阿立哌唑光降解杂质的制备方法与流程
本发明涉及一种阿立哌唑光降解杂质的制备方法,属于药物合成技术领域。
背景技术:
阿立哌唑(Aripiprazole),化学名为7-[4-[4-(2,3-二氯苯基)-1-哌嗪基]丁氧基]-3,4-二氢-2(1H)-喹啉酮,由日本大冢制药株式会社研制,2002年11月在美国上市,属于第三代非典型抗精神病药物。阿立哌唑是第一种多巴胺系统稳定剂,对精神分裂症阳性和阴性症状均有显著疗效。。阿立哌唑用于治疗精神分裂症,能够显著改善这类精神分裂症状,但却没有其他抗精神病药常见的一些副作用,例如体重的增加和非自主性肌肉活动等。
阿立哌唑光降解杂质是指在光照的条件下降解产生的杂质,其结构如图所示:
与阿立哌唑相比,哌嗪环开环脱掉了两个碳原子。
阿立哌唑光降解杂质对阿立哌唑的深入研究具有重大意义,但现有技术中未见该杂质的合成路线的公开报道。
技术实现要素:
本发明的目的在于提供一种阿立哌唑光降解杂质的制备方法,其合成路线合理,原料易得,操作简单易行,所得目标产物的收率高,纯度高。
本发明所述的阿立哌唑光降解杂质的制备方法,反应路线为以下路线中的一种:
(a)以2,3-二氯苯胺为原料,与化合物Ⅴ反应生成化合物Ⅳ,然后化合物Ⅳ经过硝基还原生成化合物Ⅱ,化合物Ⅱ与化合物Ⅲ在乙腈和碳酸钾的体系中回流反应,得到目标产物化合物I;所述化合物Ⅴ中的A为Cl、Br、COOH、CHO或COCl基团;A’为NO2基团;n=1或2;
(b)以2,3-二氯苯胺为原料,与化合物Ⅴ反应生成化合物Ⅱ,化合物Ⅱ与化合物Ⅲ在乙腈和碳酸钾的体系中回流反应,得到目标产物化合物I;所述化合物Ⅴ中的A为Cl、Br、COOH、CHO或COCl基团;A’为NH2基团;n=1或2;
反应方程式如下:
所述化合物Ⅳ合成过程中采用催化剂和配体,或者采用还原剂和碱。
所述化合物Ⅳ合成过程中采用催化剂Pd2(dba)3、Pd(OAc)2、Pd(dppf)Cl2或CuO,采用配体P(t-Bu)3、BINAP、P(o-tolyl)3或Xantphos。
所述化合物Ⅳ合成过程中采用的溶剂为甲苯、二甲苯、1,4-二氧六环、吡啶、二氯甲烷、甲醇、乙醇、丙酮、四氢呋喃、乙酸乙酯、N,N-二甲基甲酰胺或二甲基亚砜中的一种或多种。
所述化合物Ⅳ合成过程中采用的还原剂为硼氢化钠、硼氢化钾、三乙酰基硼氢化钠、氰基硼氢化钠、三乙酰基硼氢化钾或氰基硼氢化钾。
所述化合物Ⅳ合成过程中采用的碱为碳酸钾、碳酸钠、碳酸铯、叔丁醇钾、叔丁醇钠或叔丁醇锂。
所述化合物Ⅱ合成过程中采用的还原剂为铁粉、钯炭、氢气或锌粉。
所述化合物Ⅱ与化合物Ⅲ合成化合物I过程中,采用的碳酸钾的用量为化合物Ⅱ的摩尔量的1-3倍。
所述化合物Ⅱ与化合物Ⅲ合成化合物I过程中,采用的碘化钠的用量为化合物Ⅱ的摩尔量的0.5-3倍。
