和MNIP进行红外光谱扫描,并对实施例1中⒃步骤得到的磁性分子印迹聚合物进行了透射电子显微镜(TEM)和磁响应性能测试。
⑴红外光谱表征
分别对Fe3O4、Fe3O4@ SiO2、MMIP和MNIP进行红外光谱扫描,结果分别如图9、图10、图11和图12所示。
由图9可见,572 cm-1为Fe-O的特征吸收峰,说明成功制备了Fe3O4粒子。图10中563 cm-1处为Fe-O的特征峰,除此之外,1097 cm-1处为Si-O-Si的反对称伸缩振动峰,794 cm-1和468 cm-1处为Si-O基团的对称伸缩振动峰,952 cm-1处为Si-O-H的弯曲振动吸收峰,这些特征峰的呈现说明SiO2 已成功包裹在Fe3O4粒子上。
图11为磁性印迹聚合物的红外谱图,3439 cm-1处为硅胶表面的O-H基团的伸缩振动峰,1096 cm-1处为Si-O-Si反对称伸缩振动峰,800 cm-1和462 cm-1处为Si-O基团的对称伸缩振动峰,950 cm-1处为Si-O-H的弯曲振动吸收峰,584 cm-1处为Fe-O的特征峰,除此之外,2989 cm-1处为饱和C-H的伸缩振动吸收峰,1735 cm-1处为C=O伸缩振动吸收峰, 1630 cm-1处对应的C=C伸缩振动吸收峰很小,说明交联剂和功能单体大部分进行了交联聚合反应,这些特征峰的呈现说明在包裹了SiO2的Fe3O4表面有聚合物生成。图12为磁性非印迹聚合物的红外光谱,该图中吸收峰位置与图11的吸收峰位置基本一致,但1735 cm-1处C=O伸缩振动吸收峰相对于1100 cm-1处Si-O-Si反对称伸缩振动峰,相对峰值大为减小,说明在磁性微球表面发生聚合的非印迹聚合物较少(试验过程中,非印迹聚合物的产率同样低于印迹聚合物)。结果表明,功能单体MAA与模板分子预聚合形成复合物时,在磁性微球表面有利于聚合物的聚合。磁性非印迹聚合物的吸收峰位置与图11磁性印迹聚合物的吸收峰位置基本一致,说明磁性印迹聚合物与磁性非印迹聚合物两种交联共聚物骨架结构的化学组成相同。经过红外光谱的测定结果,可以看到在包覆有SiO2的磁性Fe3O4载体的表面发生了印迹聚合反应。
⑵TEM表征
通过透射电子显微镜对实施例1中⒃步骤得到的磁性印迹聚合物进行了表征,结果如图13所示。
从图13中左图可以看出,Fe3O4为大小约10 nm的球形粒子,且有一定的团聚,说明制备所得Fe3O4为纳米级粒子。由图13中右图可见,印迹聚合物分散性较好,粒径约为100 nm,其粒径较Fe3O4粒子大很多,说明在Fe3O4表面确实印迹上了聚合物层。
⑶磁响应性能分析
为了考察印迹聚合物的磁响应性能,将其置于乙腈溶剂中,超声使其均匀分散,将比色管置于磁铁的一侧,每隔1 s拍照一次,记录其在外加磁场下与溶液的分离过程。
试验发现,磁性印迹聚合物在磁场作用下,能迅速聚集在与磁铁接触的右端管壁上,与磁铁接触10s时完全聚集在与磁铁接触的管壁上,溶液澄清。当撤走外加磁场后,聚合物粒子很快会重新分散在乙腈中,得到稳定的磁流体。试验结果表明,在乙腈溶剂中,磁性印迹聚合物具有良好的磁响应和再分散性能。
实施例12 混合四模板分子印迹聚合物的吸附性能研究
本实施例对实施例1制备的混合四模板磁性分子印迹聚合物进行了选择性吸附的研究。
⑴对模板物质及其结构类似物选择性吸附能力的测定
为了进一步研究实施例1制备的印迹聚合物对三唑类、菊酯类、三嗪类和氨基甲酸酯类物质的选择吸附性能,将实施例1制备的印迹聚合物及无模板分子的非印迹聚合物分散在含有三唑类、菊酯类、三嗪类和氨基甲酸酯类农药的一定浓度溶液中,研究其对模板物质及模板物质结构类似物的选择性吸附能力。
具体的结果见图14。
由图14可知,磁性分子印迹聚合物对11种农药均有吸附,吸附效果依次为灭多威>莠去津≈克百威>莠灭净>扑草净>联苯三唑醇>戊唑醇>烯唑醇>胺菊酯>三氟氯氰菊酯>四氟甲醚菊酯,说明多种物质存在时磁性分子印迹聚合物对其吸附性能存在差异,主要原因是11种农药的结构与分子量的大小不同。分子量最小的灭多威吸附量最大,莠去津与克百威同为模板分子,分子量又相差不多,因此吸附量相似,而四氟甲醚菊酯的分子量虽比模板分子三氟氯氰菊酯小,但因结构有差异,竞争性吸附过程四氟甲醚菊酯的吸附量小于三氟氯氰菊酯。
实施例13 混合四模板磁性分子印迹聚合物微球在小麦样品检测中的应用
⑴ 样品处理
① 样品的提取
以小麦样品为例,称取10.00 g小麦样品(来自超市),捣碎、研磨,用20 mL乙腈溶解,进行提取(分两次),搅匀,超声20 min,在1000 rpm的离心机上离心10 min,合并两次上清液,浓缩后,用乙腈定容至5 mL。
②样品的净化