具体的反应路线举例如下:
1、第一种反应路线:
在反应器中加入2,3-二氯苯胺、1-溴-2-硝基乙烷及叔丁醇钾和甲苯,在少量钯催化剂及配体的作用下,回流反应12h,得化合物Ⅳ;化合物Ⅳ经硝基还原生成化合物Ⅱ;化合物Ⅱ和Ⅲ在乙腈、碳酸钾的体系中回流反应4h,得化合物Ⅰ。
2、第二种反应路线:
在反应器中加入2,3-二氯苯胺、2-硝基乙醛及三乙酰基硼氢化钠和二氯甲烷,回流反应6h,再经硝基还原反应得化合物Ⅱ;化合物Ⅱ和Ⅲ在乙腈、碳酸钾的体系中回流反应4h,得化合物Ⅰ。
3、第三种反应路线:
在反应器中加入2,3-二氯苯胺、2-溴乙胺及碳酸钾、新制氧化铜和吡啶,回流反应12h,经柱层析(洗脱剂为二氯甲烷:甲醇=8:1)得化合物Ⅱ;化合物Ⅱ和Ⅲ在乙腈、碳酸钾的体系中回流反应4h,得化合物Ⅰ。
本发明与现有技术相比,具有以下有益效果:
本发明首次定向设计并合成阿立哌唑光降解杂质,该杂质可作为对照品供阿立哌唑质量研究中杂质的定性定量研究使用,控制原料药有关物质的含量,保证原料药的质量;本发明合成路线合理,原料易得,使用溶剂低毒价格低廉,操作方法简单步骤少,所得目标产物的收率高,纯度高。
附图说明
图1为化合物Ⅰ的核磁1H-NMR;
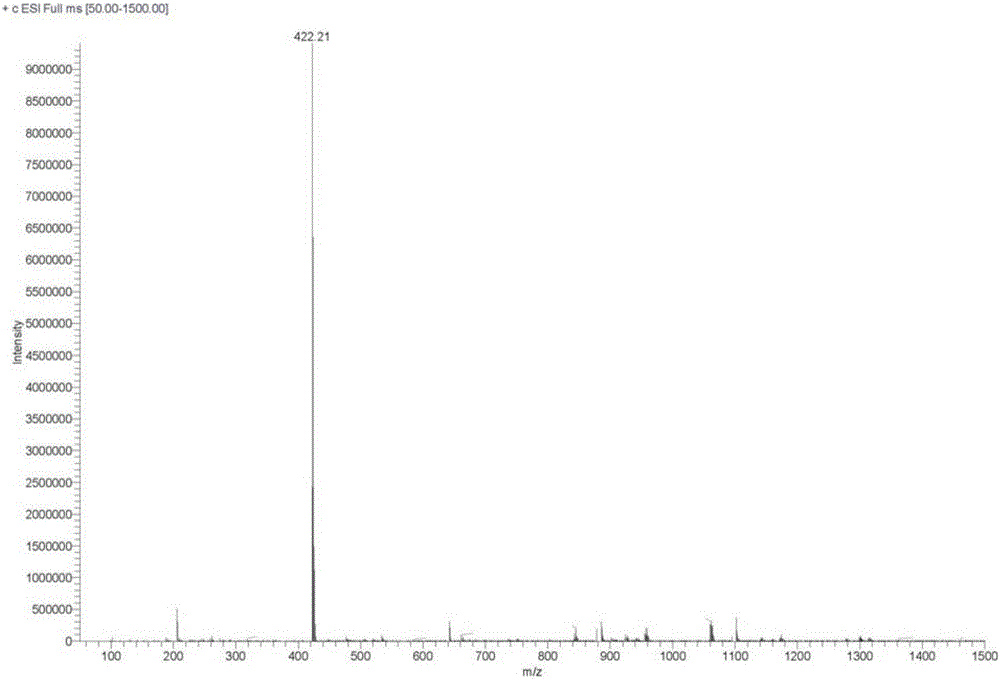
图2为化合物Ⅰ的质谱图;
图3为化合物Ⅰ的高效液相色谱图。
具体实施方式
下面结合实施例对本发明作进一步的说明,但其并不限制本发明的实施。
所用原料均为市售产品。
实施例1
(1)将16.2g2,3-二氯苯胺加入500mL反应瓶中,加入160mL甲苯溶解,搅拌下加入15.4g1-溴-2-硝基乙烷,再加入0.23g醋酸钯和1.2gBINAP,回流反应12h。将反应液冷却至室温,加入80mL甲基叔丁基醚稀释,然后抽滤,浓缩得粗产物,再经80mL乙酸乙酯和160mL精制得18.8g化合物Ⅳ,收率约为80%。
(2)取11.75g化合物Ⅳ,加入到250mL反应瓶中,加入30mL水和90mL乙醇,搅拌下加入11.2g铁粉和5.4g氯化铵,回流反应4h。然后趁热过滤,冷却降温过程中析出固体,抽滤,将所得滤饼用50mL乙醇打浆,抽滤,干燥,得8.7g化合物Ⅱ,收率约为85%。
(3)将6.2g化合物Ⅱ,9g化合物Ⅲ加入250mL反应瓶中,加入120mL乙腈,搅拌下加入8.3g碳酸钾和9g碘化钠,回流反应3h。然后冷至室温,有固体析出,抽滤,将滤饼加入到50mL水和50mL甲醇中打浆1h,抽滤,干燥得11.4g化合物Ⅰ,收率90%。
实施例2
(1)将16.2g2,3-二氯苯胺加入500mL反应瓶中,加入160mL二氯甲烷溶解,搅拌下加入8.9g硝基乙醛,再加入30g三乙酰基硼氢化钠,回流反应6h,加水淬灭反应。萃取,取有机层,加无水硫酸钠干燥,过滤,旋蒸,得19.9g化合物Ⅳ,收率85%。
(2)将14.1g化合物Ⅳ加入到250mL反应瓶中,加入140mL甲醇,搅拌下加入0.3g10%钯炭,在加氢气条件下,回流反应8h,热滤,旋蒸,得得11.1g化合物Ⅱ,收率约为90%。
(3)将6.2g化合物Ⅱ,9g化合物Ⅲ加入250mL反应瓶中,加入120mL乙腈,搅拌下加入4.15g碳酸钾和7g碘化钠,回流反应3h。然后冷至室温,有固体析出,抽滤,将滤饼加入到50mL水和50mL甲醇中打浆1h,抽滤,干燥得11.5g化合物Ⅰ,收率90%。
实施例3
(1)将16.2g2,3-二氯苯胺加入500mL反应瓶中,加入160mL吡啶溶解,搅拌下加入12.4g2-溴乙胺,再加入4g新制氧化铜和13.8g碳酸钾,回流反应12h。将反应液冷却至室温,然后抽滤,浓缩得粗产物,再经柱层析(洗脱剂为二氯甲烷:甲醇=8:1)得17.6g化合物Ⅱ,收率约为75%。
(2)将6.2g化合物Ⅱ,9g化合物Ⅲ加入250mL反应瓶中,加入120mL乙腈,搅拌下加入12.45g碳酸钾和42g碘化钠,回流反应3h。然后冷至室温,有固体析出,抽滤,将滤饼加入到50mL水和50mL甲醇中打浆1h,抽滤,干燥得11.2g化合物Ⅰ,收率88%。
化合物Ⅰ的核磁1H-NMR和质谱图测试结果如下:
1H-NMR(400MHz,DMSO-d6):δ10.039(s,1H),7.142-7.183(t,J=8Hz,1H),7.038(d,J=8Hz,1H),6.853(m,2H),6.495(m,2H),6.025-6.053(t,J=5.6Hz,1H),3.916(m,2H),3.368(m,2H),3.067(m,4H),2.762-2.799(t,J=7.2Hz,2H),2.394-2.431(t,J=7.6Hz,2H),1.788(br s,4H);
MS:(ESI)m/z:422.2。
化合物Ⅰ的纯度测定:
以乙腈-甲醇-水-乙酸(30:10:60:1)为稀释剂。取本品适量,精密称定,加稀释剂溶解并稀释制成每1ml中约含0.1mg的溶液作为供试品溶液;采用照高效液相色谱法测定。见表1。以YMC-Pack ODS-A(4.6×100mm,3μm)为色谱柱,以乙腈-0.05%三氟乙酸水溶液(10:90)为流动相A,乙腈-0.05%三氟乙酸水溶液(90:10)为流动相B;按下表进行梯度洗脱;流速1.2ml/min;检测波长为254nm;柱温为35℃。
表1化合物Ⅰ的纯度测定
精密量供试品溶液20μl,注入液相色谱仪,化合物Ⅰ的出峰时间为14.6min,记录色谱图见附图3。
制得的化合物Ⅰ的HPLC纯度检测为97.9%。
- 还没有人留言评论。精彩留言会获得点赞!