吸取1 mL样品提取液,用磁性印迹聚合物进行萃取,用20 mL水淋洗,再用10 mL甲醇洗脱,10 mL洗脱液过0.45μm的微孔滤膜过滤,待高效液相色谱仪检测。
③检测波长的选择
11种农药的紫外光谱扫描图见图15。由图15可见,11种农药的最大吸收波长在199nm到230nm之间,综合考虑各物质及流动相溶剂的紫外吸收情况,检测波长范围选定为200~300nm。
④流动相的选择
流动相的组成与比例对样品的分离效果和分离时间有很大的影响。若以纯甲醇作为流动相,11种农药不能实现分离。于是向流动相中加入水,当甲醇:水=90:10(V/V)时,11种农药的分离有所改善,但仍未完全分离。当甲醇:水=80:20时,11种农药虽然可完全分离,但是分离时间过长,浪费时间。因此,采用梯度洗脱技术, 11种农药色谱分离图如图16所示。由图16看出,11种农药色谱峰尖锐对称,分离效果好、速度快。
色谱柱:安捷伦C18(4.6mm×150mm ,5μm);检测波长:200~300nm;流动相为甲醇-水,梯度洗脱程序见表4 ;柱温为25℃;进样量为20μL。
表4 梯度洗脱程序
其中,小麦样品提取液未经MMIP的高效液相色谱图如图17,小麦提取液经MMIP处理后的洗脱液的高效液相色谱图如图18所示,小麦提取液加标经MMIP处理后的洗脱液高效液相色谱图见图19。
磁性分子印迹萃取材料利用非共价键对目标物特异性吸附,通过淋洗将样品中杂质成分去除,然后用洗脱剂将目标物洗脱下来。从图17和18中对比可知,磁性分子印迹萃取材料对杂质不具有吸附能力,样品净化效果好。从图19可知,磁性分子印迹萃取材料只对模板物质及其结构类似物有吸附能力,表明聚合物从组成复杂的样品中特异性分离富集目标物的优越性能。
⑵线性关系与检测限
将灭多威、克百威、莠灭净、莠去津、扑草净、戊唑醇、联苯三唑醇、烯唑醇、胺菊酯、三氟氯氰菊酯和四氟甲醚菊酯分别配制成浓度为0.01μg/mL~200μg/mL的标准混合溶液,在上述色谱条件下测定,以峰面积为纵坐标,浓度为横坐标绘制标准曲线,结果见表5。
表5 线性关系与检测限
由表5可知,11种农药有良好的线性相关性。11种农药的线性范围在0.05~100 μg/mL之间,检出限(由S/N=3确定)为3~20 ng/mL之间,11种农药的线性相关系数在0.9996~0.9999之间。
⑵ 小麦、玉米、苹果、白菜中11种被测物的加标回收率试验
分别在小麦、玉米、苹果、白菜样品中同时加入含有一定量灭多威、克百威、莠灭净、莠去津、扑草净、戊唑醇、联苯三唑醇、烯唑醇、胺菊酯、三氟氯氰菊酯和四氟甲醚菊酯的标准品,进行样品提取,加标提取液在磁性微球萃取分离后洗脱,其中小麦、玉米、苹果、白菜样品提取液的制备与本实施例14中①的制备步骤相同,不同之处仅在于当小麦样品换为苹果、白菜样品时,样品捣碎后须加入5.000g无水硫酸钠,样品净化过程与本实施例14中②相同。
小麦、玉米、苹果、白菜样品在0.1 μg/mL和10 μg/mL2个添加水平下,进行加标回收率试验,分析结果如表6所示。结果表明,11种农药的平均回收率在82.3%~99.8%之间,相对标准偏差(RSD)在1.34%~3.54%之间(n=5)。由此说明制备的混合四模板磁性分子印迹聚合物对样品中三唑类、菊酯类、三嗪类和氨基甲酸酯类农药的回收率高,精密度好。
表6 样品回收率及精密度试验
实施例14-17 混合四模板磁性分子印迹聚合物微球的制备方法
实施例14-17 分别为一种混合四模板磁性分子印迹聚合物微球的制备方法,制备步骤以及相应的技术参数与实施例1相同,不同之处仅在于实施例14中的三唑类模板分子为戊唑醇,菊酯类模板分子为四氟甲醚菊酯,三嗪类模板分子为莠灭净,氨基甲酸脂类模板分子为灭多威;实施例15中的三唑类模板分子为烯唑醇,菊酯类模板分子为三氟氯氰菊酯,三嗪类模板分子为扑草净,氨基甲酸脂类模板分子为克百威;实施例16中的三唑类模板分子为联苯三唑醇,菊酯类模板分子为胺菊酯,三嗪类模板分子为莠去净,氨基甲酸脂类模板分子为克百威;实施例17中的三唑类模板分子为烯唑醇,菊酯类模板分子为四氟甲醚菊酯,三嗪类模板分子为莠去津,氨基甲酸脂类模板分子为灭多威。
实施例14-17中所制备的混合四模板磁性分子印迹聚合物的制备方法简单,过程易于控制,成本低,聚合物耐酸碱,且无需研磨和装柱,微球具有特异性的空间位点,可实现特异性吸附。
实施例1-5及实施例14-17,仅是本发明的较佳实施例而已,并非是对本发明所作的其它形式的限定,任何熟悉本专业的技术人员可能利用上述技术内容作为启示加以变更或改型为等同变化的等效实施例。但凡是未脱离本发明权利要求的技术实质,对以上实施例所作出的简单修改、等同变化与改型,仍属于本发明权利要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!